

Abstract
Using information theory, it is argued that from among possible definitions of what an atom is when it is in a molecule, a particular one merits special attention. Namely, it is the atom defined by the “stockholders partitioning” of a molecule invented by Hirshfeld [(1977) Theor. Chim. Acta 44, 129]. The theoretical tool used is the minimum entropy deficiency principle (minimum missing information principle) of Kullback and Liebler [(1951) Ann. Math. Stat. 22, 79]. A corresponding analysis is given of the problem of assessing similarity between molecules or pieces of molecules.
Fundamental to chemistry is the understanding of molecules as combinations of atoms. It is not surprising, then, that the concept of atoms in molecules (AIM) has been much discussed in the literature (1). Chemistry mainly involves small changes among atoms and molecular fragments, with reasonably well-understood molecular invariants, e.g. AIM, functional groups, molecular subsystems, etc., which tend to maintain their identity. Most molecular systems may be thought of as consisting of slightly perturbed atoms (or atomic ions), possibly deformed by the presence of molecular remainders and exhibiting modified net charges arising from charge transfers and/or the formation of chemical bonds. These chemical atoms therefore are open subsystems.
The imposing variety of published theoretical methods for partitioning a molecular density into AIM contributions, e.g. refs. 1–15, testifies to the importance of this theme for chemistry. The different methods are based on different principles, some to a degree arbitrary (5) or heuristic (15), which can produce conflicting trends in the associated atomic net charges (effective oxidation states). Methods differ in the theoretical technique used, e.g., topological analysis of the density, wave-function description, or density-functional description. They also differ in the physical/heuristic principles invoked, e.g., electronegativity equalization, zero flux, and minimum-promotion energy rules. They can have specific disadvantages, e.g., basis set dependence. The well-known and appealing quantum-topological approach (1–4) suffers from the fact that its defined atomic densities are not “υ-representable.” [An atomic density is υ-representable when there exists an external potential that has this density as a ground-state density.]
Appropriate isolated atom densities constitute good references for defining properties of chemical atoms in terms of fast convergent Taylor series in the external potential and charge transfer displacements relative to the potentials and charges of the free atoms. The sum of the isolated atom/ion densities, with nuclear cusps at the actual positions of the nuclei, defines the density distribution of the promolecule (15). The promolecule is a key ingredient in the density difference analysis of the chemical bond done by Hirshfeld (15), in which the assumption is made that in forming a molecule each atom partakes of a local gain or loss in proportion to its local contribution to the promolecule density. In the present paper, we will recover this Hirshfeld “stockholder partitioning” of a molecular density into atomic components.
One would hope to find that a chemical atom, like its free analog, would possess a single cusp in its electron density, linked to the effective atomic number of the nucleus (16). AIM densities should be related to both promolecule and molecular densities, as representing the atomic fragments in a particular molecular system. One would want some degree of overlap between the densities of these chemical atoms, to reflect the presence of chemical bonds (1, 17, 18).
That atomic ground states, and/or small perturbations thereof, are uniquely appropriate reference states for a detailed description of AIM, was well understood by the pioneers, for instance Pauling and Mulliken, in their use of the concepts of hybridization, promotion, polarization, and ionic character. Ideally, an AIM definition will preserve as much information as possible about the separated atoms. [Here and later, the word atoms usually stands for atoms or ions.]
In defining AIM, how can one preserve, to the extent possible, the information content of ground-state atoms? It is natural to use some information—theoretic principle (19–26) for this purpose. And here is where density-functional theory (DFT) (27, 28) helps. For, DFT states that the electron density itself carries all of the information about a ground state. So, we may define AIM in a way that makes the atomic densities resemble as much as possible the isolated atom densities, and thereby achieve the “best” atoms we can have in a molecule in an information theoretic sense.
Kullback–Leibler Entropy Deficiency Functional
Suppose that P0(r⃗) is a given (reference) well-behaved probability distribution and that P(r⃗) is some trial probability distribution, the information content of which we want to make as close as possible to that of P0, subject to one or more constraints. Define (25) the entropy deficiency (missing information) functional by
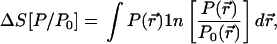 |
1 |
and let there be constraints of the form
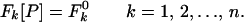 |
2 |
Then the P which is the “best” approximation to P0 that satisfies the constraints is obtained by solving the minimum entropy deficiency (missing information) principle
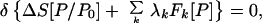 |
3 |
where {λk} are Lagrange multipliers. ΔS and the λk are found by solving Eqs. 2 and 3 simultaneously. A given Lagrange multiplier measures how sensitive the entropy deficiency is to the corresponding constraint. If there are several solutions for ΔS, one takes the minimum one.
This constrained entropy extremization (24) constitutes the most unbiased manner to assimilate (as completely as possible) the information included in the reference distribution and the auxiliary conditions. For specific P and P0, Eq. 1 gives a non-negative number, which defines the information distance between the two distributions, or the entropy deficiency of P relative to P0.
Example: To begin to approach the AIM problem, suppose that an accurate AB electron density is known, ρ, and that we want to represent it as the sum of two densities ρA and ρB, with electron numbers NA0 and NB0, both fixed, and that we want to keep ρA as close to ρA0 and ρB as close to ρB0 as possible. We take
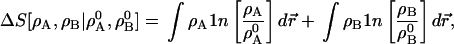 |
4 |
and the constraints
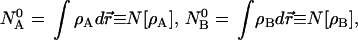 |
5 |
with the Lagrange multipliers λA and λB, respectively. Eqs. 2 and 3 then give
 |
6 |
 |
That is, if the “atoms” A and B are closed (not allowed to transfer electrons), entropy does not drive any density change. One will need to implicate electron transfer to define the atom in a molecule.
Derivation of the Hirshfeld AIM Stockholder Partitioning
An elementary modification of the example just given produces what we want, open atoms A and B, with electron numbers NA and NB not necessarily equal to NA0 and NB0 but still conforming with NA + NB = NA0 + NB0 = N0. We can require exhaustive allocation of the whole electronic distribution in AB to either A or B,
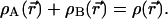 |
7 |
This constraint requires using a Lagrange multiplier λ(r⃗) that is a function of r⃗. Here ρ denotes the accurate (presumed known) density of the AB system, which is normalized to N0 electrons. Also normalized to N0 is the isoelectronic promolecule density ρ0 = ρA0 + ρB0. Implementing the minimum entropy deficiency principle of Eq. 3, with the constraint of Eq. 7, gives the equation for the AIM densities ρA and ρB,
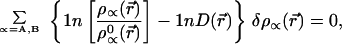 |
8 |
where 1n D(r⃗) = λ (r⃗) −1. Hence
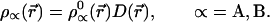 |
9 |
The proportionality factor D(r⃗) is determined from the constraint of Eq. 7:
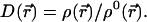 |
10 |
We therefore find
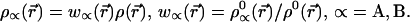 |
11 |
This is the “stockholder partition” of the electron density proposed long ago by Hirshfeld (7).
The sharing factor w∝(r⃗) determines the relative share of atom ∝ in the promolecule density ρ0(r⃗). The result is trivially generalized to any number of constituent atoms. The “atoms” could be neutral atoms, ions, or functional groups.
The Kullback–Liebler entropy deficiency minimization of Eq. 8 is seen to provide a solid theoretical basis for the Hirshfeld prescription, which is known to yield fairly transferable charge distributions and moments (15,30), which can be used e.g. in calculations of the electrostatic potential and the interaction energy. The AIM fragments defined by Eq. 11 have continuous but well-localized densities. This partitioning procedure is basis set independent and it can be used for either theoretical or experimental densities.
The AIM densities of Eq. 11 possess correct behavior at the separated (isolated) atoms limit, where all internuclear distances R∝β → ∞, because D(r⃗) → 1 when ρ(r⃗) → ρ0 (r⃗). The AIM densities fulfill, quite well through not perfectly, the proper nuclear cusp and long-range decay conditions. These densities are most probably υ-representable or at least ensemble υ-representable. [Recent unpublished work by Paul Ayers on the υ-representability problem implies that Hirshfeld atomic densities are either υ-representable or infinitely close to υrepresentable.]
The bonding factor D(r̄) of Eq. 10 indicates how the free atom density has been modified in the chemical atom. In the usual case where there is density accumulation in the molecular bonding region and depletion in the nonbonding regions of atoms bonded in a molecule, relative to ρ0, bonded atoms are polarized toward their bonding partners.
The chemical potentials of the bonded atoms each must be equal to the molecular chemical potential μ (8, 9, 31). The argument follows. For the fixed molecular external potential υ we have:
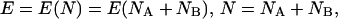 |
12 |
so that
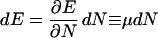 |
13 |
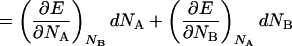 |
14 |
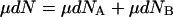 |
15 |
Subtracting Eq. 15 from Eq. 14 gives:
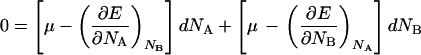 |
16 |
Therefore, if we define
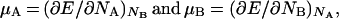 |
17 |
we find:
 |
18 |
AIM from a Modified Entropy Functional
To illustrate the many possibilities for extensions and variations of this analysis, we examine another entropy deficiency functional, in which in addition we include the information distance between ρ(r̄) and the resultant molecular density ρ̄A(r⃗) + ρ̄B (r⃗) ≡ ρ̄(r⃗), which is not fixed but is to be determined. The modified Eq. 4 becomes
 |
19 |
We minimize this subject to the constraint on the fixed overall number of electrons,
 |
 |
20 |
where λ is a global Lagrange multiplier. Setting 1nc = λ −2 we obtain the optimum AIM densities,
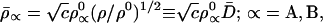 |
21 |
and the optimum overall density
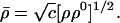 |
22 |
The proportionality constant can be determined from the global constraint in Eq. 20:
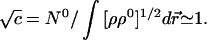 |
23 |
Thus, the variational principle of Eq. 20 gives the optimum molecular density ρ̄(r⃗), as the geometric mean of the promolecule and the true ground-state density of the molecule.
Comparison of Eq. 21 with Eqs. 9 and 10 indicates that this modified entropy deficiency minimization also gives chemical atoms, which are properly polarized toward the bonding region of a covalent bond. However, this effect is weaker in Eq. 21 because D̄(r⃗) = [D(r⃗)]1/2, where D(r⃗) (Eq. 10) stands for the bonding factor of the Hirshfeld stockholder atoms. Nevertheless, it is reassuring that the last “similarity” term in Eq. 19, which drives ρ̄ toward ρ, is seen to modify the promolecule density ρ0 in the qualitatively correct direction, toward ρ, thus generating a partial bonding in a molecule. The density ρ̄ is a transition density between ρ0 and ρ.
Both the Hirshfeld atoms of Eq. 11 and the modified chemical atoms of Eq. 21 represent open atomic fragments in a molecule, with the effective charges
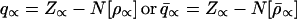 |
24 |
generally different from the free atom (ion) charge, q∝0 = Z∝ −N[ρ∝0], caused by the charge transfer component of the chemical bond. Equal chemical potentials do not generally obtain in this case, however.
Information-Theoretic Measure of Molecular Similarity
A molecular similarity concept sometimes is invoked to characterize structural resemblance of different molecules or their fragments. Measures that have been used have included overlap integrals for electronic density, electrostatic potential, and Fukui function (32). Here, we assume that similarity of electronic structure implies a closeness of the information content of electronic density distributions. This again calls for a measure of the information distance as provided by the entropy deficiency functional of Eq. 1.
In a typical screening search through candidate species with densities {ρi}, which are being tested for similar chemical activity to the reference system with density ρ0, one would select those with the minimum value of the entropy deficiency ΔS[ρi|ρ0]. When reactivity of a specific “active site” is required, the densities of the relevant molecular fragments should be chosen as the input information carriers.
The simplest variational criterion for the minimum entropy deficiency between the test molecular/fragment density ρi and the reference density ρ0 normalized to N[ρ0] = N0, subject to the normalization constraint N[ρi] = Ni0, would be
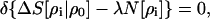 |
25 |
giving
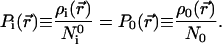 |
26 |
The most favorable matching therefore is achieved when the shape functions Pi(r⃗) and P0(r⃗) are as close as possible. This result is reminiscent of the recent extension of the Hohenberg–Kohn theorems (26) by Ayers (33), demonstrating that the shape function of the bound states of a molecule determines all properties of the system under consideration. Requiring the maximum amount of common information in ρi and ρ0 indeed calls for the same shape functions for the two compared densities.
As a final example, consider the optimum matching criterion of a trial electron density ρ, with two reference densities ρ10(N[ρ10] = N10) and ρ20(N[ρ20] = N20), subject to the usual normalization constraint, N[ρ] = N0:
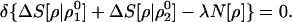 |
27 |
Solution of this equation gives
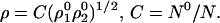 |
28 |
The optimum match is the normalized geometric mean.
One encounters such a problem in heterogeneous catalysis, when matching the adsorbate density (ρ) with the two surface active sites (ρ10, ρ20) involved in bonding the adsorbate. Another example is a molecule binding to two sites of another molecule. The maximum value of ρ(r⃗) is expected in the overlapping region between ρ10 and ρ20, because the least biased information distance criterion of Eq. 27 calls for ρ to resemble (appreciably overlap with) the overlap density between the two reference densities.
Summary and Concluding Remarks
In this paper we have used the information theory to decompose a molecular electron density into its component atomic densities. More particularly, we have selected the entropy deficiency (informative distance) approach of Kullback and Leibler (25), using the free atom densities as the reference distributions, to obtain AIM densities. Such a reference is both natural and unique in chemistry, because by the Hohenberg–Kohn theorem the isolated atom densities carry all the information of the periodic table, which is the fountainhead of chemical thought.
We have demonstrated that the minimum entropy deficiency (missing information) principle subject to the constraint of the exhaustive partitioning of the molecular density recovers the “stockholder” definition of bonded atoms done by Hirshfeld (15), thus providing it with a fundamental theoretical derivation. These Hirshfeld chemical atoms are unbiased pieces of the molecular density, which are the least distant in their information content from their isolated atom analogs. The chemical atoms have equalized chemical potentials at the global, molecular level, and they are probably υ-representable. Their overlap in a molecule accords with the familiar classical interpretation of the origin of the chemical bond (17). The quantum-topological atoms, in contrast, have zero overlap (1).
In an extension, we have demonstrated how the entropy deficiency functional can be used to generate the bonding character of chemical atoms. The resulting optimum density of a molecule then is given by the geometric mean of the true ground-state density and that of the promolecule (having free atom densities centered at the actual positions of nuclei in a molecule). Both the intermediate polarization (promoted) and final charge transfer stages of the atomic density reorganization in a molecule have been discussed. We also have illustrated the use of the entropy deficiency minimization principle in molecular similarity problems.
Finally, we note that the same ideas apply to excited electronic states. In this case, however, the reference, ground-state densities of free atoms might not be the ones that give the minimum value of the informative distance. Using the excited-state densities of separated atoms as reference densities may give more realistic excited-state (promoted) AIM densities, while still linking the resulting chemical atoms to the isolated atoms (periodic table) information. The information-theoretic approach should be helpful in quantitative studies of charge transfer and effective oxidation states.
Acknowledgments
We have had helpful discussions with several people, most importantly Paul Ayers, Richard Bader, Peter Politzer, and Vedene Smith. This research has been supported by a grant from the Committee for Scientific Research in Poland and by a grant from the Petroleum Research Fund of the American Chemical Society.
Abbreviation
- AIM
atoms in molecules
References
- 1.Bader R F W. An Introduction to the Electronic Structure of Atoms and Molecules. Toronto: Clarke; 1970. [Google Scholar]
- 2.Bader R F W. Atoms in Molecules. New York: Oxford; 1994. [Google Scholar]
- 3.Bader R F, Nguen-Dang T F. Adv Quant Chem. 1981;14:63–124. [Google Scholar]
- 4.Bader R F W, Becker P. Chem Phys Lett. 1998;148:452–458. [Google Scholar]
- 5.Moffitt W. Proc R Soc London Ser A. 1951;210:245–268. [Google Scholar]
- 6.Mulliken R S. J Chem Phys. 1935;3:573–585. [Google Scholar]
- 7.Mulliken R S. J Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 8.Parr R G, Donnelly R A, Levy M, Palke W E. J Chem Phys. 1978;68:3801–3807. [Google Scholar]
- 9.Parr R G. Int J Quantum Chem. 1984;26:687–692. [Google Scholar]
- 10.Rychlewski J, Parr R G. J Chem Phys. 1986;84:1696–1703. [Google Scholar]
- 11.Li L, Parr R G. J Chem Phys. 1986;84:1704–1711. [Google Scholar]
- 12.Bader R F W. J Chem Phys. 1986;85:3133. [Google Scholar]
- 13.Parr R G. J Chem Phys. 1986;85:3135. [Google Scholar]
- 14.Cedillo A, Chattaraj P K, Parr R G. Int J Quantum Chem. 2000;71:403–407. [Google Scholar]
- 15.Hirshfeld F L. Theo Chim Acta. 1977;44:129–138. [Google Scholar]
- 16.Kato T. Commun Pure Appl Math. 1957;10:151–171. [Google Scholar]
- 17.Ruedenberg K. Rev Mod Phys. 1962;34:326–376. [Google Scholar]
- 18.Feinberg M J, Ruedenberg K. J Chem Phys. 1971;54:1495–1511. [Google Scholar]
- 19.Shannon C F. Bell System Tech J. 1948;27:379–493. , 623–656. [Google Scholar]
- 20.Kullback S. Information Theory and Statistics. New York: Wiley; 1959. [Google Scholar]
- 21.Mathai A M, Rathie P N. Basic Concepts in Information Theory and Statistics. New York: Wiley; 1975. [Google Scholar]
- 22.Sears S B. Ph.D. thesis. Chapel Hill: University of North Carolina; 1980. [Google Scholar]
- 23.Sears S B, Parr R G, Dinur U. Isr J Chem. 1980;19:165–173. [Google Scholar]
- 24.Jaynes E T. Phys Rev. 1957;106:620–630. [Google Scholar]
- 25.Kullback K, Leibler R A. Ann Math Stat. 1951;22:79–86. [Google Scholar]
- 26.Esquivel R O, Rodriquez A L, Sagar R P, H, Smith V H., Jr Phys Rev A. 1996;54:259–265. doi: 10.1103/physreva.54.259. [DOI] [PubMed] [Google Scholar]
- 27.Hohenberg P, Kohn W. Phys Rev. 1959;136:B864–B871. [Google Scholar]
- 28.Parr R G, Yang W. Density Functional Theory of Atoms and Molecules. New York: Oxford; 1989. [Google Scholar]
- 29.Hõ M, Sagar R B, Schmider H, Weaver D F, Smith V H., Jr Int J Quantum Chem. 1995;53:627–633. [Google Scholar]
- 30.Brook C P, Dunitz J P, Hirshfeld F L. Acta Crystallogr B. 1991;47:789–797. [Google Scholar]
- 31.Sanderson R T. Science. 1951;114:670–672. doi: 10.1126/science.114.2973.670. [DOI] [PubMed] [Google Scholar]
- 32.Boon G, De Proft F, Langenaeker W, Geerliings P. Chem Phys Lett. 1998;295:122–128. [Google Scholar]
- 33.Ayers P. Proc Natl Acad Sci USA. 2000;97:1959–1964. doi: 10.1073/pnas.040539297. [DOI] [PMC free article] [PubMed] [Google Scholar]