

Abstract
Calcineurin (CN), a calcium- and calmodulin-dependent protein phosphatase, plays a significant role in the central nervous system. Previously, we reported that forebrain-specific CN knockout mice (CN mutant mice) have impaired working memory. To further analyze the behavioral effects of CN deficiency, we subjected CN mutant mice to a comprehensive behavioral test battery. Mutant mice showed increased locomotor activity, decreased social interaction, and impairments in prepulse inhibition and latent inhibition. In addition, CN mutant mice displayed an increased response to the locomotor stimulating effects of MK-801. Collectively, the abnormalities of CN mutant mice are strikingly similar to those described for schizophrenia. We propose that alterations affecting CN signaling could comprise a contributing factor in schizophrenia pathogenesis.
Calcineurin (CN), also called protein phosphatase 2B, is a heterodimeric Ca2+/calmodulin-dependent serine/threonine protein phosphatase composed of CNB regulatory and CNA catalytic subunits (1). Originally identified in the brain, CN was later found to play a critical role in T cell function, through activation of nuclear factor of activated T cell-mediated transcription of cytokine genes, including the IL-2 gene (2, 3). This action of CN comprises a target for the immunosuppressants, cyclosporin A and FK506, which associate with immunophilins and bind to and inactivate CN. More recently, CN has been shown to play an important role in CNS functions, including neurite extension, synaptic plasticity and learning and memory (4, 5).
We previously reported a severe and specific working memory deficit of forebrain specific CNB-deficient mice (CN mutant mice) as assessed by delayed matching to place Morris water maze and eight-arm radial maze paradigms (5). To further investigate the behavioral significance of CN, CN mutant mice were subjected to a comprehensive behavioral test battery (6, 7). CN mutant mice display a spectrum of abnormalities that is strikingly similar to those observed in schizophrenia patients. In addition, a number of supporting lines of evidence are consistent with the possibility that alterations in CN function occur in schizophrenia. Furthermore, in our accompanying paper, we investigated the potential involvement of altered CN signaling in genetic susceptibility to schizophrenia, and we report evidence supporting association of the PPP3CC gene encoding the CNAγ catalytic subunit with disease (8). Based on these findings, we propose that alterations affecting CN signaling could comprise an important contributing factor in schizophrenia pathogenesis.
Materials and Methods
Animals and Experimental Design. The generation of the CN mutants is detailed elsewhere (5). The background strain used to generate the mutation was C57BL/6J. All mutant and control mice were in a pure C57BL/6J background. All CN mutant mice consisted of homozygous floxed, heterozygous Cre recombinase transgenic mice. All control mice consisted of homozygous or heterozygous floxed, Cre transgene negative littermates. All behavioral tests were carried out with male mice that were 10 weeks old at the start of the testing. Mice were housed in a room with a 12-hr light/dark cycle (lights on at 7:00 a.m.) with access to food and water ad libitum. Behavioral testing was performed between 9:00 a.m. and 5:00 p.m. except for the home cage social interaction test. All procedures relating to animal care and treatment conformed to Massachusetts Institute of Technology and National Institutes of Health guidelines. Sequences of tests, housing conditions, and the number of animals used are shown in Table 1, which is published as supporting information on the PNAS web site, www.pnas.org. The methods for rotarod, hot plate, light/dark transition, elevated plus maze, object exploration, social interaction (in a novel environment), Porsolt forced swim tests, in vivo microdialysis, and HPLC assessment of brain content of monoamines and metabolites are described in Supporting Text, which is published as supporting information on the PNAS web site.
Open Field Test. Each subject was placed in the center of an open field apparatus (40 × 40 × 30 cm; Accuscan Instruments, Columbus, OH). Total distance traveled (in cm), vertical activity, time spent in the center, and the beam–break counts for stereotyped behaviors were recorded. Data were collected over a 60-min period.
Social Interaction in Home Cage. To monitor social behavior between two mice in a familiar environment, a system that automatically analyzes social behavior in home cages of mice was developed (Fig. 7, which is published as supporting information on the PNAS web site). The system contains a home cage (29 × 18 × 12 cm) and a filtered cage top, separated by a 13-cm-high metal stand containing an infrared video camera, which is attached at the top of the stand. Two genetically identical mice that had been housed separately were placed together in a home cage. Their social behavior was then monitored for 3 days. Outputs from the video cameras were fed into a Macintosh computer. Images from each cage were captured at a rate of one frame per second. Social interaction was measured by counting the number of particles in each frame: two particles indicated the mice were not in contact with each other; and one particle demonstrated contact between the two mice. We also measured locomotor activity during these experiments by quantifying the number of pixels changed between each pair of successive frames. For details of the image analysis, see Supporting Text.
Latent Inhibition Test. This experiment was performed in a similar manner to that reported by others (9) with the equipment used by Zeng et al. (5). On the first day, each mouse was placed in a conditioning chamber (Coulbourn Instruments, Allentown, PA). The mice were divided into two groups: preexposed (P) group and non-preexposed (NP) group. The P group received 40 white noise tones (68 dB, 5-sec duration, 25-sec interstimulus interval), whereas the NP group received no stimulus during an equivalent period. Immediately after the tone preexposure or the exposure to the chamber, tone–shock pairs consisting of a 5-sec tone coterminating with a 2-sec foot shock at 0.40 mA were delivered to both groups with a 25-sec interstimulus interval. Afterward, mice remained in the chamber for 25 sec before being returned to the home cage. On day 2, the mice were placed back in the conditioning chamber for 5 min for the measurement of freezing to the context. On day 3, the mice were put in a white Plexiglas chamber scented with vanilla essence, and after 180 sec, a 180-sec tone was delivered to measure cued freezing.
Prepulse Inhibition Task. A startle reflex measurement system was used (MED Associates, St. Albans, VT) and the experiment was performed in a manner identical to that of Miyakawa et al. (7). See Supporting Text for details.
Sensitivity to MK-801 and Amphetamine. To test the locomotor activating effects of MK-801 or amphetamine (Sigma), mice were habituated to an open field for 1 h, and then drugs were administered i.p. Amphetamine or MK-801 were dissolved in saline and administered at 0.1 ml per 10 g of body weight. Because there was a difference in baseline locomotor activity between genotypes, the ratio of activity during the hour after injection to the activity during the hour before injection was used as an index of the locomotor activating effects of the drugs.
Quantification of Nesting. One piece of nesting material (Nestlets, Ancare, Bellmore, NY), made of cotton fiber, was introduced into a cage in which a mouse was individually housed. Pictures of the nests were taken by a digital camera (Olympus, Melville, NY) and exported into a computer. The number of scattered particles of the nestlets was counted for each cage by the NIH image program.
In Vivo Microdialysis. In vivo microdialysis measurements of extracellular dopamine and metabolites were performed in freely moving mice in an identical manner to that described in Mohn et al. (10). See Supporting Text for details.
Image Analysis. All applications used for the behavioral studies (Image SI, Image OE, Image LD4, Image PS, Image OF, Image HA, and Image FZ) were run on Macintosh computers. Applications were based on the public domain NIH image program (developed by Wayne Rasband at the National Institute of Mental Health, Bethesda) and were modified for each test by Tsuyoshi Miyakawa (7) (available through O'Hara & Co., Tokyo).
Statistical Analysis. Statistical analysis was conducted by using statview (SAS Institute, Cary, NC). Data were analyzed by two-tailed t test, ANOVA, or repeated-measures ANOVA. Values in graphs were expressed as mean ± SEM.
Results
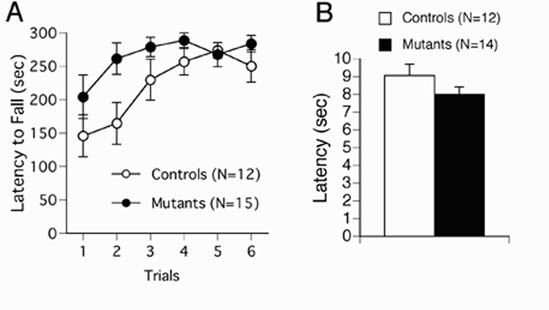
General Characteristics. CN mutants weighed ≈12% less than their wild-type littermates (controls, 30.3 ± 0.7; CN mutants, 26.6 ± 0.7; P < 0.01). There were no significant differences in motor coordination (accelerating rotarod test) and pain sensitivity (hot plate test) (Fig. 8 A and B, respectively, which is published as supporting information on the PNAS web site). Although CN mutant mice appear grossly normal and healthy, they are prone to sudden and acute deterioration of health leading to death within several days of onset, such that ≈50% of CN mutants die by 6 months of age. Although we cannot formally exclude a possible relation between this health effect and the behavioral abnormalities (see below), we do not believe it is a major contributing factor for several reasons. First, the onset of this deterioration is sudden and obvious, and therefore, affected mice were excluded from behavioral testing. Second, as previously reported, CN mutant mice perform normally in a number of behavioral tasks including the hidden platform version of the Morris water maze and cued and contextual fear conditioning (5). Thus, it is unlikely that specific behavioral abnormalities in these mice are a reflection of general health effects.
Increased Locomotor Activity. CN mutants consistently showed a pronounced increase in locomotor activity in several different tests. Total distance traveled by CN mutants was significantly greater than that of controls during an open field test (Fig. 1A; P < 0.0001), elevated plus maze test (Fig. 9E, which is published as supporting information on the PNAS web site; P < 0.0001), object exploration test (Fig. 10A, which is published as supporting information on the PNAS web site), social interaction test (Fig. 11A, which is published as supporting information on the PNAS web site), and in the home cage (Fig. 2B, P = 0.0082). The number of vertical activities in the open field test (Fig. 1B) and the stereotypy counts (Fig. 1C) were also significantly increased in the CN mutants relative to controls (P = 0.010, P = 0.001, respectively).
Fig. 1.

Increased locomotor activity of CN mutant mice in the open field test. Distance traveled (cm) (A), vertical activity (B), and stereotypic behavior counts (C) were significantly greater in CN mutants than in controls. Time spent in center (D) was shorter during the first 30 min in CN mutants relative to controls.
Fig. 2.

Decreased social interaction in home cage. Number of particles in home cage (A), an index of social activity, and activity level (B) are shown. (C and D) All frames taken during an hour were averaged. Control and CN mutants were active during the dark cycle (C; 11:00 p.m. to 12:00 p.m.). During the light cycle (D; 11:00 a.m. to 12:00 a.m.), control and CN mutants were inactive. During inactive periods, control mice stayed in contact with their cage mates, whereas mutant mice tended to stay separated from each other.
During the Porsolt forced swim test, there were no significant differences between genotypes in the distance traveled, although the time spent in immobile posture was significantly shorter in CN mutants than in controls (Fig. 10C), which may reflect general hyperactivity of CN mutants. The increase in activity or flight behavior of CN mutant mice complicated the assessment of their anxiety-like behavior (see Fig. 9 and Supporting Text for a discussion of abnormal anxiety-like behavior of CN mutant mice).
Decreased Social Interaction. During a 10-min social interaction test in a novel environment, the number of contacts between CN mutants did not differ significantly from that of control mice, except for the first minute, during which mutant mice made less contacts than controls (Fig. 11B; genotype × time interaction, P = 0.0031; genotype effect during the first minute, P = 0.0008). The total duration of contacts and the mean duration per contact of CN mutants were significantly shorter than those of control mice (Fig. 11 D and C; P = 0.0088 and P = 0.0005, respectively). However, because locomotor activity of the CN mice was increased in this test, it is possible that their decreased duration of contacts was a consequence of increased locomotion. To confirm that the decrease in social interaction was not simply the result of hyperactivity in CN mutants, we monitored social interaction in the home cage over a 3-day period. Overall, the time that CN mutants spent separated from each other was significantly greater than that of controls (Fig. 2A, P < 0.0001). In fact, even during the light cycle, when mice are usually sleeping, the time spent in separation was significantly greater in CN mutants compared with controls (Fig. 2 A, C, and D; P < 0.0001, P = 0.0037, and P = 0.0073 for light phase of day 2, day 3, and day 4, respectively). Overall, locomotor activity was also significantly greater in CN mutants (Fig. 2B).
Impaired Prepulse Inhibition. The startle amplitudes were not significantly different between genotypes at “startle stimulus only” trials for any stimulus amplitudes (Fig. 3A). The percent prepulse inhibition, an index of sensorimotor gating, was significantly lower in CN mutants than in controls (Fig. 3B; overall ANOVA including all four conditions, P = 0.0015). The difference between genotypes was greater when the startle stimulus intensity was 110 dB (P < 0.0001). At the stimulus intensity of 120 dB, a significant difference between genotypes was not observed (P = 0.0922), probably because of a ceiling effect caused by the strong intensity of the startle stimulus.
Fig. 3.

Impaired prepulse inhibition (A and B) and latent inhibition (C–F) of CN mutant mice. Startle amplitude was not different between genotypes (A), but the percentage prepulse inhibition was significantly smaller in CN mutants compared with controls (B). Percent freezing during the conditioning phase was significantly less in CN mutants (D) compared with controls (C), most likely due to their hyperactivity. CN mutants traveled longer distances in response to shocks than controls (Insets in D and C, respectively). Freezing during contextual testing was not significantly different between genotypes (C and D). In cued testing, the percent freezing for the P group was significantly lower than that of the NP group in control mice, indicating significant latent inhibition in control mice (E). In contrast, the CN mutants failed to show a significant latent inhibition (F).
Impaired Latent Inhibition. Percent immobility during the preshock period of the conditioning phase was significantly lower in mutant mice than in controls (Fig. 3 C and D; P = 0.0030), reflecting their hyperactivity. The percent freezing during the postshock period of the conditioning trial and during contextual testing were not statistically different between genotypes (P = 0.4721 and P = 0.6419, respectively). The distance traveled during shock presentations was significantly greater in the CN mutants compared with controls (Fig. 3 C and D Insets; P < 0.0001), probably because of their increased locomotor activity and flight behavior. In cued testing, the percent freezing for the P group was significantly lower than that of the NP group (Fig. 3E; P = 0.010) in control mice, indicating significant latent inhibition in control mice. In contrast, the CN mutants failed to show a significant latent inhibition (Fig. 3F, P = 0.9248). No significant difference in freezing between genotypes was observed in the pretone period of cued testing.
Impaired Nesting Behavior. We noticed, by casual observation, that nests of CN mutants were poorly formed. Normal mice usually form a clean and identifiable nest in a distinct location in the cage (Fig. 4 A and B). However, the CN mutants did not generally form distinguishable discrete nests and tended to scatter pieces of nesting material over the floor of the cage (Fig. 4 C and D). Therefore, pictures of the nests were taken and the number of scattered particles of nesting material was counted for each cage. The number of particles in the cages of CN mutants was significantly larger than that of controls (controls: 9.5 ± 2.3, CN mutants: 18.2 ± 3.2; P = 0.037).
Fig. 4.

Impaired nesting behavior. Representative pictures of the cages of control mice (A and B) and CN mutant mice (C and D).
Enhanced Sensitivity to the Locomotor Stimulatory Effect of MK-801. CN is activated after N-methyl-d-aspartate (NMDA) receptor activation and is a downstream element in dopaminergic signaling. To investigate the effects of NMDA receptor antagonism and dopamine receptor activation in the CN mutant mice, we assessed the locomotor stimulatory effects of a NMDA receptor blocker, MK-801, and an indirect dopaminergic agonist, amphetamine. CN mutants showed dramatically enhanced locomotor stimulatory effects of MK-801, a competitive blocker of the NMDA receptor (Fig. 5 C, D, and F; P < 0.0001 compared with control mice on ratio index). Interestingly, the dose response of MK-801 seems to be shifted, because 0.1 mg/kg of this drug did not affect wild-type mice, but potently activated mutant mice. In contrast, the locomotor stimulatory effects of amphetamine did not seem to differ between genotypes (Fig. 5 A, B, and E). Although CN mutants were “less sensitive” in terms of ratio index, this apparent decrease of ratio might be due to the increased baseline locomotor activity of CN mutants.
Fig. 5.

Locomotor activating effects of amphetamine and MK-801. Locomotor stimulation after amphetamine injection was observed in both genotypes (A and B), although CN mutants were less sensitive to amphetamine in terms of the ratio between activity before injection and activity after injection (E). On the other hand, the locomotor stimulatory effect of MK-801 was significantly greater in CN mutants compared with controls (C, D, and F; 7–10 animals were used for each group).
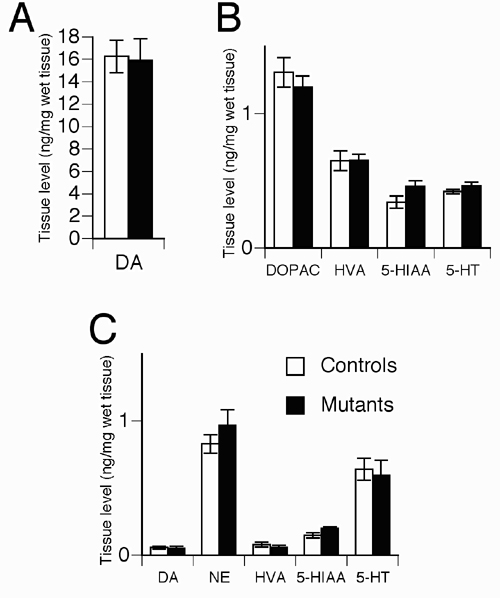
Normal Dopamine Release and Metabolism. Locomotor hyperactivity is typically associated with increased dopaminergic transmission in major motor brain areas such as the striatum. To assess the impact of CN mutation on the state of dopaminergic transmission, a detailed neurochemical analysis was performed. HPLC analysis of dopamine, serotonin, and metabolites did not detect any alterations in their tissue levels in the striatum or frontal cortex of mutant mice (Fig. 12, which is published as supporting information on the PNAS web site). Similarly, no difference in frontal cortex norepinephrine levels was observed (Fig. 12). Furthermore, a low perfusion rate quantitative microdialysis in freely moving mice did not reveal any significant difference in striatal extracellular levels of dopamine or its metabolites DOPAC and HVA (Fig. 6). Thus, locomotor hyperactivity and other aberrant behaviors in CN mutant mice are not associated with observable alterations in monoaminergic transmission.
Fig. 6.

Normal dopamine release. Despite their pronounced locomotor hyperactivity, CN mutants had no increase in either extracellular levels of dopamine (A) or its metabolites (B) in striatum compared with controls (n = 5 and n = 4, respectively).
Discussion
We have performed a comprehensive behavioral analysis of forebrain-specific CN mutant mice. These mice were found to have increased locomotor activity, decreased social interaction, impaired attentional function as assessed by prepulse (PPI) inhibition and latent inhibition (LI) tests and impaired nesting behavior. Previously, we reported a severe working memory deficit in these mutant mice (5). In addition to these innate behavioral abnormalities, CN mutant mice displayed increased sensitivity to the locomotor stimulatory effects of MK-801. Impairments in working memory (11, 12), PPI (13), LI (14), and social withdrawal (15) are prominent features of schizophrenia symptomatology. Hyperactivity is characteristic of rodent models of schizophrenia (16) and could correspond to psychomotor agitation present in schizophrenic patients. Furthermore, symptoms of schizophrenic patients are exacerbated by drugs with NMDA receptor antagonistic properties (17). Thus, the spectrum of abnormalities in CN mutant mice is strikingly similar to that observed in schizophrenia patients. These findings identify the CN mutant mice as a novel schizophrenia mouse model, and suggest that alterations in CN function could contribute to schizophrenia etiology.
Other Rodent Models. The behavioral abnormalities of the CN mutant mice are similar to those observed in a number of currently available rodent models that recapitulate certain aspects of schizophrenia, such as animals injected with indirect dopamine agonists (18, 19), dopamine transporter knockout and knockdown mice (16, 20), mice lacking Dvl1 (21), NMDA receptor knockdown mice (10), and mice carrying targeted point mutations in the glycine binding site of NMDA receptors (22). The CN mutant mice differ from other models in the regionally restricted nature of the lesion. In CN mutant mice, the gene encoding CNB is disrupted specifically in the cortex, hippocampal formation and amygdala, whereas expression in the basal ganglia, including the striatum, is intact. It is notable that this limited ablation leads to such a comprehensive spectrum of behavioral abnormalities, and supports the idea that higher brain regions such as the cortex could comprise a primary site of dysfunction in schizophrenia pathogenesis. Consistent with this notion, rats with neonatal hippocampal lesions have also been considered to comprise a rodent schizophrenia model (19, 23).
Relation to Glutamatergic and Dopaminergic Mechanisms. Our electrophysiological analysis of CN mutant mice has so far been limited to the hippocampus (5). In that study we found a deficiency in NMDA receptor-dependent long-term depression with a consequent decrease in the range of bidirectional synaptic modifiabilty. It is of interest to extend this analysis to determine whether similar electrophysiological deficits occur in the cortex. A major explanatory hypothesis implicating glutamatergic dysfunction in schizophrenia pathogenesis is based on the observation that compounds with NMDA-receptor antagonistic properties evoke behavioral abnormalities similar to those observed in schizophrenia patients (17). The nature of affected pathways downstream of the NMDA receptor remains to be determined. Our findings raise the interesting possibility that alterations in NMDA-receptor-dependent, CN-mediated phosphatase signaling could be of particular relevance.
Another explanatory hypothesis proposes that alterations in dopaminergic signaling comprise a major contributing factor in schizophrenia pathogenesis (18). More recently, this concept has evolved to propose that decreased cortical and increased mesolimbic dopaminergic transmission could account for various cognitive and behavioral manifestations of schizophrenia (24). One key function of CN is dephosphorylation of DARPP-32/inhibitor 1 leading to activation of protein phosphatase 1 (25). Because DARPP-32 phosphorylation is a consequence of dopamine D1 receptor activation, CN could in this way act as a brake in D1-mediated signaling. Thus, although striatal dopaminergic tone is normal in CN mutant mice, absence of CN activity in the amygdala, hippocampus, and entorhinal cortex could recapitulate some aspects of increased mesolimbic dopaminergic transmission. Moreover, because CN is activated by dopamine D2 receptor signaling (25), absence of CN could mimic certain aspects of decreased D2-mediated dopaminergic transmission. Because CN is involved in NMDA and dopamine receptor signaling pathways, its absence could disrupt critical interactions between the glutamatergic and dopaminergic neurotransmitter systems.
Normal Striatal Monoamine Levels and Response to Amphetamine. Although evidence for increased dopaminergic transmission in schizophrenia is not conclusive (26), several studies have detected increased striatal dopaminergic transmission in schizophrenia patients (27–29). Our analysis of monoamine levels revealed normal levels of dopamine and its metabolites in the striatum of CN mutant mice. There are several explanations for this possible inconsistency with schizophrenia pathophysiology. First, because schizophrenia is a complex genetic disease, it is unlikely that all behavioral and biochemical aspects would be recapitulated by a single genetic modification. For example, NMDA receptor knockdown mice also display a number of rodent correlates of schizophrenia symptomatology, and yet have normal striatal dopaminergic transmission (10). Second, unlike naturally occurring genetic variations, the mutation in these mice is restricted to the cortex, hippocampus, and amygdala, leaving CNB expression in the basal ganglia intact. It is possible that deletion of CNB in the striatum or elsewhere in the basal ganglia, particularly in the substantia nigra or ventral tegmentum, could lead to increased dopaminergic transmission or sensitivity to amphetamine. Such studies are of considerable interest.
The locomotor response to amphetamine in the CN mutant mice was smaller than that of controls when normalized to basal levels. However, it should be noted that the absolute level of locomoter activity is higher in CN mutant mice than controls after amphetamine administration.
Supportive Evidence. As CN is a key molecule in the immune system (3), the CN inhibitors, cyclosporin A and FK-506, are widely used as immunosuppressants. Psychotropic effects of these compounds including agitation, restlessness, anxiety, insomnia, confusion, visual/auditory hallucination, delusion, paranoia, depression, apathy, and flattened affect have been reported (30–32). In addition, the lower incidence of rheumatoid arthritis in schizophrenic patients (33) and a variety of comorbidities, including diabetes (34) and cardiovascular problems (35), are consistent with therapeutic (36) or side effects of CN inhibitors (3, 37, 38). Moreover, various immune abnormalities observed in schizophrenia, particularly alterations in levels of cytokines such as Il-2, are consistent with altered CN signaling (39, 40).
Recently, DNA microarray analysis was used to assay gene expression levels in postmortem brains of schizophrenic patients (41, 42). Expression of 10 genes with functions known to be related to CN was found to be altered (see Supporting Text for details). In addition, transcripts encoding proteins involved in the regulation of presynaptic function have been found to be decreased in postmortem brains of schizophrenic patients (41). CN is critically involved in presynaptic function of neurons (43–45). Caution is required in interpreting such studies as schizophrenic patients are often subject to chronic medication with possible effects on gene expression. In this regard, a recent study showed that administration of the antipsychotic, clozapine, induced an increased expression of the CNA gene in the prefrontal cortex (46) of rats. Clozapine and haloperidol have also been found to increase IL-2 induction (47), suggesting their possible activating effects on the CN signaling pathway. Our findings raise the possibility that antipsychotic drugs may exert their effects, at least in part, by modulating CN-signaling.
CN Genes and Schizophrenia Susceptibility. Based on the behavioral abnormalities observed in CN mutant mice, we have directly investigated the possibility that alterations in CN signaling could contribute to schizophrenia susceptibility. In the accompanying article (8), we report our initial analysis of potential association of genes encoding CN subunits and CN-interacting molecules with schizophrenia and provide direct genetic evidence supporting the association of the PPP3CC gene encoding the CNAγ catalytic subunit with disease.
Conclusion
We have assembled several lines of evidence indicating that altered CN signaling could comprise an important contributing factor in schizophrenia pathogenesis. Further analysis of the various pathways downstream of CN activity is of considerable interest. Application of conditional gene targeting technology should be particularly useful for this line of investigation. We have demonstrated that the strategy of using a comprehensive behavioral test battery on genetically engineered mice is a useful tool to identify schizophrenia susceptibility genes.
Supplementary Material
Acknowledgments
We thank Hiroki Hamanaka for his technical assistance, Takeshi Yagi for helping T.M. to develop home cage social interaction analysis system, and Junji Ichikawa and Akinori Nishi for insightful discussion. This research was supported by funding from National Institutes of Health Grant MH58880-03 (to S.T.), the Howard Hughes Medical Institute (S.T.), RIKEN (S.T.), and a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (to T.M.).
Abbreviations: CN, calcineurin; NMDA, N-methyl-d-aspartate; P, preexposed; NP, non-preexposed.
References
- 1.Rusnak, F. & Mertz, P. (2000) Physiol. Rev. 80, 1483-1521. [DOI] [PubMed] [Google Scholar]
- 2.Snyder, S. H., Sabatini, D. M., Lai, M. M., Steiner, J. P., Hamilton, G. S. & Suzdak, P. D. (1998) Trends Pharmacol. Sci. 19, 21-26. [DOI] [PubMed] [Google Scholar]
- 3.Crabtree, G. R. (2001) J. Biol. Chem. 276, 2313-2316. [DOI] [PubMed] [Google Scholar]
- 4.Winder, D. G. & Sweatt, J. D. (2001) Nat. Rev. Neurosci. 2, 461-474. [DOI] [PubMed] [Google Scholar]
- 5.Zeng, H., Chattarji, S., Barbarosie, M., Rondi-Reig, L., Philpot, B. D., Miyakawa, T., Bear, M. F. & Tonegawa, S. (2001) Cell 107, 617-629. [DOI] [PubMed] [Google Scholar]
- 6.Crawley, J. N. (2000) What's Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice (Wiley, New York).
- 7.Miyakawa, T., Yamada, M., Duttaroy, A. & Wess, J. (2001) J. Neurosci. 21, 5239-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerber, D. J., Hall, D., Miyakawa, T., Demars, S., Gogos, J. A., Karayiorgou, M. & Tonegawa, S. (2003) Proc. Natl. Acad. Sci. USA 100, 8993-8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldarone, B. J., Duman, C. H. & Picciotto, M. R. (2000) Neuropharmacology 39, 2779-2784. [DOI] [PubMed] [Google Scholar]
- 10.Mohn, A. R., Gainetdinov, R. R., Caron, M. G. & Koller, B. H. (1999) Cell 98, 427-436. [DOI] [PubMed] [Google Scholar]
- 11.Goldman-Rakic, P. S. (1994) J. Neuropsychiatry Clin. Neurosci. 6, 348-357. [DOI] [PubMed] [Google Scholar]
- 12.Elvevag, B. & Goldberg, T. E. (2000) Crit. Rev. Neurobiol. 14, 1-21. [PubMed] [Google Scholar]
- 13.Braff, D. L. & Geyer, M. A. (1990) Arch. Gen. Psychiatry 47, 181-188. [DOI] [PubMed] [Google Scholar]
- 14.Weiner, I. (2003) Psychopharmacology (Berlin), in press.
- 15.American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders (Am. Psychiatr. Assoc., Washington, DC), 4th Ed.
- 16.Gainetdinov, R. R., Mohn, A. R., Bohn, L. M. & Caron, M. G. (2001) Proc. Natl. Acad. Sci. USA 98, 11047-11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai, G. & Coyle, J. T. (2002) Annu. Rev. Pharmacol. Toxicol. 42, 165-179. [DOI] [PubMed] [Google Scholar]
- 18.Carlsson, A., Hansson, L. O., Waters, N. & Carlsson, M. L. (1997) Life Sci. 61, 75-94. [DOI] [PubMed] [Google Scholar]
- 19.Lipska, B. K. & Weinberger, D. R. (2000) Neuropsychopharmacology 23, 223-239. [DOI] [PubMed] [Google Scholar]
- 20.Giros, B., Jaber, M., Jones, S. R., Wightman, R. M. & Caron, M. G. (1996) Nature 379, 606-612. [DOI] [PubMed] [Google Scholar]
- 21.Lijam, N., Paylor, R., McDonald, M. P., Crawley, J. N., Deng, C. X., Herrup, K., Stevens, K. E., Maccaferri, G., McBain, C. J., Sussman, D. J. & Wynshaw-Boris, A. (1997) Cell 90, 895-905. [DOI] [PubMed] [Google Scholar]
- 22.Ballard, T. M., Pauly-Evers, M., Higgins, G. A., Ouagazzal, A. M., Mutel, V., Borroni, E., Kemp, J. A., Bluethmann, H. & Kew, J. N. (2002) J. Neurosci. 22, 6713-6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Amin, H. A., Weinberger, D. R. & Lipska, B. K. (2000) Behav. Pharmacol. 11, 269-278. [DOI] [PubMed] [Google Scholar]
- 24.Weinberger, D. R. (1987) Arch. Gen. Psychiatry 44, 660-669. [DOI] [PubMed] [Google Scholar]
- 25.Greengard, P., Allen, P. B. & Nairn, A. C. (1999) Neuron 23, 435-447. [DOI] [PubMed] [Google Scholar]
- 26.Carlsson, A., Waters, N., Holm-Waters, S., Tedroff, J., Nilsson, M. & Carlsson, M. L. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 237-260. [DOI] [PubMed] [Google Scholar]
- 27.Abi-Dargham, A., Rodenhiser, J., Printz, D., Zea-Ponce, Y., Gil, R., Kegeles, L. S., Weiss, R., Cooper, T. B., Mann, J. J., Van Heertum, R. L., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 8104-8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laruelle, M., Abi-Dargham, A., van Dyck, C. H., Gil, R., D'Souza, C. D., Erdos, J., McCance, E., Rosenblatt, W., Fingado, C., Zoghbi, S. S., et al. (1996) Proc. Natl. Acad. Sci. USA 93, 9235-9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breier, A., Su, T. P., Saunders, R., Carson, R. E., Kolachan, B. S., de Bartolomeis, A., Weinberger, D. R., Weisenfeld, N., Malhotra, A. K., Eckelman, W. C., et al. (1997) Proc. Natl. Acad. Sci. USA 94, 2569-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Groen, P. C., Aksamit, A. J., Rakela, J., Forbes, G. S. & Krom, R. A. (1987) N. Engl. J. Med. 317, 861-866. [DOI] [PubMed] [Google Scholar]
- 31.Craven, J. L. (1991) Psychosomatics 32, 94-102. [DOI] [PubMed] [Google Scholar]
- 32.Bechstein, W. O. (2000) Transpl. Int. 13, 313-326. [DOI] [PubMed] [Google Scholar]
- 33.Torrey, E. F. & Yolken, R. H. (2001) Brain Behav. Immun. 15, 401-410. [DOI] [PubMed] [Google Scholar]
- 34.Henderson, D. C. & Ettinger, E. R. (2002) Int. Rev. Neurobiol. 51, 481-501. [DOI] [PubMed] [Google Scholar]
- 35.Davidson, M. (2002) J. Clin. Psychiatry 63 (Suppl. 9), 5-11. [PubMed] [Google Scholar]
- 36.Cush, J. J., Tugwell, P., Weinblatt, M. & Yocum, D. (1999) J. Rheumatol. 26, 1176-1186. [PubMed] [Google Scholar]
- 37.Doyle, M. E. & Egan, J. M. (2003) Pharmacol. Rev. 55, 105-131. [DOI] [PubMed] [Google Scholar]
- 38.Olson, E. N. & Williams, R. S. (2000) Cell 101, 689-692. [DOI] [PubMed] [Google Scholar]
- 39.Hinze-Selch, D. & Pollmacher, T. (2001) Brain Behav. Immun. 15, 282-318. [DOI] [PubMed] [Google Scholar]
- 40.Rothermundt, M., Arolt, V. & Bayer, T. A. (2001) Brain Behav. Immun. 15, 319-339. [DOI] [PubMed] [Google Scholar]
- 41.Mirnics, K., Middleton, F. A., Marquez, A., Lewis, D. A. & Levitt, P. (2000) Neuron 28, 53-67. [DOI] [PubMed] [Google Scholar]
- 42.Hakak, Y., Walker, J. R., Li, C., Wong, W. H., Davis, K. L., Buxbaum, J. D., Haroutunian, V. & Fienberg, A. A. (2001) Proc. Natl. Acad. Sci. USA 98, 4746-4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakel, J. L. (1997) Trends Pharmacol. Sci. 18, 124-134. [DOI] [PubMed] [Google Scholar]
- 44.Lai, M. M., Hong, J. J., Ruggiero, A. M., Burnett, P. E., Slepnev, V. I., De Camilli, P. & Snyder, S. H. (1999) J. Biol. Chem. 274, 25963-25966. [DOI] [PubMed] [Google Scholar]
- 45.Cousin, M. A. & Robinson, P. J. (2001) Trends Neurosci. 24, 659-665. [DOI] [PubMed] [Google Scholar]
- 46.Kontkanen, O., Toronen, P., Lakso, M., Wong, G. & Castren, E. (2002) J. Neurochem. 83, 1043-1053. [DOI] [PubMed] [Google Scholar]
- 47.Rudolf, S., Peters, M., Rothermundt, M., Arolt, V. & Kirchner, H. (2002) Neuropsychobiology 46, 180-185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}