

Abstract
Acetylation is a post-translational modification that influences the activity of numerous proteins in vitro. Among them, the myogenic transcription factor MyoD shows an increased transcriptional activity in vitro when acetylated on two lysines (K): lysines 99 and 102. Here, we have investigated the biological relevance of this acetylation in vivo. Using specific antibodies, we show that endogenous MyoD is acetylated on lysines 99 and 102 in myoblasts. Moreover, we show the functional importance of acetylation in live animals by using a mutant of MyoD in which lysines 99 and 102 were replaced by arginines (R). Knock-in embryos homozygous for the MyoDR99,102 allele expressed slightly reduced levels of MyoD but developed normally. However, the knock-in homozygous adult mice showed a phenotype that was almost identical to that of MyoD-knockout animals, including delayed muscle regeneration in vivo and an increased number of myoblasts but with reduced differentiation potential in vitro. Together, these results show the importance of MyoD acetylation for adult myogenesis.
Keywords: MyoD, acetylation, knock-in, muscle, regeneration
Introduction
Acetylation of lysines (K) in histones and non-histone proteins by histone acetyltransferases (HATs)—GCN5 (Brownell et al, 1996), PCAF (Yang et al, 1996) or CREB-binding protein (CBP)/p300 (Ogryzko et al, 1996)—is an essential process in transcriptional activation (Bayle & Crabtree, 1997). Non-histone protein acetylation can influence the stability or subcellular localization of proteins and activities such as DNA binding or transcriptional repression/activation (Soutoglou et al, 2000). Acetylation generally modifies the interaction between proteins and other components (Gu & Roeder, 1997; Kiernan et al, 1999; Martinez-Balbas et al, 2000; Polesskaya et al, 2000, 2001). The functional importance of non-histone protein acetylation has been extensively explored in vitro, but comparable in vivo data are lacking.
The myogenic transcription factor MyoD is a substrate for HATs in vitro (Sartorelli et al, 1999; Polesskaya et al, 2000). MyoD is a transcription factor of the basic helix–loop–helix (bHLH) family (Lassar et al, 1989) that converts non-muscle cells to myoblasts; it functions by transactivating muscle-specific promoters (Weintraub et al, 1991). MyoD is not essential for muscle formation during development in mice, and knockout embryos develop normally owing to compensation by overexpression of other members of the same family, such as myogenic factor 5 (Myf-5; Rudnicki et al, 1992). MyoD-null myoblasts, however, have a reduced differentiation potential in vitro (Sabourin et al, 1999). Moreover, adult MyoD-null mice show impaired skeletal muscle regeneration, indicating that MyoD is specifically required for this function (Megeney et al, 1996). In vitro, MyoD is acetylated by two HATs: CBP/p300 and PCAF (Sartorelli et al, 1999; Polesskaya et al, 2000; Polesskaya & Harel-Bellan, 2001). Acetylation occurs on lysines 99 and 102, located at the vicinity of the DNA-binding/dimerization domain—the bHLH domain. There is evidence indicating that acetylation of MyoD increases its affinity for the HAT CBP/p300 (Polesskaya et al, 2001). Acetylation of recombinant MyoD before its introduction into cultured myoblasts results in increased transcriptional activity, as monitored by using a reporter assay (Polesskaya et al, 2000). Conversely, mutation of lysines 99, 102 and 104 strongly reduces the myogenic activity of MyoD (Sartorelli et al, 1999; Polesskaya et al, 2000). Endogenous MyoD is acetylated in live cells (Sartorelli et al, 1999; Polesskaya et al, 2000), but the residues that are acetylated in vivo have not been characterized. We now show, for the first time, that acetylation of MyoD is important for skeletal muscle regeneration in vivo and terminal differentiation in vitro.
Results And Discussion
MyoD is acetylated on lysines 99 and 102 in live cells
Endogenous MyoD was previously shown to be acetylated on uncharacterized lysines in myoblast cell lines (Sartorelli et al, 1999; Polesskaya et al, 2000). In vitro, recombinant MyoD is acetylated by HATs on lysines 99 and 102, but so far the lysines acetylated in vivo have not been identified. To address this issue, we raised a polyclonal antibody that specifically recognizes MyoD acetylated on K99 and K102 but does not bind to non-acetylated MyoD (Fig 1) or to an irrelevant protein acetylated in vitro (supplementary information S1 online). This antibody was used to detect acetylated MyoD in extracts from myogenic cells (C2C12) or from the non-muscle cell line HeLa used as a negative control. Equivalent amounts of recombinant MyoD, non-acetylated or acetylated in vitro, were used as internal controls. The acetyl-MyoD antibody detected a protein that migrates at the position of MyoD in C2C12 extracts but which is absent from HeLa extracts. Together, these results show that MyoD is acetylated on lysines 99 and 102 in C2C12 cells.
Figure 1.
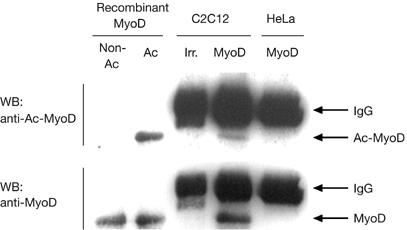
MyoD is acetylated on lysines 99 and 102 in myoblasts. Proteins were extracted from trichostatin A-treated cells, using either myoblasts (C2C12) or, as a negative control, non-muscle cells (HeLa). Extracts were immunoprecipitated using MyoD antibody (MyoD) or irrelevant antibodies (Irr.) and then analysed by western blot using purified anti-acetyl-K99,102-MyoD serum (WB: anti-Ac-MyoD) or anti-non-acetylated-MyoD (WB: anti-MyoD) to monitor the total level of MyoD. Recombinant MyoD, either mock acetylated (Non-Ac) or acetylated (Ac) in vitro, was run on the same gel. K, lysine.
MyoDR99,102 has reduced myogenic activity
We next addressed the functional importance of acetylation of MyoD by replacing lysines 99 and 102 by arginines (R). This lysine mutation does not affect the nuclear localization of MyoD, its DNA-binding capacity or its basic interaction with other proteins such as E12 and CBP (Polesskaya et al, 2000, 2001). The effect of the double mutation was evaluated using an in vitro myogenic assay. Totipotent mouse embryonic stem (ES) cells were chosen as recipients because a low-level expression of wild-type MyoD has been shown to convert 100% of the population to myogenesis (Dinsmore et al, 1996; see also Fig 2C). Stably transfected ES cells expressing similar amounts of MyoD, either wild type or mutant (Fig 2A), were placed under differentiation conditions. Expression of the early marker myogenin was monitored by reverse transcription–quantitative PCR (RT–Q-PCR; Fig 2B) and that of the late markers myosin heavy chain (MHC) and muscle creatine kinase (MCK) by immunofluorescence (Fig 2C) and western blot (Fig 2D). Expression of all differentiation markers was both delayed and reduced in ES-MyoDR99,102 cells as compared with their wild-type counterparts. These results confirm previously published data obtained with a triple mutant (Polesskaya et al, 2000), and show that mutation of lysines 99 and 102 is sufficient to significantly reduce MyoD activity.
Figure 2.
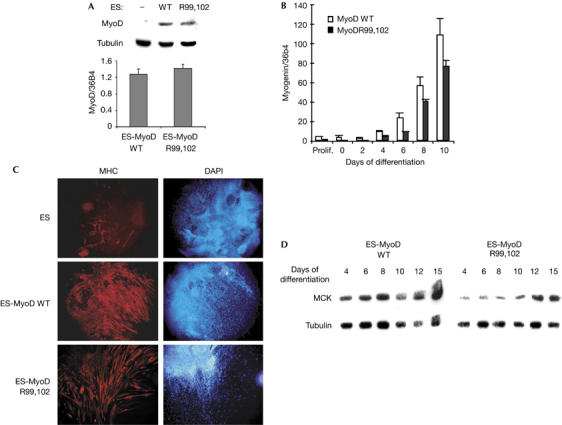
MyoDR99,102 has reduced myogenic activity. (A) MyoD expression in stably transfected embryonic stem (ES) cells. ES cells were stably transfected with plasmids expressing wild-type (WT) MyoD or MyoDR99,102 (R99,102) and analysed by western blot (upper panel) or by reverse transcription–quantitative PCR (RT–Q-PCR; lower panel). RT–Q-PCR results were normalized using the housekeeping messenger RNA 36B4; transcripts were quantified in triplicate; –, non-transfected. (B) Myogenin expression in converted ES cells. RNAs extracted from embryoid bodies expressing wild-type MyoD (MyoD WT) or MyoDR99,102 (MyoD R99,102) were analysed in triplicate by RT–Q-PCR and normalized on 36B4 mRNA as in (A). (C) Myogenic conversion of ES cells by wild-type MyoD (ES-MyoD WT) or MyoDR99,102 (ES-MyoD R99,102): ES cells were stably transfected with pEMSV-MyoD or left untreated for the controls, and placed under differentiation conditions. Embryoid bodies were analysed by immunofluorescence for myosin heavy chain (MHC) expression, and nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI). (D) Muscle creatine kinase (MCK) expression in converted ES cells. Proteins were extracted from embryoid bodies at different stages of differentiation and analysed by western blot. Tubulin is shown as a loading control. These experiments were carried out twice with similar results. R, arginine.
Establishing MyoDR99,102 knock-in mice
ES cells heterozygous for the MyoDR99,102 allele were created as described in the supplementary information S2 online. Mice heterozygous for the MyoDR99,102 allele were intercrossed, and the resulting newborn mice were genotyped by analysing MyoD intron I, the mutant allele of which is predicted to have an increased size. The proportion of homozygous mice was as expected, and this analysis did not show any embryonic or perinatal lethality associated with the mutant MyoDR99,102 allele. Homozygous or heterozygous mice carrying this allele developed into apparently normal adults, with normal weight gain (data not shown), reminiscent of MyoD-null mice, which likewise develop normally.
MyoDR99,102 homozygous embryos
The level of MyoD in embryos was evaluated by RT–Q-PCR. Results (Fig 3A) showed some individual variability between animals and suggested that, in homozygous mutants, MyoD expression was slightly reduced overall. RNAs produced from the wild-type and mutant alleles, in heterozygous embryos, were discriminated by an RT–PCR–restriction fragment length polymorphism (RFLP) assay in which PCR amplification products were subsequently digested with PstI (a PstI site is created by the MyoDR99,102 mutation; supplementary information S2 online). Specificity of the RT–PCR step is shown in Fig 3B (upper panel). Quantification of digestion products showed equal amounts of the two messenger RNA species, indicating equal expression of the two alleles and eliminating the possibility that MyoDR99,102 is impaired in its expression capacity (Fig 3B, middle and lower panels). Reduced levels of MyoD might thus reflect the existence of an autoregulatory loop for MyoD gene transcription: the non-acetylatable form of the protein is less efficient than the wild-type protein in transactivating target promoters; this is probably true for its own promoter as well.
Figure 3.
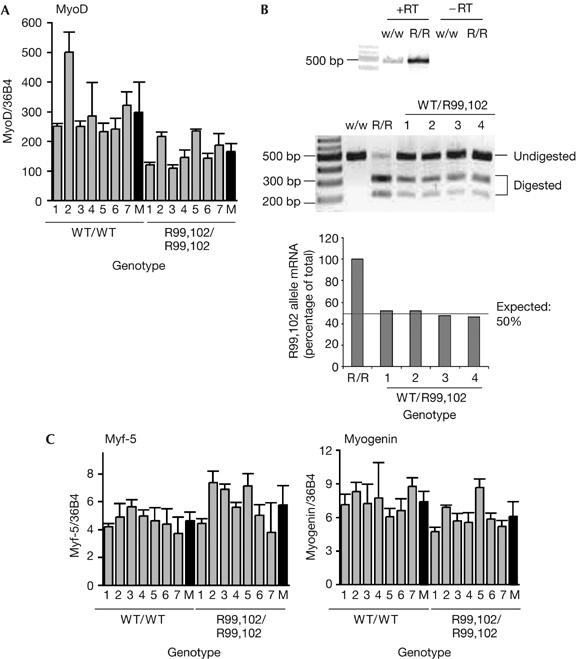
Expression of myogenic factors in embryos at embryonic day 14. (A) MyoD expression is reduced in MyoDR99,102 homozygous embryos (R99,102/R99,102). MyoD RNA was extracted from seven individual embryonic day (E) 14 embryos for each genotype, and MyoD was detected by reverse transcription–quantitative PCR (RT–Q-PCR); results are normalized with reference to 36B4 messenger RNA; M, mean values; WT, wild type; P<0.01. (B) MyoDR99,102 and WT MyoD are equally expressed in heterozygous embryos. RNAs from four wild-type (w/w), heterozygous (WT/R99,102) or homozygous MyoDR99,102 (R/R) E14 embryos were analysed by RT–PCR–restriction fragment length polymorphism; the upper panel shows that amplification is dependent on RT; the centre panel shows the patterns obtained after digestion with PstI (note that it is incomplete); the lower panel shows quantification of the gel using the Image Quant software; the proportion of MyoDR99,102 allele was evaluated by comparison with embryos homozygous for the MyoDR99,102 allele (R/R). (C) Expression of myogenic factors. RNAs from individual E14 embryos were extracted and myogenic factors analysed in triplicate by RT–Q-PCR; results were normalized using 36B4 RNA. P>0.05, Student's t-test. R, arginine.
Other muscle markers were analysed in homozygous embryos by RT–Q-PCR (Fig 3C). Myf-5 seemed to be slightly overexpressed in mutants but did not seem to reach the high levels observed in knockout mice; moreover, the difference was not statistically significant. MRF4 (data not shown) and myogenin (Fig 3C) expression was normal or close to normal.
Phenotypic analyses of MyoDR99,102 homozygous adults
To assess the functional impact of MyoDR99,102 mutation in vivo, we monitored the regeneration capacity of adult mice after injection of cardiotoxin. Embryonic MHC (eMHC) acted as a marker of regeneration: in adults, eMHC is re-expressed only in the regenerating muscle. The results indicated that eMHC expression was significantly delayed (3 days) in mutant mice (maximal expression between days 5 and 10) compared with wild-type mice (days 2–7; Fig 4A,B). By contrast, no significant difference was observed with regard to MyoD expression, showing that the defect in MyoD expression observed in embryos does not persist throughout the whole lifespan, and indicating a normal activation of myoblasts on the regeneration site. These data eliminate the possibility that decreased regeneration in adult MyoDR99,102 mice is due to reduced MyoD levels. Cross-sections of the muscles were analysed on day 2 by immunofluorescence for expression of eMHC (Fig 4C): eMHC-expressing cells were either differentiating individual myoblasts or cells fused to form new fibres and showing central nuclei; muscles from MyoDR99,102 mice showed a reduced number of fibres scoring positive for eMHC (Fig 4C,D). However, on day 15, no difference could be detected between the wild-type and mutant mice, and abnormalities such as fibrosis or fat infiltration were not observed (supplementary information S3 online). Moreover, the average size of mutant fibres was similar to that of wild-type fibres (data not shown). Together, these results indicate that regeneration was not abolished but was significantly delayed in these 7-week-old mutant mice.
Figure 4.
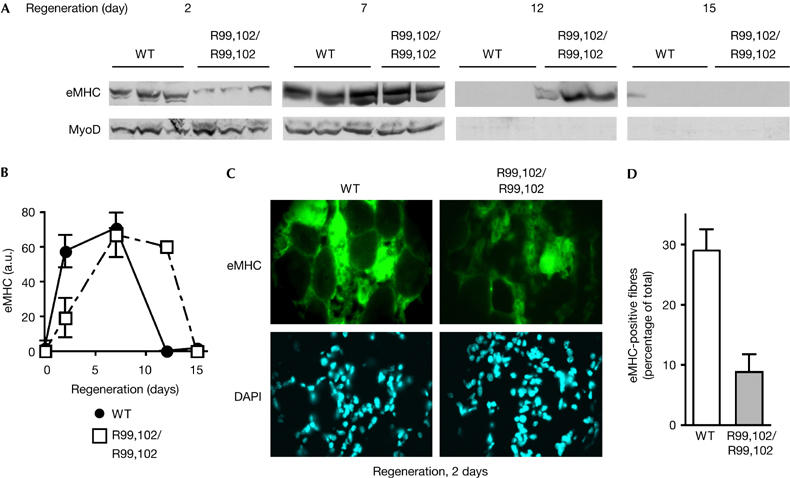
Delayed regeneration in mice homozygous for the MyoDR99,102 allele. (A) Tibialis anterior muscles of wild-type (WT) or MyoDR99,102 mice (R99,102/R99,102; the results of three individual mice, two for the 7-day time point in MyoDR99,102 mice owing to the accidental death of one animal) were injected with cardiotoxin, and regeneration was monitored at the indicated time points for a period of 15 days; embryonic myosin heavy chain (eMHC) and MyoD were detected by western blot. (B) Quantification of the western blots shown in (A) using Image Quant software. a.u., arbitrary units. (C,D) Immunohistochemical staining of cross-sections of regenerating tibialis anterior muscle with eMHC antibodies 2 days after cardiotoxin treatment. (D) Quantification of the results shown in (C). eMHC-positive fibres were counted on 15 cross-sections of three individual regenerating tibialis anterior muscles from WT and MyoDR99,102 mice. DAPI, 4,6-diamidino-2-phenylindole.
MyoDR99,102 mice had normal numbers of satellite cells, as assessed by paired-box protein 7 (Pax7) staining (data not shown) and previously observed for MyoD-knockout mice (Megeney et al, 1996). Although the yield of satellite-cell-derived myoblasts that can be obtained from skeletal muscle of MyoD-knockout mice is higher than that of wild-type controls, MyoD-knockout cells differentiate less efficiently than their wild-type counterparts. We found that, in MyoDR99,102 homozygous mice, the number of myoblasts derived from the muscles of hind leg, foreleg and diaphragm was two- to threefold higher than that of the wild-type controls (Fig 5A). However, when placed under differentiation conditions, myoblasts derived from the hind leg of MyoDR99,102 mice showed a strongly perturbed expression of early (myogenin) and late (MCK, MHC) differentiation markers (Fig 5B). In particular, in contrast to what happens in wild-type myoblasts, MyoDR99,102 levels decreased as soon as differentiation was triggered. In addition, the level of myogenin, which was higher in proliferating MyoDR99,102 myoblasts than in wild-type myoblasts, likewise decreased on differentiation, in contrast to the induction observed in normal cells. The levels of MHC and MCK were strongly decreased and only a few myotubes were formed, most likely as a consequence of these perturbations (Fig 5C). Moreover, with MyoDR99,102 myoblasts, the absolute number of cells decreased after several days under differentiation conditions, suggesting a high level of cell death in this population.
Figure 5.
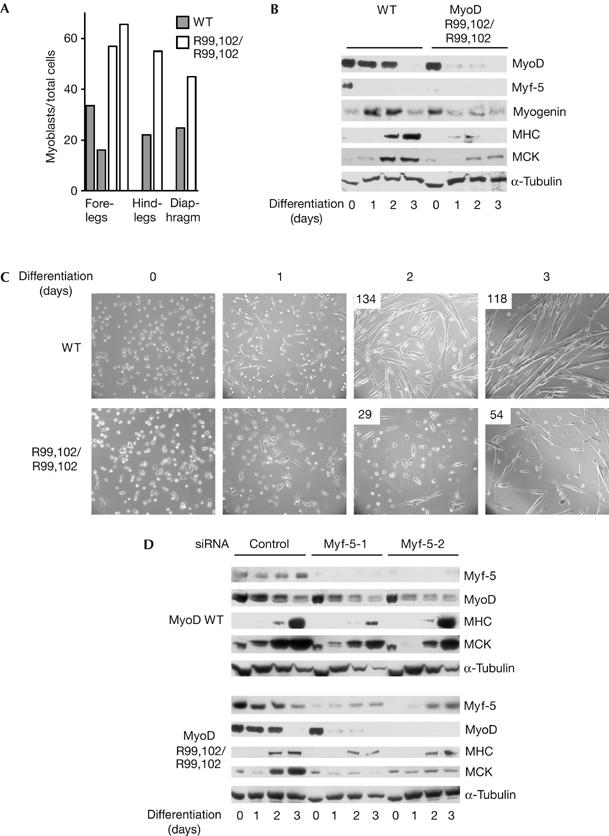
Increased proliferation and delayed differentiation of adult primary myoblasts derived from MyoDR99,102 mice. (A) Primary myoblasts were isolated from the indicated skeletal muscle of wild-type (WT) and MyoDR99,102 (R99,102/R99,102) mice. After 5 days in culture, cells were fixed and stained for MyoD and desmin. The percentage of MyoD/desmin-positive cells in the total number of nuclei from individual mice is presented. (B,C) Impaired differentiation of passage-3 myoblasts, from the muscles of the hind leg of WT and MyoDR99,102 mice, placed under differentiation conditions and monitored for 72 h. (B) Western blot; (C) phase contrast; upper left corner: number of myotubes per field. (D) Downregulation of Myf-5 further decreases differentiation. Passage-5 myoblasts from the muscles of the hind leg of WT and MyoDR99,102 mice were depleted of Myf-5 by RNA interference, placed under differentiating conditions and analysed by western blot as in (B); the results obtained with two distinct Myf-5 short interfering RNAs (siRNAs) or a control scrambled siRNA are shown. (Phase contrast of the same samples is given in supplementary information S4 online.) MCK, muscle creatine kinase; MHC, myosin heavy chain; R, Arg, arginine; α-Tubulin, used as a loading control.
Neither wild-type nor MyoDR99,102 myoblasts expressed high amounts of Myf-5. In both cell types, however, Myf-5 expression increased with the number of passages. This phenomenon did not restore the normal expression of muscle markers in differentiating MyoDR99,102 myoblasts (Fig 5D), suggesting that Myf-5 is not able to fully compensate for MyoD modification in these cells. However, Myf-5 did seem to affect the differentiation process, as its downregulation by two distinct short interfering RNA (siRNA) oligonucleotides further reduced differentiation efficiency (Fig 5D; supplementary information S4 online). Interestingly, differentiating myoblasts derived from the diaphragm of MyoDR99,102 mice also showed a marked defect in fusion, indicating the functional importance of MyoD acetylation in distinct skeletal muscles in vivo (supplementary information S5 online).
Together, our data support a model in which acetylation of MyoD serves as an important mechanism for enhancing its transcriptional activity both in vivo and in vitro. Regeneration of adult skeletal muscle is delayed in the absence of MyoD acetylation on lysines 99 and 102. Satellite-cell-derived primary myoblasts from the limbs and diaphragm of MyoDR99,102 mice are more abundant, but their differentiation is significantly reduced. Thus, MyoD acetylation is important for repression of proliferation, for myoblast fusion, and for muscle marker expression. However, myogenesis is not completely abolished by the introduction of non-acetylatable MyoD, which is consistent with previous in vitro studies in which acetylation of MyoD was shown to increase its transcriptional activity considerably, but was not indispensable for its basic function (Sartorelli et al, 1999; Polesskaya et al, 2000). Our data indicate the importance of non-histone protein acetylation in live mice.
Methods
Plasmids. A detailed description of plasmid construction is given in the supplementary information online.
Cell cultures. C2C12 and HeLa cells were maintained under standard conditions. The AT1 ES cell line was provided by Dr Muriel Vernet (CEA, Grenoble, France). ES cells were cultured on a feeder layer of inactivated primary mouse embryonic fibroblasts in standard medium supplemented with 100 μM β-mercaptoethanol (Sigma, St Quentin Fallavier, France) and 500 IU/ml leukaemia inhibitory factor (Esgro, Euromedex, Mundolsheim, France). ES-MyoD cell lines were established by electroporating ES cells with 15–60 μg of linearized wild-type pEMSV-MyoD or pEMSV-MyoDR99,102 together with 2 μg of linearized pGK-Neo and culturing under conditions of selection (geneticin 100–250 μg/ml) for 8 days. Embryoid bodies were generated according to Rohwedel et al (1994).
Primary myoblasts were obtained from the dissected muscle tissue, as described previously (Megeney et al, 1996), and cultured in Ham's media supplemented with 20% FCS and 2.5 ng/ml basic fibroblast growth factor (Invitrogen, Cergy-Pontoise, France) on collagen-coated dishes. Differentiation was induced by transferring to Dulbecco's modified Eagle medium supplemented with 5% horse serum (Invitrogen).
RNA interference assays on primary myoblasts were carried out using Hiperfect (Qiagen, Courtaboeuf, France) according to the manufacturer's protocol.
PCR and Southern blotting. All primers are described in the supplementary information online. DNA from ES cells, mouse tails or the yolk sac of embryos at embryonic day (E) 14 was prepared using standard procedures. Genomic PCRs were performed using Dynazyme Ext DNA polymerase (Finnzymes, Ozyme, St Quentin en Yvelines, France) according to the manufacturer's recommendations, with the addition of 1% dimethylsulphoxide.
Southern blot analyses were carried out using standard procedures, and genomic DNA (5–10 mg) was digested with 15 U of restriction enzymes overnight. Probe A corresponds to the NcoI–XhoI fragment (1.1 kb) adjacent to the 5′ flanking region of the transgene. Probe B is a 1 kb fragment corresponding to the −600 to +450 region. Probe C corresponds to the BamHI–EcoRI fragment (1.2 kb) located 700 bp downstream of the 3′ flanking region.
RT–PCR–RFLP, RT–Q-PCR and RNA interference. RNA was isolated from ES cells, embryoid bodies or E14 embryos using TRIzol® Reagent (Invitrogen) according to the manufacturer's recommendations and then treated with DNase RQ1 (Promega, Lyon, France).
For RT–PCR–RFLP, RT was performed using Superscript III (Invitrogen). PCR products were digested with PstI (New England Biologicals, Ozyme, St Quentin en Yvelines, France).
For RT–Q-PCR, RT was performed as described above, followed by quantitative PCR on a LightCycler (Roche Diagnostics, Meylan, France).
SiRNA oligonucleotides were obtained from Qiagen. Myf-5 siRNA sequences are given in the supplementary information online.
Immunoprecipitation and western blotting. Immunoprecipitations were carried out on 10 mg of whole-cell lysates using standard procedures. For detection of acetylated MyoD in C2C12, cells were treated with a deacetylase inhibitor, trichostatin A (Sigma), at 50 nM for 15 h before lysis. The rabbit antibody against acetylated MyoD was raised using a mouse MyoD peptide containing acetylated lysines at positions 99 and 102 (AC-acK-AC-acK-RKTTNADRR) and coupled to diphtheria toxin. Total IgGs were purified from the serum on a protein-G–Sepharose column (Mab-trap kit, Amersham Biosciences, Orsay, France). Antibodies against acetyl lysine and non-acetylated MyoD were eliminated by adsorption onto non-acetylated recombinant MyoD, followed by an irrelevant peptide containing one acetylated lysine, both coupled to cyanogen–bromide-activated beads. Specificity was verified by western blot. Other antibodies used for western blot were specific for MyoD (C20, Santa Cruz, Heidelberg, Germany), α-tubulin (DM 1A, Sigma), MCK (gift from K. Kato, Aichi, Japan), eMHC (F1.652, Stanford University School of Medicine, Stanford, CA, USA), or horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Sigma).
Immunofluorescence. Embryoid bodies were analysed by immunofluorescence using standard procedures after fixation in 4% paraformaldehyde, permeabilization with 0.4% Triton X-100 and incubation in a blocking solution (5% FCS and 1% BSA). Antibodies were mouse MHC antibodies (MY-32, Sigma) and Cy3-coupled anti-mouse IgG secondary antibody.
Cardiotoxin-induced regeneration assay. Regeneration assays were carried out by injecting 10 μl of cardiotoxin into the tibialis anterior muscle of 7- to 8-week-old mice, as described previously (Polesskaya et al, 2003). Each tibialis anterior muscle was weighed before lysis. Tissue extracts were normalized by total protein content and further controlled by staining with Ponceau red (an example is shown in supplementary Fig S1 online).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information S1
Supplementary Information S2
Supplementary Information S3
Supplementary Information S4
Supplementary Information S5
Acknowledgments
We thank M. Rudnicki, M. Vernet and K. Kato for the kind gift of materials, Hélène Humbertclaude and Sylvie Journier for help with ES cell culture. This work was supported by grants from the Association Française contre les Myopathies and grant no. LSHG-CT-2004-502950 from the European Union 6th Framework program to A.H.-B.
References
- Bayle JH, Crabtree GR (1997) Protein acetylation: more than chromatin modification to regulate transcription. Chem Biol 4: 885–888 [DOI] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD (1996) Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84: 843–851 [DOI] [PubMed] [Google Scholar]
- Dinsmore J, Ratliff J, Deacon T, Pakzaban P, Jacoby D, Galpern W, Isacson O (1996) Embryonic stem cells differentiated in vitro as a novel source of cells for transplantation. Cell Transplant 5: 131–143 [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- Kiernan RE et al. (1999) HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J 18: 6106–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassar AB, Buskin JN, Lockshon D, Davis RL, Apone S, Hauschka SD, Weintraub H (1989) MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell 58: 823–831 [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T (2000) Regulation of E2F1 activity by acetylation. EMBO J 19: 662–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA (1996) MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev 10: 1173–1183 [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Harel-Bellan A (2001) Acetylation of MyoD by p300 requires more than its histone acetyltransferase domain. J Biol Chem 276: 44502–44503 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Duquet A, Naguibneva I, Weise C, Vervisch A, Bengal E, Hucho F, Robin P, Harel-Bellan A (2000) CREB-binding protein/p300 activates MyoD by acetylation. J Biol Chem 275: 34359–34364 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Naguibneva I, Duquet A, Bengal E, Robin P, Harel-Bellan A (2001) Interaction between acetylated MyoD and the bromodomain of CBP and/or p300. Mol Cell Biol 21: 5312–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesskaya A, Seale P, Rudnicki MA (2003) Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113: 841–852 [DOI] [PubMed] [Google Scholar]
- Rohwedel J, Maltsev V, Bober E, Arnold HH, Hescheler J, Wobus AM (1994) Muscle cell differentiation of embryonic stem cells reflects myogenesis in vivo: developmentally regulated expression of myogenic determination genes and functional expression of ionic currents. Dev Biol 164: 87–101 [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Braun T, Hinuma S, Jaenisch R (1992) Inactivation of MyoD in mice leads to upregulation of the myogenic HLH gene Myf 5 and results in apparently normal muscle development. Cell 71: 383–390 [DOI] [PubMed] [Google Scholar]
- Sabourin LA, Girgis-Gabardo A, Seale P, Asakura A, Rudnicki MA (1999) Reduced differentiation potential of primary MyoD−/− myogenic cells derived from adult skeletal muscle. J Cell Biol 144: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, Wang JY, Kedes L (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell 4: 725–734 [DOI] [PubMed] [Google Scholar]
- Soutoglou E, Katrakili N, Talianidis I (2000) Acetylation regulated transcription factor activity at multiple levels. Mol Cell 5: 745–751 [DOI] [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991) Muscle-specific transcriptional activation by MyoD. Genes Dev 5: 1377–1386 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382: 319–324 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information S1
Supplementary Information S2
Supplementary Information S3
Supplementary Information S4
Supplementary Information S5