

Abstract
The effects of 2-Methoxyestradiol (2ME)-induced apoptosis was examined in human leukemia cells (U937 and Jurkat) in relation to mitochondrial injury, oxidative damage, and perturbations in signaling pathways. 2ME induced apoptosis in these cells in a dose-dependent manner associated with release of mitochondrial proteins (cytochrome c, AIF), generation of reactive oxygen species (ROS), downregulation of Mcl-1 and XIAP, and inactivation (dephosphorylation) of Akt accompanied by activation of JNK. In these cells, enforced activation of Akt by a constitutively active myristolated Akt construct prevented 2ME-mediated mitochondrial injury, XIAP and Mcl-1 downregulation, JNK activation, and apoptosis, but not ROS generation. Conversely, 2ME lethality was potentiated by the phosphatidylinostol 3-kinase (PI3K) inhibitor LY294002. Furthermore, in U937 cells, the hydrogen peroxide scavenger catalase and a superoxide dismutase (SOD) mimetic, TBAP, blocked these events, as well as Akt inactivation. Interruption of the JNK pathway by pharmacologic or genetic (e.g. siRNA) means attenuated 2ME-induced mitochondrial injury, XIAP and Mcl-1 downregulation, and apoptosis. Collectively, these findings suggest a hierarchical model of 2ME-related apoptosis induction in human leukemia cells in which 2ME-induced oxidative injury represents a primary event resulting in Akt inactivation, leading, in turn, to JNK activation, and culminating in XIAP and Mcl-1 downregulation, mitochondrial injury, and apoptosis. They also suggest that in human leukemia cells, the Akt pathway plays a critical role in mediating the response to oxidative stress induced by 2ME.
Keywords: 2ME, apoptosis, leukemia, Akt, ROS
Introduction
2-Methoxyestradiol (2ME) is a physiological metabolite of the endogenous estrogen estradiol-17b which fails to bind the estrogen receptor (LaVallee et al., 2002). Estradiol is hydroxylated by NADPH-dependent cytochrome P450 enzymes mainly in the liver to catechol estrogens, for example, 2- or 4-hydroxyestradiol (Martucci and Fishman, 1993). These catechol estrogens are metabolically O-methylated to monomethyl ethers such as 2ME by catechol-O-methyltransferase. The estrogen metabolite 2ME appears to have anticarcinogenic activity. In addition, 2ME exerts antiproliferative and antiangiogenic effects (Hughes et al., 2002; Tinley et al., 2003). It has also been reported that 2ME induces apoptosis in various cell types (Zoubine et al., 1999; Lin et al., 2001; Bu et al., 2002; Qanungo et al., 2002). In a recent communication, Huang et al. (2000) demonstrated that 2ME potently induces apoptosis in human leukemia cells through a mechanism that involves the selective inhibition of superoxide dismutase (SOD) and accumulation of reactive oxygen species (ROS). Since 2ME and related compounds are relatively nontoxic to normal tissues (Huang et al., 2000), an important implication of these findings is that such agents might play a useful role in the therapy of leukemia and possibly other hematologic malignancies.
The phosphatidylinositol 3-kinase (PI3K)/Akt signal transduction pathway plays an important role in cell survival decisions (Franke et al., 2003). Mutations in this pathway, particularly those related to the PTEN phosphatase, are frequently observed in human cancers (Paez and Sellers, 2003). In addition to effects on multiple downstream targets, including mTOR, Bad, and caspase-9 among numerous others (Datta et al., 1999; Mitsiades et al., 2004), Akt has been implicated in the response of cells to oxidative stress (Deora et al., 1998; Ozaki et al., 2003). Specifically, redox-related perturbations in PTEN activity have been documented (Leslie et al., 2003), and members of the FOXO3 family, downstream targets of Akt, have been shown to regulate cell death responses to oxidative injury (Kops et al., 2002). Furthermore, activation of the stress-related JNK (c-Jun N-terminal kinase) pathway is known to play a key role in mediating the response of tumor cells to oxidative injury (Holgado-Madruga and Wong, 2003). In contrast, the MEK1/2/ERK1/2 pathway generally plays a cytoprotective function, particularly in response to redox-related stresses (Yu et al., 2004). Collectively, these findings suggest a model in which cell fate decisions are decided by the net output of cytoprotective versus stress-related signaling pathways (Martindale and Holbrook, 2002).
Studies investigating the role of signaling cascades in 2-ME-related lethality have primarily focused on those related to oxidative injury, including the HIF-1α and JNK pathways (Mooberry, 2003). For example, in Ewing’s sarcoma cells, 2-ME has been shown to induce apoptosis in association with hydrogen peroxide (H2O2) generation and JNK activation (Djavaheri-Mergny et al., 2003). In prostate cancer cells, the PI3K pathways has been implicated in 2-ME-induced engagement of the extrinsic apoptotic pathway (Shimada et al., 2004), suggesting that certain cytoprotective signal transduction cascades might also influence 2-ME lethality. Currently, the role of the Akt pathway in the response of human leukemia cells to agents such as ME has not yet been explored, nor have the relationships between this pathway, oxidative damage, and other survival signaling events been examined in depth. The purpose of the present study was to characterize the functional role of Akt and related pathways, using genetically engineered model systems, on the lethality of ME toward human leukemia cells. Our results indicate that downregulation/inactivation of Akt plays a key role in mediating the cytotoxic effects of ME in these cells, and suggest that interruption of the PI3K/Akt pathway can dramatically potentiate ME-related antileukemic actions. They also indicate a hierarchical model of 2ME-induced lethality in human leukemia cells characterized by initial oxidative injury, leading to downregulation of the cytoprotective Akt pathway, resulting in JNK activation, and culminating in cytochrome c release as well as Mcl-1 and XIAP downregulation.
Results
Dose-response and time course analysis of 2ME-mediated apoptosis and mitochondrial injury (loss of Dcm) in U937 cells are shown in Figure 1a and b. Modest degrees of apoptosis and mitochondrial injury were noted at 1 μM 2ME concentrations (24 h), which increased substantially at concentrations ≥2 μM. These events became apparent after 12 h of drug exposure, and reached near-maximal levels after 24 h (Figure 1b).
Figure 1.

2ME markedly induces apoptosis and mitochondrial injury in U937human leukemia cells in a dose- and time-dependent manner. (a) U937cells were treated without or with various concentrations of 2ME as indicated for 24 h. (b) U937cell were treated with 4 μM 2ME for 3, 6, 12, and 24 h. Cells were stained with annexin V/propidium iodide (PI), and apoptosis was determined using flow cytometry as described in Materials and methods. In separate experiment, cells were stained with DiOC6, and reduction in Ψm was determined by monitoring uptake of DiOC6 using flow cytometry as described in Materials and methods.‘Low’Ψm values are expressed as the percentage of cells exhibiting a diminished mitochondrial membrane potential. The values obtained from annexin V/PI and DiOC6 assays represent the mean±s.d. for three separate experiments. (c) U937cells were treated without or with various concentrations of 2ME as indicated for 24 h. (d) U937cell were treated without or with 4 μM 2ME for 3, 6, 12, and 24 h. After treatment of U937cells with the indicated 2ME concentration or the indicated interval, total cellular extracts, cytosolic S-100 fractions (cytochrome c, AIF), and nuclear extracts (AIF) were prepared and subjected to Western blot assay using antibodies against PARP, cleaved-caspase-3, caspase-8, caspase-9, AIF, and cytochrome c. AIF(N) 1/4 AIF in the nuclear fraction. Each lane was loaded with 30 μg protein. Blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading and transfer. Two additional studies yielded equivalent results
Consistent with these findings, the same 2ME concentrations and exposure intervals resulted in cleavage/activation of caspases-9, -8, and -3, as well as PARP degradation (Figure 1c and d). These events were also accompanied by release of cytochrome c and AIF into the cytosolic S-100 cell fraction, and a corresponding increase in nuclear AIF accumulation (Figure 1c and d).
Concentration- and time-dependent effects of 2ME were then examined in relation to expression of various Bcl-2 family members. 2-ME concentrations ≥2 μM induced downregulation of Mcl-1 and XIAP which became apparent after 12 h of exposure (Figure 2a and b). The appearance of a Bcl-2 cleavage fragment was noted as a relatively late event (24 h). On the other hand, total levels of Bcl-xL and Bax remained largely unchanged with treatment. In contrast, translocation of Bax from the cytosol to the mitochondria was noted with toxic 2ME concentrations beginning at 12 h of drug exposure.
Figure 2.

Effects of 2ME on apoptosis-related gene expression and various signal transduction pathways. (a) U937cells were treated with the indicated concentration of 2ME for 24 h. (b) U937cell were treated with 4 μM 2ME for 3, 6, 12, and 24 h. Total cellular extract were prepared and subjected to Western blot analysis using antibodies against apoptosis-related proteins including XIAP, Mcl-1, Bcl-2, Bcl-xL, and Bax. Cytosolic and membrane extracts were also prepared and subjected to Western blot assay using antibody against Bax. U937cells were treated with 2ME at the indicated concentrations (c) or for the indicated intervals (d) as described above. Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-Akt, Akt, phosphor-ERK, ERK, phosphor-p38, p38, phosphor-JNK, JNK, phospho-mTOR, and mTOR. (e) U937cells were pretreated with the caspase inhibitor Z-VAD-FMK (10 μM) for 1 h, followed by 4 μM of 2ME for 24 h. Cells were stained with annexin V/PI. Apoptosis was determined using flow cytometry as described in Materials and methods. The values obtained from annexin V assays represent the means±s.d. for three separate experiment. *Values for cells treated with 2ME and Z-VAD-FMK were significantly reduced compared to values obtained for 2ME alone by Student’s t-test; P<0.01. (f) U937cells were pretreated with the caspase inhibitor Z-VAD-FMK (10 μM) for 1 h, follolowed by 4 μM 2ME for 12 and 24 h. Total protein extracts were prepared and subjected to Western blot assay using antibodies against phospho-Akt and Akt
Effects of 2ME in U937cells were then examined in relation to changes in various signal transduction pathways implicated in apoptosis regulation. Toxic concentrations of 2ME resulted in a reduction in levels of both total and phospho-Akt as early as 6 h of drug exposure (Figure 2c and d). Roughly parallel effects were noted in the expression of total and phosphorylated mTOR, a well-described Akt downstream target (Manning and Cantley, 2003). In contrast, 2ME had little or no effect on expression of total or phospho-p38 MAPK or ERK. Furthermore, toxic concentrations of 2ME resulted in a clear increase in activation of the stress-related kinase JNK as early as 6 h after drug exposure, but did not affect levels of total JNK. In separate studies, cotreatment of cells with the caspase inhibitor Z-VAD-FMK, which abrogated 2ME-induced apoptosis (Figure 2e), failed to prevent Akt inactivation, although it partially blocked 2ME-mediated reductions in levels of total Akt (Figure 2f). Such findings indicate that inactivation of Akt by 2ME does not simply represent a secondary, caspase-dependent event. Thus, 2ME-induced lethality was associated with downregulation of the cytoprotective Akt pathway and reciprocal activation of the stress-related JNK pathway.
To determine whether these events were restricted to myeloid leukemia cells, parallel studies were performed in Jurkat T-lymphoblastic leukemia cells. These cells exhibited dose-response effects of 2ME similar to those observed in U937 cells (Figure 3a), and comparable degrees of caspase-3 and -8 activation and PARP degradation (Figure 3b). As in the case of U937cells, 2ME induced XIAP and Mcl-1 downregulation, but had little effect on expression of Bcl-2 or Bcl-xL (Figure 3c). Lastly, the ability of 2ME to trigger downregulation of phospho- and total Akt levels, and activation of JNK was essentially identical to effects observed in U937cells (Figure 3c).
Figure 3.
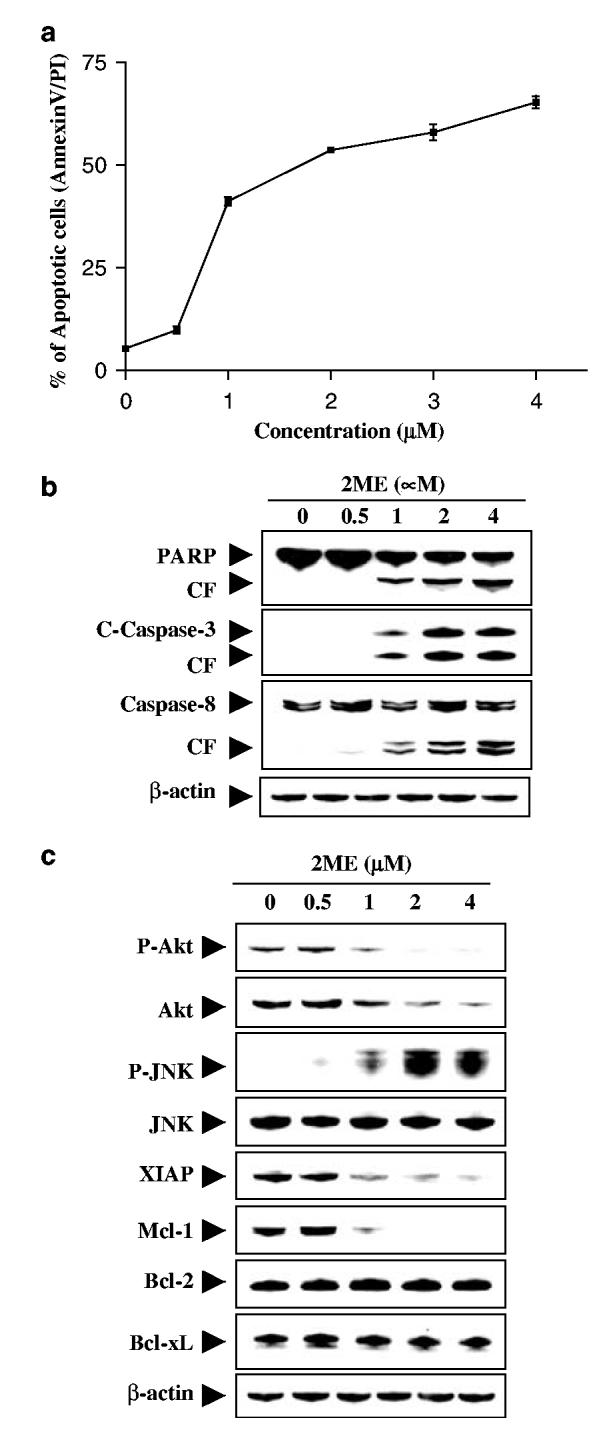
2ME markedly induces apoptosis in Jurkat acute T-cell leukemia cells in a dose-dependent manner. Jurkat cells were treated without or with various concentrations of 2ME as indicated for 24 h. (a) Cells were stained with annexin V/PI, and apoptosis was determined using flow cytometry as described in Materials and methods. The values obtained from annexin V/PI assays represent the mean±s.d. for three separate experiments. (b) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against PARP, cleaved caspase-3, and caspase-8. (c) Total cellular extracts were also prepared and subjected to Western blot assay using antibodies against phosphor-Akt, Akt, phospho-JNK, JNK, XIAP, and Mcl-1. For Western blot analysis, each lane was loaded with 30 μg of protein; blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results
The preceding findings implied that downregulation of Akt might play an important role in 2ME-mediated lethality. To test this possibility, cells were coexposed to 2ME and the PI3K inhibitor LY294002, and apoptosis was monitored. As shown in Figure 4a, coadministration of a nontoxic concentration of LY294002 (i.e. 20 μM) with a modestly toxic concentration of 2ME (1 μM; ∼23%) resulted in a pronounced increase in apoptosis (i.e. to ∼60%). Western blot analysis revealed that coadminstration of 2ME and LY294002 at concentrations that were ineffective or marginally effective by themselves resulted in pronounced increase in activation of caspases-3, -8, and -9, PARP degradation, and AIF and cytochrome c release into the cytosol (Figure 4b). Combined treatment also resulted in potentiation of Mcl-1 and XIAP downregulation, and the appearance of a Bcl-2 cleavage product, but had little effect on Bcl-xL levels (Figure 4c). In addition, coadministration of 2ME and LY294002 resulted in the virtual abrogation of Akt expression/activation, and a pronounced increase in JNK activation (Figure 4d). Together, these findings suggest that downregulation of Akt plays a critical role in regulating the lethality of 2ME in human leukemia cells.
Figure 4.

Effects of the pharmacological inhibitor of PI3K, LY294002 (LY) on apoptosis induced by 2ME in U937cells. U937cells were pretreated with 20 μM of LY for 1 h, followed by the addition of 1 μM of 2ME for 24 h. (a) Cells were stained with annexin V/PI, and apoptosis was determined using flow cytometry as described in Materials and methods. The values obtained from annexin V/PI assays represent the means±s.d. for three separate experiments. *Values for cells treated with 2ME and LY in combination were significantly greater than those for cells treated with 2ME alone by Student’s t-test; P<0.01. (b) Total cellular or cytosolic extracts were prepared as described in Materials and methods, and subjected to Western blot analysis using antibodies against PARP, cleaved-caspase-3, caspase-8, caspase-9, AIF, and cytochrome c. Total cellular extracts were also prepared and subjected to Western blot assays using antibodies against apoptosis-related proteins including XIAP, Mcl-1, Bcl-2, and Bcl-xL (c), and cell signaling proteins including phospho-Akt, Akt, phospho-JNK, and JNK (d). For Western blot assays, each lane was loaded with 30 μg of protein; blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results
To assess the functional signficance of Akt inactivation in 2ME lethality more definitively, U937 cells ectopically expressing constitutively active, myristolated Akt were employed (Rahmani et al., 2005). Three separate clones (AktCA-3, AktCA-6, and AktCA-11) displaying variable degrees of constitutive Akt activation were each markedly less sensitive to 2ME-induced apoptosis (4 μM; 24 h) than their wild-type countparts (pUSE; P<0.01 in each case; Figure 5a). Consistent with these findings, 2ME was considerably less effective in triggering PARP degradation, and cytochrome c or AIF release into the cytosol in mutant cells compared to controls (Figure 5b). Furthermore, enforced activation of Akt blocked 2ME-mediated XIAP and Mcl-1 down-regulation (Figure 5c), as well as Bcl-2 cleavage (data not shown). Western blot analysis documented the marked increase in levels of total and phospho-Akt in AktCA-3, -6 and -11 cells, and the inability of 2ME to induce downregulation/inactivation of either Akt in the mutant lines. Interestingly, the ability of 2ME to induce JNK activation was essentially abrogated in cells expressing constitutively active Akt, indicating that engagement of this stress pathway by 2ME depends, at least in part, upon inactivation of Akt. To determine whether these findings were restricted to myeloid leukemia cells, parallel studies were performed in Jurkat lymphoblastic leukemia cells expressing a doxycycline-inducible constitutively active (myristolated) Akt construct. As shown in Figure 5d, addition of doxycycline to the medium significantly reduced 2ME lethality in these cells (P<0.01 versus controls). Western blot analysis (Figure 5e) demonstrated that addition of doxycycline resulted in a marked increase in expression of total and phospho-Akt in 2ME-treated cells. Consistent with the results obtained in U937cells, induction of Akt by doxycycline also blocked 2ME-mediated JNK activation. Together, these findings indicate that downregulation of Akt plays a significant functional role in 2ME lethality in human leukemia cells, and that this phenomenon operates upstream of XIAP and Mcl-1 downregulation and JNK activation.
Figure 5.

Induction of activated Akt markedly protect cells from 2ME-induced apoptosis. U937cells were stably transfected with constitutively active forms of Akt (three clones designated CA-3, CA-6, and CA-11) or an empty vector (pUSE) as described in Materials and methods. U937(Akt-CA-3, Akt-CA6, and Akt-CA11) cells and pUSE cells were then treated for 24 h with 4 μM of 2ME. (a) After treatment, apoptosis was analysed using annexin V-FITC assay as described in Materials and methods. *Values for Akt-CA3, Akt-CA6, and Akt-CA11 cells treated with 2ME were significantly decreased compared to those for pUSE cells by Student’s t-test; P<0.01. (b) Total cellular or cytosolic extracts were prepared and subjected to Western blot analysis using antibodies against PARP, AIF and cytochrome c.(c) Total cellular extract were also prepared and subjected to Western blot analysis using antibodies against phospho-Akt, Akt, phosphor-JNK, JNK, XIAP, and Mcl-1. Jurkat cells (Akt CA16), which inducibly express myc-tagged myristoylated Akt in the presence of doxycycline, were treated with 2 μg/ml doxycycline for 24 h, followed by the addition of 4 μM of 2ME for 24 h. (d) After treatment, apoptosis was analysed using annexin V-FITC assay as described in Materials and methods. (e) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against phospho-Akt, Akt, phosphor-JNK, and JNK. For Western blot assay, each lane was loaded with 30 μg of protein; blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results
The functional significance of JNK activation in 2ME lethality was then investigated using both pharmacologic and genetic approaches. Coadministration of the JNK inhibitor SP600125 essentially abrogated 2ME-related lethality (Figure 6a) in association with inactivation of JNK, as determined by diminished phosphorylation of c-Jun (Figure 6b). Coadministration of SP600125 also blocked 2ME-induced caspase-3, -8, and -9 activation (data not shown), and PARP degradation (Figure 6c). Antagonism of 2ME-mediated XIAP and Mcl-1 downregulation and Bcl-2 cleavage by SP600125 was also observed (Figure 6c). Finally, SP600125 also largely blocked 2ME-mediated mitochondrial injury (cytocrhome c and AIF release). Since SP600125 is not completely specific for JNK (Minutoli et al., 2004), a genetic approach utilizing JNK1 siRNA was employed. As shown in Figure 6e, transient transfection of U937 cells with JNK1 siRNA reduced expression of JNK1 by over 50%, and resulted in a significant reduction in 2ME-mediated apoptosis (P<0.01 control siRNA; Figure 6d). Collectively, these findings indicate that 2ME-induced JNK activation plays an important functional role in 2ME-related lethality. They also indicate that activation of JNK operates upstream of mitochondrial injury in 2ME-mediated engagement of the apoptotic cascade.
Figure 6.

Pharmacological inhibition of JNK and transfection of JNK1 siRNA significantly protect cells from 2ME-induced apoptosis. U937cells were pretreated with 10 μM of JNK inhibitor, SP600125 (SP), for 1 h, followed by the addition of 4 μM of 2ME for 24 h. (a) Cells were stained with annexin V/PI, and apoptosis was determined using flow cytometry as described in Materials and methods. The values obtained from annexin V/PI assays represent the mean±s.d. for three separate experiments. *Values for cells treated with 2ME and SP were significantly less than those obtained for cells treated with 2ME alone by Student’s t-test; P<0.01. After treatment, total cellular and cytosolic extracts were prepared and subjected to Western blot analysis using antibodies against phospho-c-Jun, c-Jun (b), and apoptosis-related proteins including PARP, XIAP, Mcl-1, Bcl-2, Bcl-xL, cytochrome c, and AIF (cytosolic S-100 fraction) (c). U937cells were transfected with JNK1 siRNA oligonucleotides or controls and incubated for 24 h at 371C, after which cells were treated with 4 μM of 2ME for 24 h. (d) Apoptosis was determined using the annexin V-FITC assay as described in Materials and methods. *Values for cells treated with 2ME after transfection with JNK1 siRNA oligonucleotides were significantly decreased compared to those for control cells treated with 2ME by Student’s t-test; P<0.01. (e) Total cellular extracts were prepared and subjected to Western blot analysis using antibodies against JNK1 and JNK2. For Western blot assay, each lane was loaded with 30 μg of protein; blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results
In human lymphoid and other leukemic cells, superoxide dismutase has been shown to play a key role in 2ME-mediated ROS generation and lethality (Huang et al., 2000). However, the role of individual ROS species in 2ME-mediated apoptosis, mitochondrial injury, and perturbations in signaling pathways has not yet been fully characterized, particularly in human leukemia cells. To explore further the role of individual ROS on 2ME-mediated apoptosis, we employed three antioxidants, for example, TBAP, a cell permeable SOD mimetic (Faulkner et al., 1994), catalase, and sodium formate, which primarily act on superoxide radical (), H2O2, and hydroxyl radical (OH·), respectively. For these studies, HE, which is relatively specific for , was used to monitor ROS formation. As shown in Figure 7a, catalase, a H2O2 scavenger, essentially abrogated 2ME-mediated ROS generation in U937 cells, as did TBAP, the scavenger. In contrast, sodium formate, a OH · scavenger, failed to block 2ME-mediated ROS generation in these cells. Essentially equivalent results were obtained when DCFH-DA, which largely reflects levels of H2O2, was utilized (Figure 7b). Consistent with these findings, catalase and TBAP, but not sodium formate, significantly blocked 2ME-mediated apoptosis (Figure 7c; P<0.01), PARP degradation, cytochrome c, and AIF release (Figure 7d). Catalase, and TBAP, but not sodium formate, also blocked 2ME-mediated XIAP and Mcl-1 inhibition, and Bcl-2 cleavage (Figure 7e). Finally, catalase and TBAP, but not sodium formate, prevented inactivation of Akt and mTOR, as well as activation of JNK in these cells (Figure 7f). Together, these finding suggest that H2O2 and radicals are primarily responsible for perturbations in survival signaling events and lethality induced by 2ME.
Figure 7.

Effects of antioxidants on 2ME-induced ROS generation, apoptosis, and cell signaling proteins. U937cells were pretreated with antioxidants including as TBAP (200 μM), catalase (5000 U/ml), and sodium formate (SF, 2 mm) for 1 h, followed by the addition of 2ME (4 μM) for 6 h (ROS analysis) or 24 h (annexin V-FITC and Western blot assays). Cells were stained with HE (a) and DCFH-DA (b), after which ROS production was analysed by flow cytometry as described in Materials and methods. (c) Cells were stained with annexin V and apoptosis was analysed by flow cytometry as described in Materials and methods. In figures (a-c), *Values significantly decreased compared to values obtained with 2ME treatment alone by Student’s t-test; P<0.01. (d) Total cellular or cytosolic extracts were prepared and subjected to Western blot analysis using antibodies against PARP, AIF, and cytochrome c. Total cellular proteins were also prepared and subjected to Western blot analysis using antibodies directed against apoptosis-related proteins including XIAP, Mcl-1, and Bcl-2 (e), or cell signaling proteins including phospho-Akt, Akt, phospho-JNK, JNK, phosphor-mTOR, and mTOR (f). For all Western blot assays, each lane was loaded with 30 μg of protein; blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading. Two additional studies yielded equivalent results. (g) U937cells were stably transfected with constitutively active forms of Akt (CA-6) or an empty vector (pUSE). Cells were treated with 4 μM 2ME for 6 h, after which ROS production was analysed using flow cytometry as described in Materials and methods. (h) Jurkat cells (Akt CA16) inducibly expressing myc-tagged myristoylated Akt were in the presence or absence of 2 μg/ml doxycycline for 24 h, followed by the addition of 4 μM of 2ME for 6 h. ROS production was analysed using flow cytometry as described in Materials and methodsAcknowledgements This work was supported by awards CA 63753, CA 100866, and CA 93738 from the NIH, award 6045-03 from the Leukemia and Lymphoma Society of America, and award DAMD-17-03-1-0209 from the Department of Defense.
Finally, to gain further insights into the hierarchy of events accompanying 2ME lethality, 2ME-mediated ROS generation was monitored in U937cells ectopically expressing activated Akt. As shown in Figure 7g, constitutive activation of Akt, which substantially protected cells from 2ME-induced lethality, had little effect on 2ME-induced ROS generation. Similarly, enforced activation of inducible Akt in Jurkat cells by addition of doxycycline to the medium failed to modify 2ME-induced ROS production (Figure 7h). These findings provide further support for the notion that 2ME-mediated oxidative injury represents a primary cause, rather than a consequence, of Akt inactivation in human leukemia cells.
Discussion
The present results indicate that induction of cell death by the estrogen derivative 2ME in human leukemia cells results in inactivation of Akt, and that this process plays a critical role in regulating the cell death response, at least in some human leukemia cell types. In a recent study, 2ME was shown to induce SOD in human leukemia cells, leading to the production of oxygen free radicals, mitochondrial damage, and apoptosis (Huang et al., 2000). It is also well established that signal transduction pathways represent important mediators of oxidative stress-related responses (Gao et al., 2002; Ikeyama et al., 2002). In addition, the ability of oxidative injury to trigger mitochondrial dysfunction, culminating in engagement of the apoptotic cascade, has been widely reported (Rego and Oliveira, 2003). Currently, investigation of signaling events in 2-ME-treated cells has emphasized activation of stress-related pathways (e.g. JNK, H1Fα) implicated in oxidative injury (Djavaheri-Mergny et al., 2003; Mooberry, 2003). Presently, little information is available concerning the functional role of the Akt pathway in mediating 2-ME-induced lethality, particularly in malignant hematopoietic cells. The results of the present study demonstrate that not only does Akt inactivation represent a consequence of 2ME-mediated oxidative injury but also plays a key functional contribution in amplifying mitochondrial dysfunction and subsequent lethality.
Akt is a serine-threonine kinase intimately involved in the regulation of cell survival (Kim et al., 2001). It is activated by recruitment to the cell membrane through the actions of PI3K, which in turn is regulated by the PTEN phosphatase (Cantley and Neel, 1999), mutations of which are among the most commonly encountered in human cancers (Mao et al., 2004; Raftopoulou et al., 2004). Akt is involved in activation/phosphorylation of numerous downstream targets implicated in the control of apoptosis, including GSK, mTOR, procaspase-9, and Bad, among others (Pene et al., 2002; Zhao et al., 2004). Activation of Akt occurs in response to diverse noxious stimuli, including UV radiation, osmotic stress, and oxidative damage (Mockridge et al., 2000; Gao et al., 2002; Mildner et al., 2002). In the case of oxidative injury, Akt activation generally involves PTEN inactivation (Kandel et al., 2002), and results in attenuation of lethality (Persad et al., 2000). The present findings suggest that the relationship between 2ME-mediated oxidative injury and effects on Akt activity differ from those of previous reports. Most notably, 2ME exposure resulted in diminished, rather than increased, Akt phosphorylation. While it would be tempting to attribute this phenomenon to PTEN activation, the fact that Jurkat and U937 cells do not express wild-type PTEN (Shan et al., 2000; Lin et al., 2001). argue against this notion. A more likely possibility is that 2ME, through a mechanism yet to be elucidated, blocks the actions of PI3K. The finding that LY29004, an inhibitor of PI3K (Curnock and Knox, 1998), enhanced the lethality of 2ME is potentially consistent with this concept, as well as with evidence that PI3K opposes 2-ME-induced activation of the extrinsic apoptotic path-way in prostate cancer cells (Shimada et al., 2004). It should also be noted that 2ME resulted in reduced expression of total Akt, an action that was partially reversed by coadministration of the pan-caspase inhibitor Z-VAD-FMK, raising the possibility that Akt inactivation might represent a consequence of engagement of the caspase cascade. In fact, the caspasedependent downregulation of Akt is a well-described phenomenon (Yang and Widmann, 2002). However, Z-VAD failed to restore levels of phospho-Akt, in 2ME-treated cells, arguing strongly that factors other than caspase-mediated events are involved in this phenomenon. It is also of interest that 2ME failed to modify expression/activation of the cytoprotective ERK1/2 pathway in human leukemia cells. ERK1/2 activation is often triggered in response to noxious stimuli, including oxidative stress (Li et al., 2004), and such a response has been shown to attenuate apoptosis (Yu et al., 2004). The finding that 2ME modified Akt rather than ERK1/2 activation in these cells suggests that the signaling pathways regulating oxidative stress responses are highly stimulus-dependent.
The bulk of evidence suggests that in human leukemia cells, 2ME-induced Akt inactivation plays a critical functional role in mediating 2ME lethality. Significantly, enforced activation of Akt, and prevention of 2ME-induced Akt downregulation, largely reversed the lethal consequences of 2ME exposure, including mitochon-drial injury (e.g. cytochrome c and AIF release), caspase activation, PARP cleavage, and apoptosis. On the other hand, enforced activation of Akt did not block 2ME-induced increases in ROS generation, effectively ruling out the possibility that Akt prevents or attenuates 2ME-mediated oxidative injury. It is of interest that 2ME exposure resulted in downregulation of both Mcl-1 and XIAP, anti-apoptotic proteins that may play a particularly important role in regulating apoptosis in malignant hematopoietic cells (Kobayashi et al., 2002; Bae et al., 2003). The finding that enforced activation of Akt largely blocked 2ME-induced downregulation of XIAP and Mcl-1 may therefore be significant. It is tempting to speculate that 2ME-mediated Akt downregulation involves diminished phosphorylation of Bad, an important downstream Akt target (Brunet et al., 1999) which acts by antagonizing the ability of antiapoptotic proteins such as Bcl-2 to maintain mitochondrial integrity (Peruzzi et al., 1999). However, it has been shown that phosphorylation of Bad results in enhanced proteasomal degradation, and that inference with the former process leads to Bad accumulation (Kausalya et al., 2001). The finding that inactivation of Akt by 2ME did not lead to diminished Bad phosphorylation or altered levels of total Bad argues against the possibility that perturbations in Bad are responsible for the response of these cells to 2ME-induced oxidative injury.
Induction of mitochondrial injury and apoptosis by 2ME was also associated with activation of the stressrelated JNK pathway. Engagement of the SEK/JNK pathway has been shown to play a key functional role in the lethal effects of diverse cytotoxic stimuli, including ceramide (Basu and Kolesnick, 1998) as well as oxidative injury (Mansat-de Mas et al., 1999). In fact, the net balance between cytoprotective (e.g. ERK) and stress-related (e.g. JNK) signaling may play a critical role in cell survival and death decisions (Johnson and Lapadat, 2002). The finding that pharmacologic and genetic interruption of the JNK pathway attenuated 2ME-mediated lethality indicates that stress pathways play a critical functional role in apoptosis induction by this agent. The mechanism by which oxidative stress triggers JNK activation is not known with certainty, but may involve release from GSH-mediated inhibitory effects (Kim et al., 2004) or, alternatively, perturbations in thioredoxin, leading to activation of ASK-1 (apoptosis signal-regulating kinase-1), of which JNK is a downstream target (Zhang et al., 2004). Interestingly, ectopic expression of Akt not only blocked 2ME-mediated mitochondrial injury and apoptosis but also prevented the striking increase in JNK activation, raising the possibility that one of the mechanisms by which Akt protects cells from 2ME lethality is by opposing JNK activation. Evidence that ASK-1, which activates JNK, is a target of Akt inhibitory phosphorylation (Kim et al., 2001) provides a possible explanation for this phenomenon. Lastly, the observation that pharmacologic or genetic interruption of the JNK pathway attenuated 2ME-mediated mitochondrial injury and lethality demonstrates an important functional role for this stress pathway in triggering the cell death program. Such findings are compatible with previous reports suggesting a direct role for JNK activation in promoting cytochrome c release from the mitochondria (Tournier et al., 2000). Collectively, these observations suggest a hierarchy of events in 2ME-induced lethality in which oxidative injury represents the primary insult, leading in turn to Akt inactivation, resulting in JNK activation, and culminating in mitochondrial injury and apoptosis.
ROS play critical roles in the regulation of diverse functional pathways involved in proliferation, apoptosis, and transformation (Hei et al., 1998; Adler et al., 1999; Pei et al., 2000; Gao et al., 2002). ROS, including , H2O2, and OH ·, are recognized as signaling molecules that are mobilized in reponse to various noxious stimuli. SOD, a scavenger, catalyses the conversion of into O2 and H2O2, whereas catalases, which act as H2O2 scavengers, convert H2O2 into O2 and H2O (Fridovich, 1999). Finally, sodium formate (SF), a hydroxyl radical (OH ·) scavenger, catalyses the conversion of OH · to H2O. It has recently been reported that ROS formation is intimately involved in 2ME apoptosis in certain human leukemia cells through an SOD-related process (Huang et al., 2000). In this study, we employed three antioxidants, for example, TBAP, a cell permeable SOD mimetic, catalase, and sodium formate, which primarily act on , H2O2, and OH, respectively, to investigate the involvement of individual ROS species in 2-ME-mediated apoptosis as well as perturbations in signaling events. Our results suggest that both H2O2 and play essential roles in 2ME-mediated apoptosis in U937leukemia cells, based on several lines of evidence. First, catalase, a H2O2 scavenger, and TBAP, a cell permeable SOD mimetic and a scavenger, essentially abrogated 2ME-mediated ROS generation in U937 leukemia cells, whereas SF, a OH · scavenger, failed to do so. Second, catalase and TBAP significantly inhibited 2-ME-induced apoptosis, whereas SF did not. Third, catalase and TBAP, but not SF, blocked 2ME-mediated PARP degradation and cytochrome c and AIF release. Fourth, catalase and TBAP, but not SF, blocked 2ME-mediated XIAP and Mcl-1 downregulation and Bcl-2 cleavage. Finally, catalase and TBAP also prevented inactivation of Akt and mTOR, as well as activation of JNK. Together, these findings argue that H2O2 and are primarily responsible not only for 2ME lethality in human leukemia cells, consistent with previous studies (Huang et al., 2000; Sawada et al., 2001), but also various perturbations in survival signaling pathways. They also indicate that Akt downregulation represents a critical signaling event that operates downstream of 2-ME-mediated oxidative injury to trigger mitochondrial injury, alterations in other signaling and survival proteins, and engagement of the apoptotic caspase cascade.
The results of this study could have implications for the incorporation of agents such as 2ME into the therapeutic armamentarium against leukemia and possibly other hematologic malignancies. First, the observation that 2ME downregulates the Akt pathway raises the possibility that this agent might prove useful in neoplasms characterized by PTEN mutations, in which inappropriate Akt activation plays a role in enhanced survival (Wang et al., 2000). Furthermore, efforts to unravel the molecular pathways regulating apoptotic response to this agent could help in the development of more rational combination therapies. For example, the finding that interruption of the PI3K and Akt pathways lower the threshold for 2ME-mediated lethality suggests possible strategies for enhancing 2ME antitumor activity. In this regard, examination of antileukemic interactions between 2ME and novel inhibitors of Akt or PDK1 (Harris, 2003) deserves consideration. Lastly, the ability of 2ME to interrupt Akt signaling, at least in some human leukemia cell types, raises the possibility that this agent might be effective in potentiating the activity of other novel agents whose lethal effects are regulated by this pathway. For example, recent studies suggest that the antileukemic potential of several histone deacetylase (HDAC) inhibitors is dependent upon Akt activation status (Fuino et al., 2003). Thus, the possibility exists that 2ME might interact synergistically with these agents, a number of which are currently in clinical evaluation (Zhou et al., 2002; Reddy et al., 2004). Significantly, HDAC inhibitor lethality has, like 2ME, been attributed to generation of ROS (Rosato et al., 2003). Accordingly, efforts to test this possibility are currently underway.
Materials and methods
Materials
2ME was purchased from Steraloids (Newport, RI, USA). LY294002, SP600125, and Z-VAD-FMK were purchased from EMD Biosciences (La Jolla, CA, USA). Hydroethidine (HE) and DCFH-DA were obtained from Molecular Probes (Eugene, OR, USA). Mn-TBAP was purchased from Calbiochem (La Jolla, CA, USA). SF was purchased from Sigma (St Louis, MO, USA). Catalase was purchased from Roche Molecular Biochemicals (Indianapolis, IN, USA). Antibodies against cytochrome c, AIF, Bcl-xL, Akt, phosphor-ERK, phosphor-JNK, JNK, and β-actin were purchased from Santa Cruz (Santa Cruz, CA, USA), cleaved caspase-3, phosphor-Akt, ERK, phospho-p38, p38, phospho-mTOR, and mTOR were from Cell Signaling (Beverly, MA, USA), caspase-9, XIAP, Mcl-1 were from PharMingen (San Diego, CA, USA), PARP was from Biomol (Plymouth Meeting, PA, USA), caspase-8 was from Alexis (Carlsbad, CA, USA), and Bcl-2 was from DAKO (Carpinteria, CA, USA).
Cell culture and transfection
U937human leukemia cells and Jurkat acute T-cell leukemia cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI1640 medium supplemented with sodium pyruvate, essential vitamins, l-glutamine, penicillin, streptomycin, and 10% fetal bovine serum (FBS). U937 cells were stably transfected with a constitutively active form of Akt (Rahmani et al., 2005), and clones were selected with 400 μg/ml of geneticin as we have previously reported (Rahmani et al., 2003). Jurkat cells that inducibly expressing constitutively active forms of Akt have been described previously (Rahmani et al., 2003) and selected with 400 μg/ml of hygromycin.
RNA interference and transfection
U937 cells (1.5 × 106) were transfected with 1 mg JNK1-annealed dsRNAi oligonucleotide 5′CGUGGGAUUUAUGGUCUGUGTT-3′/3′-TTGCACCUAAAUACCAGACAC-5′ (Orbigen, San Diego, CA, USA) using the Amaxa nucleofector™ (Koeln, Germany) as recommended by the manufacturer. After incubation at 37°C for 24 h, transfected cells were treated with 2ME, and subjected to determination of apoptosis and Western blot.
Annexin V/PI assays for apoptosis
For Annexin V/PI assays, cells were stained with Annexin V-FITC and PI, and evaluated for apoptosis by flow cytometry according to the manufacturer’s protocol (BD PharMingen, San Diego, CA, USA). Briefly, 1 × 106 cells were washed twice with phosphate-buffered saline (PBS), and stained with 5 mlof Annexin V-FITC and 10 mlofPI(5 μg/ml) in 1 binding buffer (10 mm HEPES, pH 7.4, 140 mm NaOH, 2.5 m m CaCl2) for 15 min at room temperature in the dark. The apoptotic cells were determined using a Becton-Dickinson FACScan cytoflurometer (Mansfield, MA, USA). Both early apoptotic (annexin V-positive, PI-negative) and late (annexin V-positive and PI-positive) apoptotic cells were included in cell death determinations.
Detection of intracellular ROS
Intracellular production of ROS was measured by using HE and DCFH-DA. HE and DCFH-DA have been shown to be relatively specific for and H2O2, respectively (Sawada et al., 2001). is able to oxidize HE to yield ethidium, and H2O2 is able to oxidize DCFH to the fluorescent DCF. To determine ROS production, control and drug-treated cells were incubated with HE (2 μM) or DCFH-DA (5 μM) for 60 min, washed twice with cold PBS and analysed within 1 h using a Becton-Dickinson FACScan flow cytometer (Hialeah, FL, USA). The effects of free radical scavengers (Mn-TBAP, catalase, formate) on ROS generation was determined as we have previously described in detail (Gao et al., 2002).
Western blot analysis
Western blot analysis was performed using the NuPAGE Bis-Tris electrophoresis system (Invitrogen, Carlsbad, CA, USA). The total cellular samples were washed twice with cold PBS and lysed in 1× NuPAGE LDS sample buffer supplemented with 50 mm dithiothreitol (DTT, Fisher Biotech, Pittsburgh, PA, USA). The protein concentration was determined using Coomassie Protein Assay Reagent (Pierce, Rockford, IL, USA). The total cellular protein extracts were separated by SDS-PAGE, and transferred to nitrocellulose membrane in 20 mm Tris-HCl (pH 8.0) containing 150 mm glycine and 20% (v/v) methanol. Membranes were blocked with 5% nonfat dry milk in 1 TBS containing 0.05% Tween 20 and incubated with antibodies described in Materials and methods. Protein bands were detected by incubating with horseradish peroxidase-conjugated antibodies (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA), and visualized with enhanced chemiluminescence reagent (PerkinElmer Life Sciences, Boston, MA, USA).
Analysis of cytosolic cytochrome c, AIF and Bax, and membrane Bax
After treatment, cells (2 × 106) were washed twice with cold PBS and lysed by incubating for 5 min in lysis buffer (75 mm NaCl, 8 mm Na2HPO4, 1mm NaH2PO4, 1mm EDTA, and 350 μg/ml digitonin). Cytosolic (supernatant) and membrane (pellet) fractions were separated by centrifugation at 12 000 g for 1 min. The protein concentration was determined using Coomassie Protein Assay Reagent (Pierce, Rockford, IL, USA). In all, 30 mg of cytosolic extract was separaed by SDS-PAGE, transferred to nitrocellulose membrane, and incubated with antibodies against cytochrome c (PharMingen) and AIF (Santa Cruz) and Bax (PharMingen). Membrane extract (30 μg) was subjected to Western blot analysis using antibody against Bax.
Analysis of AIF in nuclear extracts
After treatment, cells (2 × 106) were washed twice with cold PBS and resuspended in hypotonic buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10mm KCl, 0.5 mm DTT, 0.5 mm phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin, and 1 mg/ml aprotinin) for 15 min on ice and then added 5 μl of 10% Nonidet P-40. After centrifugation at 2000g for 10 min at 4°C, nuclear pellets were resuspended in buffer B (20 mm HEPES, pH 7.9, 1.5 mm MgCl2, 450 mm NaCl, 0.2 mm EDTA, 25% glycerol, 0.5 mm DTT, 0.5 mm phenylmethylsulfonyl fluoride, 1 mg/ml leupeptin, and 1 μg/ml aprotinin) for 30 min on ice. After centrifugation at 20 000 g for 15 min, the supernatant was collected as the nuclear extracts. The protein concentration was determined using Coomassie Protein Assay Reagent (Pierce, Rockford, IL, USA). In all, 30 μg of nuclear extract was separated by SDS-PAGE, transferred to nitrocellulose membrane, and Western blot analysis employing antibodies against AIF (Santa Cruz) was used to monitor nuclear expression of AIF.
Statistical analysis
For analysis of apoptosis, values were presented as means±s.d. Statistical differences between control and treated groups were determined by Student’s t-test. Differences were considered statistically significant for values of P<0.05, P<0.01.
References
- Adler V, Yin Z, Tew KD, Ronai Z. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- Bae J, Donigian JR, Hsueh AJ. J. Biol. Chem. 2003;278:5195–5204. doi: 10.1074/jbc.M201988200. [DOI] [PubMed] [Google Scholar]
- Basu S, Kolesnick R. Oncogene. 1998;17:3277–3285. doi: 10.1038/sj.onc.1202570. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Bu S, Blaukat A, Fu X, Heldin NE, Landstrom M. FEBS Lett. 2002;531:141–151. doi: 10.1016/s0014-5793(02)03478-6. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. Proc. Natl. Acad. Sci. USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnock AP, Knox KA. Cell Immunol. 1998;187:77–87. doi: 10.1006/cimm.1998.1335. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Deora AA, Win T, Vanhaesebroeck B, Lander HM. J. Biol. Chem. 1998;273:29923–29928. doi: 10.1074/jbc.273.45.29923. [DOI] [PubMed] [Google Scholar]
- Djavaheri-Mergny M, Wietzerbin J, Besancon F. Oncogene. 2003;22:2558–2567. doi: 10.1038/sj.onc.1206356. [DOI] [PubMed] [Google Scholar]
- Faulkner KM, Liochev SI, Fridovich I. J. Biol. Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Ann. NY Acad. Sci. 1999;893:13–18. doi: 10.1111/j.1749-6632.1999.tb07814.x. [DOI] [PubMed] [Google Scholar]
- Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, Wang HG, Atadja P, Bhalla K. Mol. Cancer Ther. 2003;2:971–984. [PubMed] [Google Scholar]
- Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, Shi X, Jiang BH. J. Biol. Chem. 2002;277:31963–31971. doi: 10.1074/jbc.M200082200. [DOI] [PubMed] [Google Scholar]
- Harris TK. IUBMB Life. 2003;55:117–126. doi: 10.1080/1521654031000115951. [DOI] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C. Proc. Natl. Acad. Sci. USA. 1998;95:8103–8107. doi: 10.1073/pnas.95.14.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgado-Madruga M, Wong AJ. Mol. Cell. Biol. 2003;23:4471–4484. doi: 10.1128/MCB.23.13.4471-4484.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Harris T, Altmann E, Mcallister D, Vlahos R, Robertson A, Cushman M, Wang Z, Stewart AG. Mol. Pharmacol. 2002;61:1053–1069. doi: 10.1124/mol.61.5.1053. [DOI] [PubMed] [Google Scholar]
- Ikeyama S, Kokkonen G, Shack S, Wang XT, Holbrook NJ. FASEB J. 2002;16:114–116. doi: 10.1096/fj.01-0409fje. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Mol. Cell. Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausalya S, Somogyi R, Orlofsky A, Prystowsky MB. J. Immunol. 2001;166:4721–4727. doi: 10.4049/jimmunol.166.7.4721. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Mol. Cell. Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Jhon DY, Lee KY. Exp. Mol. Med. 2004;36:157–164. doi: 10.1038/emm.2004.22. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Yamashita K, Takeoka T, Ohtsuki T, Suzuki Y, Takahashi R, Yamamoto K, Kaufmann SH, Uchiyama T, Sasada M, Takahashi A. J. Biol. Chem. 2002;277:33968–33977. doi: 10.1074/jbc.M203350200. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. Cancer Res. 2002;62:3691–3697. [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SY, Gomelsky M, Duan J, Zhang Z, Gomelsky L, Zhang X, Epstein PN, Ren J. J. Biol. Chem. 2004;279:11244–11252. doi: 10.1074/jbc.M308011200. [DOI] [PubMed] [Google Scholar]
- Lin HL, Liu TY, Wu CW, Chi CW. Cancer. 2001;92:500–509. doi: 10.1002/1097-0142(20010801)92:3<500::aid-cncr1348>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Biochem. Soc. Trans. 2003;31:3–578. doi: 10.1042/bst0310573. [DOI] [PubMed] [Google Scholar]
- Mansat-de Mas V, Bezombes C, Quillet-Mary A, Bettaieb A, D’orgeix AD, Laurent G, Jaffrezou JP. Mol. Pharmacol. 1999;56:867–874. doi: 10.1124/mol.56.5.867. [DOI] [PubMed] [Google Scholar]
- Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Genes Dev. 2004;18:1800–1805. doi: 10.1101/gad.1213804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. J. Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Martucci CP, Fishman J. Pharmacol. Ther. 1993;57:237–257. doi: 10.1016/0163-7258(93)90057-k. [DOI] [PubMed] [Google Scholar]
- Mildner M, Eckhart L, Lengauer B, Tschachler E. J. Biol. Chem. 2002;277:14146–14152. doi: 10.1074/jbc.M110687200. [DOI] [PubMed] [Google Scholar]
- Minutoli L, Altavilla D, Marini H, Passaniti M, Bitto A, Seminara P, Venuti FS, Famulari C, Macri A, Versaci A, Squadrito F. Life Sci. 2004;75:2853–2866. doi: 10.1016/j.lfs.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Koutsilieris M. Curr. Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- Mockridge JW, Benton EC, Andreeva LV, Latchman DS, Marber MS, Heads RJ. Biochem. Biophys. Res. Commun. 2000;273:322–327. doi: 10.1006/bbrc.2000.2934. [DOI] [PubMed] [Google Scholar]
- Mooberry SL. Drug Resist. Updates. 2003;6:355–361. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Haga S, Zhang HQ, Irani K, Suzuki S. Cell Death Differ. 2003;10:508–515. doi: 10.1038/sj.cdd.4401172. [DOI] [PubMed] [Google Scholar]
- Paez J, Sellers WR. Cancer Treat Res. 2003;115:145–167. [PubMed] [Google Scholar]
- Pei ZM, Murata Y, Benning G, Thomine S, Klusener B, Allen GJ, Grill E, Schroeder JI. Nature. 2000;406:731–734. doi: 10.1038/35021067. [DOI] [PubMed] [Google Scholar]
- Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, Dreyfus F, Lacombe C, Bouscary D. Oncogene. 2002;21:6587–6597. doi: 10.1038/sj.onc.1205923. [DOI] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S. Proc. Natl. Acad. Sci. USA. 2000;97:3207–3212. doi: 10.1073/pnas.060579697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi F, Prisco M, Dews M, Salomoni P, Grassilli E, Romano G, Calabretta B, Baserga R. Mol. Cell. Biol. 1999;19:7203–7215. doi: 10.1128/mcb.19.10.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanungo S, Basu A, Das M, Haldar S. Oncogene. 2002;21:4149–4157. doi: 10.1038/sj.onc.1205508. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P, Spiegel S, Grant S. Cancer Res. 2005;65:2422–2432. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Yu C, Reese E, Ahmed W, Hirsch K, Dent P, Grant S. Oncogene. 2003;22:6231–6242. doi: 10.1038/sj.onc.1206646. [DOI] [PubMed] [Google Scholar]
- Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, Ferrara JL. Proc. Natl. Acad. Sci. USA. 2004;101:3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego AC, Oliveira CR. Neurochem. Res. 2003;28:1563–1574. doi: 10.1023/a:1025682611389. [DOI] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Grant S. Cancer Res. 2003;63:3637–3645. [PubMed] [Google Scholar]
- Sawada M, Nakashima S, Kiyono T, Nakagawa M, Yamada J, Yamakawa H, Banno Y, Shinoda J, Nishimura Y, Nozawa Y, Sakai N. Oncogene. 2001;20:1368–1378. doi: 10.1038/sj.onc.1204207. [DOI] [PubMed] [Google Scholar]
- Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, Wange RL. Mol. Cell. Biol. 2000;20:6945–6957. doi: 10.1128/mcb.20.18.6945-6957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Nakamura M, Ishida E, Kishi M, Matsuyoshi S, Konishi N. Mol. Carcinog. 2004;39:1–9. doi: 10.1002/mc.10158. [DOI] [PubMed] [Google Scholar]
- Tinley TL, Leal RM, Randall-Hlubek DA, Cessac JW, Wilkens LR, Rao PN, Mooberry SL. Cancer Res. 2003;63:1538–1549. [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Wang X, McCullough KD, Franke TF, Holbrook NJ. J. Biol. Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- Yang JY, Widmann C. J. Biol. Chem. 2002;277:14641–14646. doi: 10.1074/jbc.M111540200. [DOI] [PubMed] [Google Scholar]
- Yu C, Rahmani M, Almenara J, Sausville EA, Dent P, Grant S. Oncogene. 2004;23:1364–1376. doi: 10.1038/sj.onc.1207248. [DOI] [PubMed] [Google Scholar]
- Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Circ. Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- Zhao S, Konopleva M, Cabreira-Hansen M, Xie Z, Hu W, Milella M, Estrov Z, Mills G, Andreeff M. Leukemia. 2004;18:267–275. doi: 10.1038/sj.leu.2403220. [DOI] [PubMed] [Google Scholar]
- Zhou DC, Kim SH, Ding W, Schultz C, Warrell RP, Jr, Gallagher RE. Blood. 2002;99:1356–1363. doi: 10.1182/blood.v99.4.1356. [DOI] [PubMed] [Google Scholar]
- Zoubine MN, Weston AP, Johnson DC, Campbell DR, Banerjee SK. Int. J. Oncol. 1999;15:639–646. doi: 10.3892/ijo.15.4.639. [DOI] [PubMed] [Google Scholar]