

Abstract
By comparing the low-, intermediate-, and high-frequency parts of the frequency spectrum, we gain information on the evolutionary forces that influence the pattern of polymorphism in population samples. We emphasize the high-frequency variants on which positive selection and negative (background) selection exhibit different effects. We propose a new estimator of θ (the product of effective population size and neutral mutation rate), θL, which is sensitive to the changes in high-frequency variants. The new θL allows us to revise Fay and Wu's H-test by normalization. To complement the existing statistics (the H-test and Tajima's D-test), we propose a new test, E, which relies on the difference between θL and Watterson's θW. We show that this test is most powerful in detecting the recovery phase after the loss of genetic diversity, which includes the postselective sweep phase. The sensitivities of these tests to (or robustness against) background selection and demographic changes are also considered. Overall, D and H in combination can be most effective in detecting positive selection while being insensitive to other perturbations. We thus propose a joint test, referred to as the DH test. Simulations indicate that DH is indeed sensitive primarily to directional selection and no other driving forces.
DETECTING the footprint of positive selection is an important task in evolutionary genetic studies. Many statistical tests have been proposed for this purpose. Some of them use only divergence data between species (see Nei and Kumar 2000 and Yang 2003 for reviews) while others use both divergence and polymorphism data (for example, Hudson et al. 1987; McDonald and Kreitman 1991; Bustamante et al. 2002). Those that rely only on polymorphism data can be classified into either “haplotype” tests (for example, Hudson et al. 1994; Sabeti et al. 2002; Voight et al. 2006) or the site-by-site “frequency spectrum” tests (for example, Tajima 1989; Fu and Li 1993).
The frequency spectrum is the distribution of the proportion of sites where the mutant is at frequency x and is the focus of this study. We may divide the spectrum broadly into three parts: the low-, intermediate-, and high-frequency variant classes. Comparing the three parts of the spectrum, we shall have a fuller view of the configuration of polymorphisms. Tajima's (1989) D and Fu and Li's (1993) D ask whether there are too few or many more rare variants than common ones. Fay and Wu's (2000) H takes into consideration the abundance of very high-frequency variants relative to the intermediate-frequency ones. Thus far, there is no test that addresses the relative abundance of the very high- and very low-frequency classes, although this comparison might be the most informative about new mutations. After all, new mutations are most likely to be found in the very low class and least abundant in the very high class.
In this article, we study the power to detect selection by applying spectrum tests to a linked neutral locus. In the first section, we briefly summarize some classical results of the frequency spectrum theory. In the second section, we introduce a new estimator of θ (θ = 4Nμ, where N is the effective population size and μ is the neutral mutation rate of the gene). On the basis of this new estimator, we revise Fay and Wu's H-test which was not normalized and introduce a new test (E) that contrasts the high- and low-frequency variants. We further propose a joint test, DH, which is a combination of D and H. In the rest of the article, we use computer simulation to compare the powers of the tests to detect positive selection. We also consider their sensitivities to balancing selection, background selection, and demographic factors.
GENERAL BACKGROUND
Let ξi be the number of segregating sites where the mutant type occurs i times in the sample. Following Fu (1995), we refer to the class of mutations with i occurrence as mutations of size i. Given the population mutation rate θ, Fu (1995) showed that
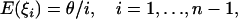 |
(1) |
for a sample of size n. Since each class of mutations contains information on θ, there can be many linear functions of ξi that are unbiased estimators of θ, depending on how each frequency class is weighted. A general form is
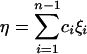 |
(2) |
with E(η) = θ, where ci's are weight constants. A few well-known examples are Watterson's (1975) θW, Tajima's (1983) θπ, Fu and Li's (1993) ξe, and Fay and Wu's (2000) θH:
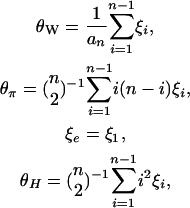 |
(3) |
where 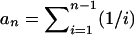 . Theoretical variances of these estimators are given in Figure 1. Among them, θW has the smallest variance.
. Theoretical variances of these estimators are given in Figure 1. Among them, θW has the smallest variance.
Figure 1.—

Variance of the five estimators of θ. Sample size (n) is 50.
Different estimators have varying sensitivities to changes in different parts of the frequency spectrum. For example, ξe and θW are sensitive to changes in low-frequency variants, θπ to changes in intermediate-frequency variants, and θH to high-frequency ones. When the level of variation is influenced by different population genetic forces, different parts of the spectrum are affected to different extents (Slatkin and Hudson 1991; Fu 1996, 1997; Fay and Wu 2000; Griffiths 2003). The difference between two θ-estimators can thus be informative about such forces. For example, rapid population growth tends to affect low-frequency variants more than it affects high-frequency ones. As a result, θW tends to be larger than θπ. The first test to take advantage of the differences between estimators is Tajima's D-test (Tajima 1989), as shown below:
 |
(4) |
Others have since been proposed (Fu and Li 1993; Fu 1996, 1997; Fay and Wu 2000). Among the evolutionary forces that may cause the frequency spectrum to deviate from the neutral equilibrium, hitchhiking (Maynard Smith and Haigh 1974) has attracted much attention. A salient feature of positive selection is the excess of high-frequency variants (Fay and Wu 2000).
It should be noted that θW and θπ can also be written as
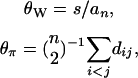 |
(5) |
where s is the number of segregating sites and dij is the number of differences between the ith and jth sequences. Equation 5 suggests that θW and θπ can be calculated without using an outgroup sequence to determine the mutant alleles. In contrast, ξe and θH do require knowledge of an outgroup sequence. As consequences, Tajima's D can be calculated without using an outgroup sequence, while the H-test needs one (see Fu and Li 1993 for a version of Fu and Li's D-test that uses ξe but requires no outgroup sequence in the calculation).
ANALYTICAL RESULTS
Measurements of variation based on very high-frequency variants—θH and θL:
To capture the dynamics of high-frequency variants, we need to put most weight on high-frequency variants in the estimation of θ. Fay and Wu (2000) used θH (3). Its variance term, however, is not easy to obtain. Here we propose a new estimator, θL. The variance of θL can be easily obtained and then used to calculate the variance of θH.
Consider a genealogy of n genes from a nonrecombining region. The mean number of mutations accumulated in each gene since the most recent common ancestor (MRCA) of the sample can be calculated as
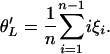 |
(6) |
Since E(ξi) = θ/i, it follows that
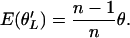 |
(7) |
Although 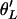 is an asymptotically unbiased estimator of θ, it is more convenient to work with its canonical form
is an asymptotically unbiased estimator of θ, it is more convenient to work with its canonical form
 |
(8) |
which has E(θL) = θ. It can be shown that
 |
(9) |
where 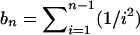 . Since i2 = n · i − i(n − i), it is easy to see that θH = 2θL − θπ. From this property, we obtain
. Since i2 = n · i − i(n − i), it is easy to see that θH = 2θL − θπ. From this property, we obtain
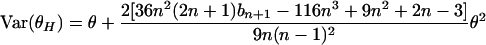 |
(10) |
(see appendix).
The normalized Fay and Wu's H-statistic—contrasting high- and intermediate-frequency variants:
Recall that Fay and Wu's (2000) H is defined as H = θπ − θH = 2(θπ − θL). Now we can write the normalized H-statistic as
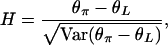 |
(11) |
where
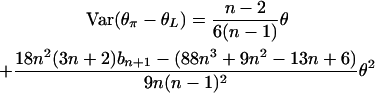 |
(12) |
(see appendix). In practice, θ in (12) can be estimated by θW, and θ2 can be estimated by 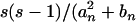 ) (Tajima 1989).
) (Tajima 1989).
A new E-test—contrasting high- and low-frequency variants:
As mentioned before, there is no test contrasting the low- and high-frequency parts of the spectrum. Both D and H use intermediate-frequency variants as a benchmark for comparison. Nevertheless, there are occasions when it is informative to contrast high- and low-frequency variants. For example, when most variants are lost (e.g., due to selective sweep), the recovery of the neutral equilibrium is most rapid for the low-frequency ones and slowest for the high-frequency ones (e.g., Figure 2B). Taking advantage of the newly derived θL, we propose a new test statistic
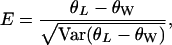 |
(13) |
where
 |
(14) |
(see appendix). In practice, θ and θ2 in (14) can be estimated by methods described after (12). Thus, the three tests, D, H, and E, contrast the three parts of the frequency spectrum, low-, intermediate-, and high-frequency variants, in a pairwise manner. By properly choosing a test statistic, we can have a better chance to detect a particular driving force (e.g., Table 3).
Figure 2.—

(A) Changes in 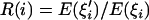 at the linked neutral locus as the advantageous mutation increases in frequency (f). (B) Changes in R(i) at different times τ (measured in units of 4N generations) after fixation of the advantageous mutation. In all simulations, the parameters are defined as follows: θ = 4Nμ, where μ is the mutation rate for the linked neutral locus; s is the selective coefficient of the advantageous mutation and c is the recombination distance (between the neutral variation under investigation and the advantageous mutation nearby), which is usually scaled by the selective coefficient. The parameter values are θ = 5, s = 0.001, c/s = 0.02, and sample size (n) is 50. In the simulation for hitchhiking, we also incorporated intragenic recombination among the neutral variants under investigation. The intragenic recombination rate of the neutral locus, multiplied by 4N, is 25 here and in Figure 3. The values of θ and intragenic recombination rate were chosen to reflect the reality of D. melanogaster; i.e., the scaled local recombination rate is about fivefold as large as the local population mutation rate. Intragenic recombination in other cases has a negligible effect on the results and was not incorporated.
at the linked neutral locus as the advantageous mutation increases in frequency (f). (B) Changes in R(i) at different times τ (measured in units of 4N generations) after fixation of the advantageous mutation. In all simulations, the parameters are defined as follows: θ = 4Nμ, where μ is the mutation rate for the linked neutral locus; s is the selective coefficient of the advantageous mutation and c is the recombination distance (between the neutral variation under investigation and the advantageous mutation nearby), which is usually scaled by the selective coefficient. The parameter values are θ = 5, s = 0.001, c/s = 0.02, and sample size (n) is 50. In the simulation for hitchhiking, we also incorporated intragenic recombination among the neutral variants under investigation. The intragenic recombination rate of the neutral locus, multiplied by 4N, is 25 here and in Figure 3. The values of θ and intragenic recombination rate were chosen to reflect the reality of D. melanogaster; i.e., the scaled local recombination rate is about fivefold as large as the local population mutation rate. Intragenic recombination in other cases has a negligible effect on the results and was not incorporated.
TABLE 3.
A qualitative summary of the powers of the four tests to detect various population genetic forces
| Driving force | D | H | E | DH |
|---|---|---|---|---|
| Positive selection (before fixation) | +++ | +++ | − | +++ |
| Positive selection (after fixation) | +++ | ++ | +++ | ++ |
| Balancing selection (initial stage) | ++ | +++ | − | +++ |
| Background selection | ++ | − | ++ | − |
| Population growth | +++ | − | +++ | − |
| Population shrinkage | + | ++ | − | + |
| Subdivision | + | ++ | − | + |
The results were drawn on the basis of Figures 3–6 and Table 2. All tests were one-sided; values falling into the lower 5% tail of the null distribution were considered significant. − denotes the lack of power under the specified condition. + denotes very weak power. ++ denotes moderately powerful. +++ denotes very powerful.
The DH test—a joint D and H test:
Simulation results (e.g., Table 3) suggest that both D and H are powerful in detecting selection, but they are sensitive to different demographic factors and are affected by background selection to different degrees. Therefore, one may conjecture that D and H in combination may be sensitive only to selection and less sensitive to other forces. For example, the sensitivity of D to population expansion may be counterbalanced by the insensitivity of H to the same factor in the joint DH test. To carry out the joint DH test, we first define
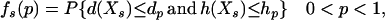 |
(15) |
where Xs is random samples (of size n) under neutrality with s (s ≥ 1) segregating sites, d(X) is the value of the D-statistic of sample X and h(·) is that of the normalized H-statistic, and dp and hp are critical values such that P{d(Xs) ≤ dp} = P{h(Xs) ≤ hp} = p. In other words, for a given s, when the significance levels of both D and H are p, that of the joint DH test is fs(p). That is, for samples with s segregating sites, the rejection region of the DH test at the significance level of fs(p) is {Xs | d(Xs) ≤ dp and h(Xs) ≤ hp}. In practice, for a given value of p, fs(p) can be evaluated using neutral simulations with the number of segregating sites s fixed (Hudson 1993). In our implementation, at least 50,000 rounds of simulations were carried out. To determine a value of p (and consequently dp and hp) such that the significance level of DH is, for example, 5%, we solved the equation fs(p) = 0.05 using the bisection algorithm (Press et al. 1992). We denote the solution as p*. We also denote dp and hp corresponding to p* as d* and h*, respectively. In Table 1, we give the p*-, d*-, and h*-values for three significance levels of DH [i.e., fs(p*)], given various observed values of s. In general, p* is close to three times the value of fs(p*).
TABLE 1.
Critical values of the D- and H-tests (d* and h*) when performed jointly as the DH test
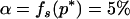
|
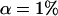
|

|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| s |  |
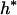 |
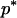 |
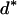 |
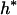 |
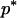 |
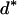 |
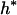 |
 |
| 10 | −0.971 | −0.535 | 0.176 | −1.468 | −1.721 | 0.053 | −1.872 | −3.302 | 0.010 |
| 20 | −1.054 | −0.766 | 0.145 | −1.536 | −2.022 | 0.039 | −1.982 | −3.537 | 0.006 |
| 30 | −1.084 | −0.820 | 0.135 | −1.568 | −2.061 | 0.034 | −1.995 | −3.644 | 0.005 |
| 40 | −1.109 | −0.867 | 0.128 | −1.562 | −2.071 | 0.033 | −2.019 | −3.489 | 0.005 |
| 50 | −1.107 | −0.885 | 0.125 | −1.579 | −2.133 | 0.030 | −2.062 | −3.656 | 0.003 |
Sample size is 50. s is the number of segregating sites in the sample. α is the significance level of the DH test. p* is the solution of the equation fs(p) = α; fs(·) is defined by (15). d* and h* are the critical values such that P{d(Xs) ≤ d*} = p* and P{h(Xs) ≤ h*} = p*, respectively; Xs is random samples under neutrality with s (s ≥ 1) segregating sites; d(·) is the value of the D-statistic of a sample and h(·) is that of the normalized H-statistic.
APPLICATIONS
Here we apply the tests discussed previously to data simulated under a variety of conditions for the purpose of evaluating their statistical powers. Such simulations have been done extensively in the past for studying existing tests (Braverman et al. 1995; Simonsen et al. 1995; Fu 1997; Przeworski 2002). Here all tests were one-sided and values falling into the lower 5% tail of the null distribution were considered significant. The reason for the one-sided test is that positive selection predictably shifts the pattern to one side of the null distribution. We report only the results of the normalized H-test, because it is always more powerful than the original one (varying from ∼1 to 8%). Details of the simulation algorithms and other related issues are described in the appendix.
The power to detect positive selection before and after fixation:
For the linked neutral locus, we define 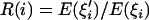 , where
, where 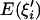 and
and 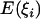 are the expected numbers of segregating sites of size i under selection and neutrality, respectively (Fu 1997). Figure 2 shows the dynamics of R(i) before and after the fixation of an advantageous mutation. The reduction in intermediate-frequency variants and the accumulation of high-frequency variants start at the early stage of the sweep (Figure 2A, f = 0.3; f is the population frequency of the advantageous mutation). When the frequency of the advantageous mutation reaches 60% in the population, the spectrum is already highly deviant from neutrality, losing many of the intermediate-frequency variants (Figure 2A, f = 0.6). By the time of fixation, the population has lost virtually all the intermediate-frequency variants, and mutations appear at either very high or very low frequency (Figure 2B, τ = 0; τ is time after fixation of the advantageous mutation and is measured in units of 4N generations). In neutral equilibrium, there should be 22.4 mutations in the sample on average for θ = 5 and n = 50. However, only 11.4 mutations, on average, are seen in the samples at τ = 0 (Figure 2B), among which 57% have a frequency <10%, and 31% have a frequency >80%. Strikingly, the mean number of mutations of size 49 is 7.2 times that of the neutral expectation. When τ > 0, these high-frequency variants quickly drift to fixation and no longer contribute to polymorphism. For a long period after fixation, fewer-than-expected numbers of intermediate- and high-frequency mutations are observed (Figure 2B).
are the expected numbers of segregating sites of size i under selection and neutrality, respectively (Fu 1997). Figure 2 shows the dynamics of R(i) before and after the fixation of an advantageous mutation. The reduction in intermediate-frequency variants and the accumulation of high-frequency variants start at the early stage of the sweep (Figure 2A, f = 0.3; f is the population frequency of the advantageous mutation). When the frequency of the advantageous mutation reaches 60% in the population, the spectrum is already highly deviant from neutrality, losing many of the intermediate-frequency variants (Figure 2A, f = 0.6). By the time of fixation, the population has lost virtually all the intermediate-frequency variants, and mutations appear at either very high or very low frequency (Figure 2B, τ = 0; τ is time after fixation of the advantageous mutation and is measured in units of 4N generations). In neutral equilibrium, there should be 22.4 mutations in the sample on average for θ = 5 and n = 50. However, only 11.4 mutations, on average, are seen in the samples at τ = 0 (Figure 2B), among which 57% have a frequency <10%, and 31% have a frequency >80%. Strikingly, the mean number of mutations of size 49 is 7.2 times that of the neutral expectation. When τ > 0, these high-frequency variants quickly drift to fixation and no longer contribute to polymorphism. For a long period after fixation, fewer-than-expected numbers of intermediate- and high-frequency mutations are observed (Figure 2B).
Figure 3 shows how the tests behave before and after the fixation of the advantageous mutation. Before fixation (Figure 3, A and B, left side), the power of D, H, and DH increases rapidly as the frequency of the advantageous mutation increases. H is generally more powerful than D in this stage. The behavior of DH resembles that of H, but note that it is often the most powerful test when the advantageous allele is in high frequency. We should note again that the purpose of the DH test is not to increase the power in detecting positive selection over either D or H but to decrease sensitivity to demographical factors and background selection. This is apparent in later sections. In contrast, the E-test has little power. This is expected as the reduction of moderate-frequency variants and the accumulation of high-frequency variants characterize this stage (Figure 2A). Sometimes, H, D, and DH reach their peak of power before fixation. This happens when too much variation is removed by the selective sweep, as is the case in Figure 3A. After fixation (Figure 3, A and B, right side) the E-test quickly becomes the most powerful test (τ ≈ 0.1 in Figure 3A; τ ≈ 0.15 in Figure 3B). From Figure 2B, we can see that after fixation the low-frequency part of the spectrum recovers first, and the high-frequency part returns to its equilibrium level last. The result of the E-test fits this observation. Furthermore, since the recovery phase is much longer than the selective phase when selection is strong, the E test can be useful for detecting sweep. The D-test performs reasonably well in the recovery stage. The power of the H-test, however, decreases quickly. As a consequence, the power of DH also decreases but at a much lower rate than that of H.
Figure 3.—

Power of the tests before and after hitchhiking is completed. The x-axis on the left represents the increase in the frequency of the advantageous mutation; on the right is the time after fixation (measured in units of 4N generations). All parameter values are the same as those of Figure 2. All tests were one-sided; values falling into the lower 5% tail of the null distribution were considered significant. Results shown in Figures 4–6 were produced by the same method. (A) c/s = 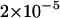 ; (B) c/s = 0.02.
; (B) c/s = 0.02.
The power to detect balancing selection:
Intuition would suggest that the results of an incomplete sweep (Figure 3) may also apply to the initial stage of balancing selection (i.e., a relatively short time period after the birth of the selected allele) under which the selected allele would reach an equilibrium frequency that is high, but <100%. Furthermore, as the selected allele does not reach fixation, the hitchhiking variants also may not have extreme frequencies. As a result, the signature of positive selection and hitchhiking may linger longer under balancing selection.
Simulation results (see supplemental Figure S1 at http://www.genetics.org/supplemental/) indeed suggest that in the phase when the selected allele is approaching the equilibrium frequency (75% in this case), the tests behave very much like the patterns in the left side of Figure 3. After the equilibrium is reached, the powers of H and DH also remain higher than in the right side of Figure 3 for a longer period of time. Nevertheless, the gain is modest as the power to detect selection generally diminishes when the selected allele stops increasing in frequency.
Sensitivity of the tests to other driving forces:
A good test of a particular population genetic force, say, positive selection, should be sensitive to that force and to that force only. Although a test that is sensitive to many different factors can be a useful general tool, it is ultimately uninformative about the true underlying force. Hence, power can in fact be a blessing in disguise. We examine the sensitivity of these tests to forces other than positive selection and balancing selection below.
Background selection:
Selection against linked deleterious mutations maintained by recurrent mutation, often referred to as background selection, can have effects on the level of genetic diversity similar to those of a selective sweep (Charlesworth et al. 1993). The distinction between selective sweep and background selection is therefore crucial. Fortunately, the two modes of selection often have very different effects on the frequency spectrum (Fu 1997). For example, background selection is not likely to have any effect on high-frequency variants (Fu 1997) and its effect on the low-frequency variants depends on U and N, where U is the deleterious mutation rate per diploid genome and N is the effective population size.
Table 2 summarizes the power of the tests to detect background selection. Both D and E are sensitive, but D is slightly more powerful than E. In general, the power increases as U increases, but this increase also depends on N. For a given U, the power of the tests decreases as N increases. H is not affected in all cases and hence is discriminatory between the two modes of selection. Significantly, DH is also not sensitive to background selection. It seems that the sensitivity of D is well counterbalanced by the insensitivity of H.
TABLE 2.
Powers of D, E, H, and DH to detect background selection
| U | N | θ | D | E | H | DH |
|---|---|---|---|---|---|---|
| 0.01 | 2,500 | 1 | 0.06 | 0.05 | 0.04 | 0.04 |
| 10 | 0.06 | 0.06 | 0.05 | 0.05 | ||
| 50,000 | 1 | 0.05 | 0.04 | 0.05 | 0.04 | |
| 10 | 0.05 | 0.06 | 0.05 | 0.06 | ||
| 0.1 | 2,500 | 1 | 0.08 | 0.02 | 0.04 | 0.01 |
| 10 | 0.34 | 0.34 | 0.02 | 0.09 | ||
| 5,000 | 1 | 0.06 | 0.01 | 0.04 | 0.01 | |
| 10 | 0.23 | 0.23 | 0.03 | 0.06 | ||
| 10,000 | 1 | 0.04 | 0.00 | 0.04 | 0.01 | |
| 10 | 0.15 | 0.14 | 0.03 | 0.06 | ||
| 20,000 | 1 | 0.02 | 0.00 | 0.04 | 0.00 | |
| 10 | 0.11 | 0.10 | 0.04 | 0.05 | ||
| 50,000 | 1 | 0.02 | 0.00 | 0.05 | 0.00 | |
| 10 | 0.07 | 0.06 | 0.05 | 0.04 | ||
| 0.2 | 2,500 | 1 | 0.09 | 0.02 | 0.02 | 0.01 |
| 10 | 0.60 | 0.55 | 0.02 | 0.07 | ||
| 5,000 | 1 | 0.07 | 0.00 | 0.02 | 0.00 | |
| 10 | 0.47 | 0.35 | 0.03 | 0.05 | ||
| 10,000 | 1 | 0.04 | 0.00 | 0.03 | 0.00 | |
| 10 | 0.35 | 0.18 | 0.02 | 0.03 | ||
| 20,000 | 1 | 0.02 | 0.00 | 0.03 | 0.00 | |
| 10 | 0.21 | 0.08 | 0.03 | 0.02 | ||
| 50,000 | 1 | 0.02 | 0.00 | 0.01 | 0.00 | |
| 10 | 0.11 | 0.02 | 0.03 | 0.01 |
Sample size is 50. A total of 5000 samples were simulated for each parameter set. All runs employed values of h (dominance coefficient) and s (selection coefficient) of 0.2 and 0.1; i.e., sh = 0.02. U is the deleterious mutation rate per diploid genome. N is the effective population size.
Population growth:
When a population increases in size, it tends to have an excess of low-frequency variants (Slatkin and Hudson 1991; Fu 1997; Griffiths 2003). Both D and E are sensitive to this type of deviation (Figure 4). E is the most sensitive test because high-frequency variants are the last to reach the new equilibrium after expansion. In contrast, H is unaffected. Strikingly, DH has little power in this case even though D exhibits tremendous power.
Figure 4.—

Sensitivity (or power) of the tests to population expansion. We assume that the effective population size increases 10-fold instantaneously at time 0 to θ = 5. Sample size (n) is 50. Time is measured in units of 4N generations.
Population shrinkage:
When the population decreases in size, the number of low-frequency variants tends to be smaller than that of the intermediate- and high-frequency ones (Fu 1996). Thus, H can be sensitive to population shrinkage, whereas D, E, and DH are largely unaffected (Figure 5).
Figure 5.—

Sensitivity (or power) of the tests to population shrinkage. We assume that the effective population size decreases 10-fold instantaneously at time 0 to θ = 2. Sample size (n) is 50. Time is measured in units of 4N generations.
Population subdivision:
When the population is structured, one commonly uses the fixation index FST as a measure of genetic differences among subpopulations. In the symmetric two-deme model, FST = 1/(1 + 16Nm), where m is the fraction of new migrants in the population and N is the population size of a deme (Nordborg 1997). When there is strong population subdivision (FST > 0.2 or 4Nm < 1, for example) and samples do not come equally from all subpopulations, the frequency spectrum is deviant from the neutral equilibrium (results not shown). Figure 6A gives an example where all genes are taken from one subpopulation. In this case, H is the most sensitive test. The D-test is sensitive only when the subdivision is strong (4Nm < 0.5 or FST > 0.33). DH has some sensitivity. Its power lies in between those of D and H, but never rises above 15%. Note that, for relatively mild population subdivisions with 4Nm > 2, none of these four tests is notably affected. Interestingly, E is completely insensitive to population subdivision. Figure 6B shows the effect of sampling on the power of the tests (4Nm = 0.1). The E-test again shows no sensitivity. Although the power of H can be as high as 55%, that of the DH test never goes above 14%. This result shows the effectiveness of using D and H in combination. However, when sampling becomes less and less biased, all tests become progressively less sensitive to population structure.
Figure 6.—

Sensitivity (or power) of the tests to population subdivision. A symmetric two-deme model with θ = 2 per deme (2N genes per deme) was simulated. Populations are assumed to be in drift–migration equilibrium with symmetric migration at a rate of m, which is the fraction of new migrants each generation. Sample size (n) is 50. (A) Sensitivity as a function of the degree of population subdivision, expressed as 4Nm on the x-axis. All genes were sampled from one subpopulation. (B) Sensitivity as a function of the sampling skewness; for example, 5/45 means 5 genes are sampled from one subpopulation and 45 from the other. In this case, 4Nm = 0.1, a value at which the tests show sensitivity to population subdivision in A.
CONCLUSION
We summarize our results in Table 3. It is clear that, among the four tests studied, DH is sensitive primarily to directional selection. In contrast, other tests are sensitive to two or more other driving forces. D is indeed a “general purpose” test as it is sensitive, to various extents, to all the processes we considered (Table 3). Being exclusive is a desirable property because it minimizes false positive results in the search for selectively favored mutations. However, no test is powerful in every stage of the selection process. Therefore, in practice, we should utilize a priori information (if available) from other sources to help us choose an appropriate test.
Acknowledgments
We thank J. Braverman and R. R. Hudson for their helpful discussions about the simulation algorithms. We also thank two anonymous reviewers for their constructive comments. K.Z. is supported by Sun Yat-sen University and the Kaisi Fund. S.S. is supported by grants from the National Natural Science Foundation of China (30230030, 30470119, 30300033, and 30500049). Y.-X. Fu is supported by National Institutes of Health (NIH) grants (GM 60777 and GM50428) and funding from Yunnan University, China. C.-I Wu is supported by NIH grants and an OOCS grant from the Chinese Academy of Sciences.
APPENDIX
Analytical results:
We list some useful properties of the θ-estimators (see Y.-X. Fu, unpublished data, for mathematical details):
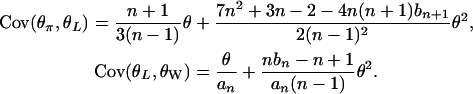 |
(A1) |
Since  , using (A1), we have
, using (A1), we have
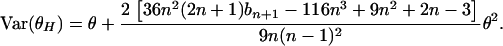 |
(A2) |
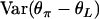 and
and 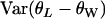 can be calculated using the above formulas.
can be calculated using the above formulas.
Simulation algorithms:
Positive selection:
We used the coalescent process with a selective phase (Kaplan et al. 1989; Stephan et al. 1992; Braverman et al. 1995) to simulate random samples and estimate the power of various tests. The model assumes that selection at the favored locus is additive and the selective pressure is strong (2Ns ≫ 1). In this case, the trajectory of the frequency of the advantageous allele in the interval with endpoints ε and 1 − ε is approximately deterministic (Stephan et al. 1992). We used ε = 1/(2N) in this study. Note that the choice of ε has little effect on the results (Braverman et al. 1995). In fact, the results were identical when we ran simulations with ε = 100/(2N) or ε = 1/(4N). Our implementation of the algorithm followed the description in Braverman et al. (1995), except for the following features: (1) we used a given value of the population mutation rate (θ = 4Nμ), rather than a fixed number of segregating sites, (2) we allowed intragenic recombination within the neutral locus to happen through the whole process (i.e., during both neutral and selective phases), (3) we assumed that only one round of selective sweep had occurred in the history of a sample rather than recurrent sweeps, and (4) the recombination distance between the selective locus and the neutral locus (c) was a given value rather than a random variable.
We extended the coalescent simulation algorithm described above to incorporate the case of incomplete hitchhiking. Suppose that the frequency of the favored allele is f. By solving the deterministic equation given by Stephan et al. (1992; Equation 1 in Braverman et al. 1995), one may obtain the time since the birth of this advantageous mutation and the corresponding frequency trajectory. In each replicate of simulation, we assigned the number of genes linked to the advantageous allele by binomial sampling with parameters n and f. Then the genealogy was constructed following the same procedure as described above.
Background selection and demographic scenarios:
We simulated background selection using the coalescent method (Hudson and Kaplan 1994; Charlesworth et al. 1995). The software package kindly provided by Hudson (2002) was used to simulate all demographic scenarios.
Critical values of the tests:
The tests, D, H, and E, were one-sided; values falling into the lower 5% tail were considered significant. The critical values of these three tests were determined by the Monte Carlo method used by Fu (1997). For a sample of size n, we first obtained an estimate 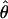 of
of 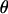 using Watterson's estimator. Then we ran 100,000 rounds of coalescent simulations with no recombination and
using Watterson's estimator. Then we ran 100,000 rounds of coalescent simulations with no recombination and 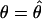 . The critical values were determined by examining the empirical distribution obtained. The supplemental table (at http://www.genetics.org/supplemental/) lists the critical values of the E-test for a number of θ-values. Another method to determine critical values was proposed by Hudson (1993). The differences between these two methods have been discussed extensively (Markovtsova et al. 2001; Wall and Hudson 2001). The results produced by both methods are quite similar; therefore, we report only those generated by the first method. The critical values of the DH test were generated using the second method.
. The critical values were determined by examining the empirical distribution obtained. The supplemental table (at http://www.genetics.org/supplemental/) lists the critical values of the E-test for a number of θ-values. Another method to determine critical values was proposed by Hudson (1993). The differences between these two methods have been discussed extensively (Markovtsova et al. 2001; Wall and Hudson 2001). The results produced by both methods are quite similar; therefore, we report only those generated by the first method. The critical values of the DH test were generated using the second method.
References
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140: 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante, C. D., R. Nielsen, S. A. Sawyer, K. M. Olsen, M. D. Purugganan et al., 2002. The cost of inbreeding in Arabidopsis. Nature 416: 531–534. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., B. Charlesworth and M. T. Morgan, 1995. The pattern of neutral molecular variation under the background selection model. Genetics 141: 1619–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1995. Statistical properties of segregating sites. Theor. Popul. Biol. 48: 172–197. [DOI] [PubMed] [Google Scholar]
- Fu, Y. X., 1996. New statistical tests of neutrality for DNA samples from a population. Genetics 143: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., 2003. The frequency spectrum of a mutation, and its age, in a general diffusion model. Theor. Popul. Biol. 64: 241–251. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1993. The how and why of generating gene genealogies, pp. 23–36 in Mechanisms of Molecular Evolution, edited by N. Takahata and A. G. Clark. Sinauer, Sunderland, MA.
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1994. Gene trees with background selection, pp. 140–153 in Non-Neutral Evolution: Theories and Molecular Data, edited by B. Golding. Chapman & Hall, London.
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., K. Bailey, D. Skarecky, J. Kwiatowski and F. J. Ayala, 1994. Evidence for positive selection in the superoxide dismutase (sod) region of Drosophila melanogaster. Genetics 136: 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovtsova, L., P. Marjoram and S. Tavaré, 2001. On a test of Depaulis and Veuille. Mol. Biol. Evol. 18: 1132–1133. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, New York.
- Nordborg, M., 1997. Structured coalescent processes on different time scales. Genetics 146: 1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 1992. Numerical Recipes in C: The Art of Scientific Computing. Cambridge University Press, Cambridge, UK.
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Simonsen, K. L., G. A. Churchill and C. F. Aquadro, 1995. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics 141: 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., and R. R. Hudson, 1991. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, W., T. H. E. Wiehe and M. W. Lenz, 1992. The effect of strongly selected substitutions on neutral polymorphism - analytical results based on diffusion-theory. Theor. Popul. Biol. 41: 237–254. [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J. D., and R. R. Hudson, 2001. Coalescent simulations and statistical tests of neutrality. Mol. Biol. Evol. 18: 1134–1135. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 2003. Adaptive molecular evolution, pp. 229–254 in Handbook of Statistical Genetics, edited by D. Balding, M. Bishop and C. Cannings. Wiley, New York.