

Abstract
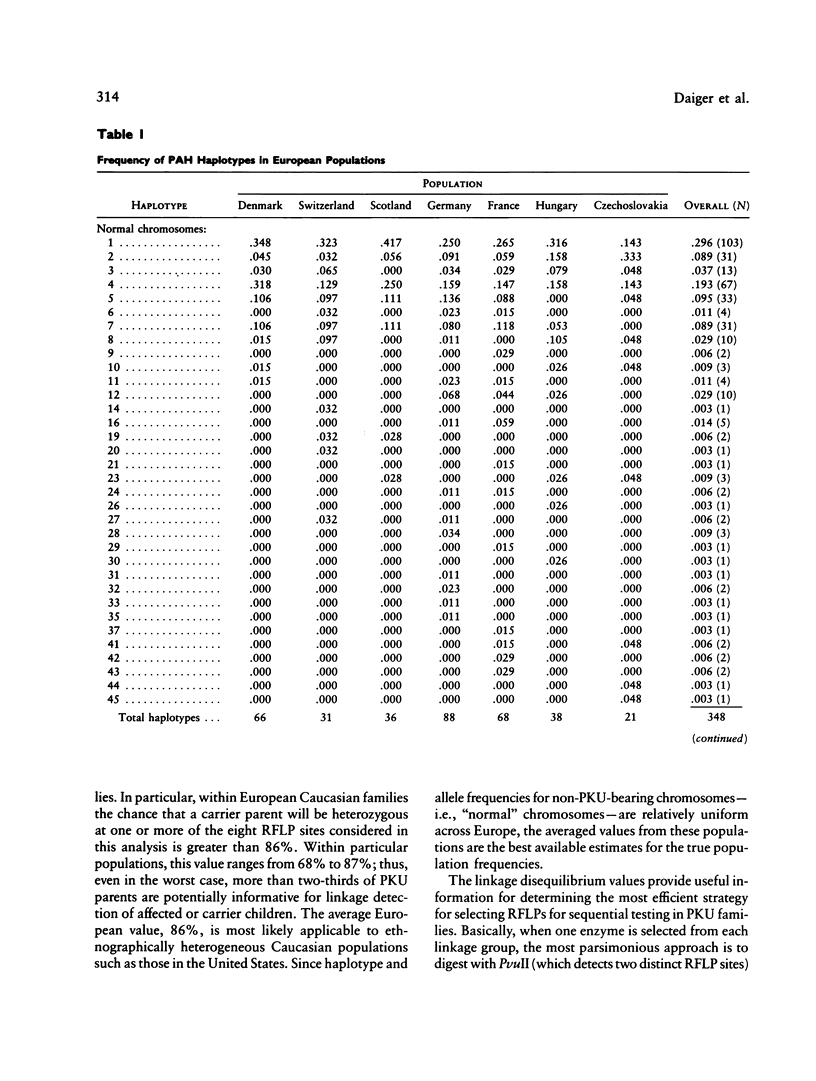
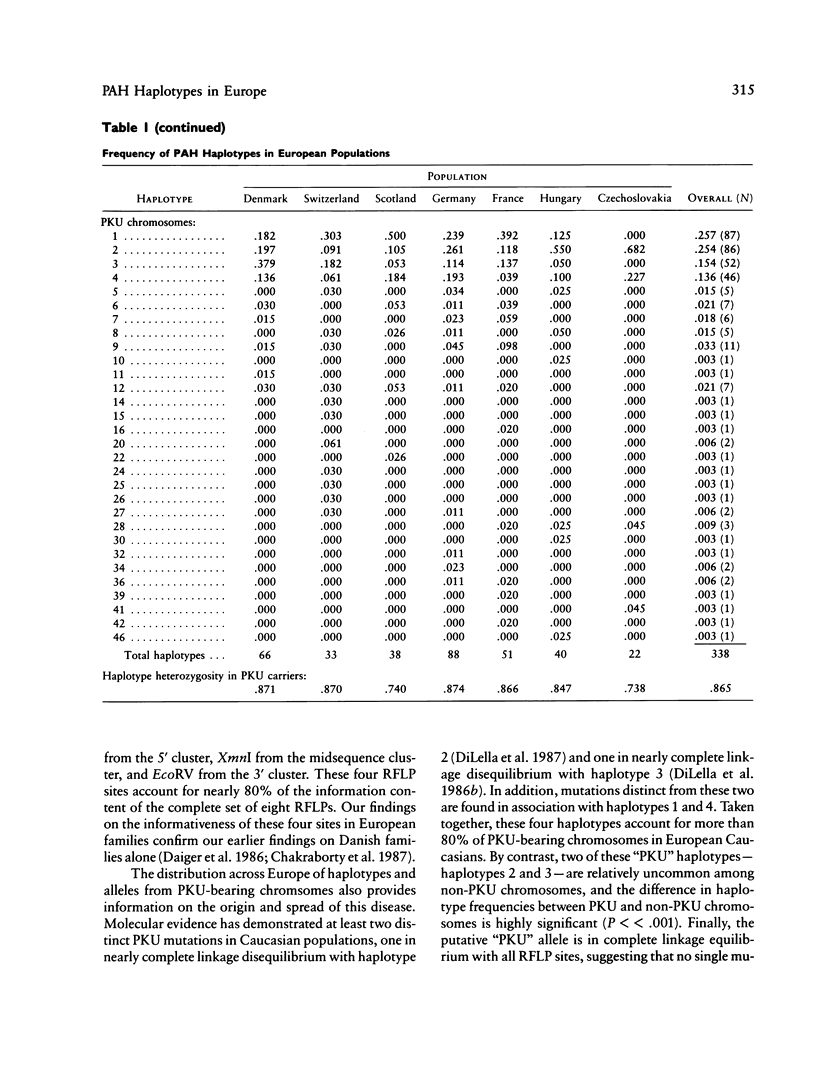
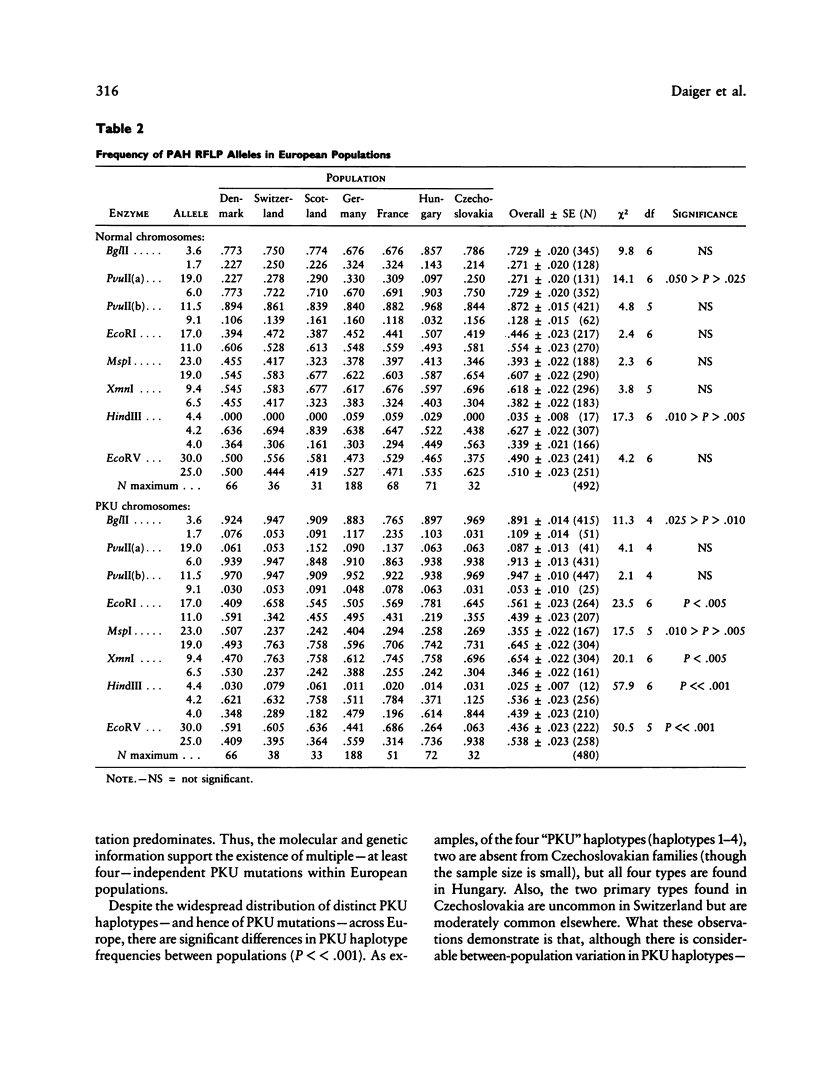
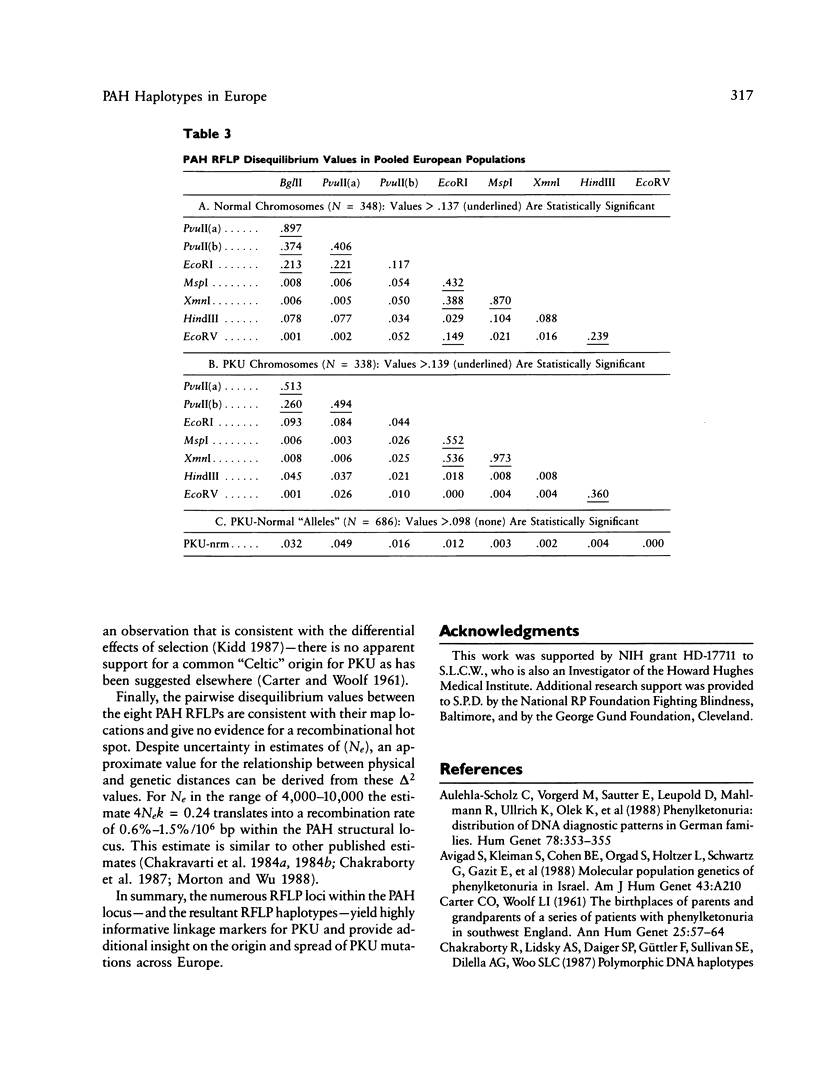
DNA haplotype data from the phenylalanine hydroxylase (PAH) locus are available from a number of European populations as a result of RFLP testing for genetic counseling in families with phenylketonuria (PKU). We have analyzed data from Hungary and Czechoslovakia together with published data from five additional countries–Denmark, Switzerland, Scotland, Germany, and France–representing a broad geographic and ethnographic range. The data include 686 complete chromosomal haplotypes for eight RFLP sites assayed in 202 unrelated Caucasian families with PKU. Forty-six distinct RFLP haplotypes have been observed to date, 10 unique to PKU-bearing chromosomes, 12 unique to non-PKU chromosomes, and the remainder found in association with both types. Despite the large number of haplotypes observed (still much less than the theoretical maximum of 384), five haplotypes alone account for more than 76% of normal European chromosomes and four haplotypes alone account for more than 80% of PKU-bearing chromosomes. We evaluated the distribution of haplotypes and alleles within these populations and calculated pairwise disequilibrium values between RFLP sites and between these sites and a hypothetical PKU “locus.” There are statistically significant differences between European populations in the frequencies of non-PKU chromosomal haplotypes (P = .025) and PKU chromosomal haplotypes (P < < .001). Haplotype frequencies of the PKU and non-PKU chromosomes also differ significantly (P < < .001. Disequilibrium values are consistent with the PAH physical map and support the molecular evidence for multiple, independent PKU mutations in Caucasians. However, the data do not support a single geographic origin for these mutations. Within these European populations a parent carrying a PKU mutation has an average probability of greater than 86% of being heterozygous–and hence informative for linkage–at one or more PAH RFLP sites. Thus these RFLP alleles and haplotypes provide an effective tool for linkage diagnosis of disease and carrier status in PKU families.
Full text
PDF




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aulehla-Scholz C., Vorgerd M., Sautter E., Leupold D., Mahlmann R., Ullrich K., Olek K., Horst J. Phenylketonuria: distribution of DNA diagnostic patterns in German families. Hum Genet. 1988 Apr;78(4):353–355. doi: 10.1007/BF00291734. [DOI] [PubMed] [Google Scholar]
- CARTER C. O., WOOLF L. I. The birthplaces of parents and grandparents of a series of patients with phenylketonuria in in south-east England. Ann Hum Genet. 1961 May;25:57–64. doi: 10.1111/j.1469-1809.1961.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Chakraborty R., Lidsky A. S., Daiger S. P., Güttler F., Sullivan S., Dilella A. G., Woo S. L. Polymorphic DNA haplotypes at the human phenylalanine hydroxylase locus and their relationship with phenylketonuria. Hum Genet. 1987 May;76(1):40–46. doi: 10.1007/BF00283048. [DOI] [PubMed] [Google Scholar]
- Chakravarti A., Buetow K. H., Antonarakis S. E., Waber P. G., Boehm C. D., Kazazian H. H. Nonuniform recombination within the human beta-globin gene cluster. Am J Hum Genet. 1984 Nov;36(6):1239–1258. [PMC free article] [PubMed] [Google Scholar]
- Chakravarti A., Phillips J. A., 3rd, Mellits K. H., Buetow K. H., Seeburg P. H. Patterns of polymorphism and linkage disequilibrium suggest independent origins of the human growth hormone gene cluster. Proc Natl Acad Sci U S A. 1984 Oct;81(19):6085–6089. doi: 10.1073/pnas.81.19.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger S. P., Lidsky A. S., Chakraborty R., Koch R., Güttler F., Woo S. L. Polymorphic DNA haplotypes at the phenylalanine hydroxylase locus in prenatal diagnosis of phenylketonuria. Lancet. 1986 Feb 1;1(8475):229–232. doi: 10.1016/s0140-6736(86)90771-3. [DOI] [PubMed] [Google Scholar]
- Daiger S. P., Reed L., Huang S. S., Zeng Y. T., Wang T., Lo W. H., Okano Y., Hase Y., Fukuda Y., Oura T. Polymorphic DNA haplotypes at the phenylalanine hydroxylase (PAH) locus in Asian families with phenylketonuria (PKU). Am J Hum Genet. 1989 Aug;45(2):319–324. [PMC free article] [PubMed] [Google Scholar]
- DiLella A. G., Kwok S. C., Ledley F. D., Marvit J., Woo S. L. Molecular structure and polymorphic map of the human phenylalanine hydroxylase gene. Biochemistry. 1986 Feb 25;25(4):743–749. doi: 10.1021/bi00352a001. [DOI] [PubMed] [Google Scholar]
- DiLella A. G., Marvit J., Brayton K., Woo S. L. An amino-acid substitution involved in phenylketonuria is in linkage disequilibrium with DNA haplotype 2. 1987 May 28-Jun 3Nature. 327(6120):333–336. doi: 10.1038/327333a0. [DOI] [PubMed] [Google Scholar]
- DiLella A. G., Marvit J., Lidsky A. S., Güttler F., Woo S. L. Tight linkage between a splicing mutation and a specific DNA haplotype in phenylketonuria. 1986 Aug 28-Sep 3Nature. 322(6082):799–803. doi: 10.1038/322799a0. [DOI] [PubMed] [Google Scholar]
- GUTHRIE R., SUSI A. A SIMPLE PHENYLALANINE METHOD FOR DETECTING PHENYLKETONURIA IN LARGE POPULATIONS OF NEWBORN INFANTS. Pediatrics. 1963 Sep;32:338–343. [PubMed] [Google Scholar]
- Güttler F. Hyperphenylalaninemia: diagnosis and classification of the various types of phenylalanine hydroxylase deficiency in childhood. Acta Paediatr Scand Suppl. 1980;280:1–80. [PubMed] [Google Scholar]
- Hertzberg M., Jahromi K., Ferguson V., Dahl H. H., Mercer J., Mickleson K. N., Trent R. J. Phenylalanine hydroxylase gene haplotypes in Polynesians: evolutionary origins and absence of alleles associated with severe phenylketonuria. Am J Hum Genet. 1989 Mar;44(3):382–387. [PMC free article] [PubMed] [Google Scholar]
- Kidd K. K. Phenylketonuria. Population genetics of a disease. 1987 May 28-Jun 3Nature. 327(6120):282–283. doi: 10.1038/327282a0. [DOI] [PubMed] [Google Scholar]
- Kwok S. C., Ledley F. D., DiLella A. G., Robson K. J., Woo S. L. Nucleotide sequence of a full-length complementary DNA clone and amino acid sequence of human phenylalanine hydroxylase. Biochemistry. 1985 Jan 29;24(3):556–561. doi: 10.1021/bi00324a002. [DOI] [PubMed] [Google Scholar]
- Ledley F. D., Koch R., Jew K., Beaudet A., O'Brien W. E., Bartos D. P., Woo S. L. Phenylalanine hydroxylase expression in liver of a fetus with phenylketonuria. J Pediatr. 1988 Sep;113(3):463–468. doi: 10.1016/s0022-3476(88)80629-2. [DOI] [PubMed] [Google Scholar]
- Lichter-Konecki U., Konecki D. S., DiLella A. G., Brayton K., Marvit J., Hahn T. M., Trefz F. K., Woo S. L. Phenylalanine hydroxylase deficiency caused by a single base substitution in an exon of the human phenylalanine hydroxylase gene. Biochemistry. 1988 Apr 19;27(8):2881–2885. doi: 10.1021/bi00408a032. [DOI] [PubMed] [Google Scholar]
- Lichter-Konecki U., Schlotter M., Konecki D. S., Labeit S., Woo S. L., Trefz F. K. Linkage disequilibrium between mutation and RFLP haplotype at the phenylalanine hydroxylase locus in the German population. Hum Genet. 1988 Apr;78(4):347–352. doi: 10.1007/BF00291733. [DOI] [PubMed] [Google Scholar]
- Lidsky A. S., Güttler F., Woo S. L. Prenatal diagnosis of classic phenylketonuria by DNA analysis. Lancet. 1985 Mar 9;1(8428):549–551. doi: 10.1016/s0140-6736(85)91208-5. [DOI] [PubMed] [Google Scholar]
- Lidsky A. S., Ledley F. D., DiLella A. G., Kwok S. C., Daiger S. P., Robson K. J., Woo S. L. Extensive restriction site polymorphism at the human phenylalanine hydroxylase locus and application in prenatal diagnosis of phenylketonuria. Am J Hum Genet. 1985 Jul;37(4):619–634. [PMC free article] [PubMed] [Google Scholar]
- Morton N. E., Wu D. Alternative bioassays of kinship between loci. Am J Hum Genet. 1988 Jan;42(1):173–177. [PMC free article] [PubMed] [Google Scholar]
- Rey F., Berthelon M., Caillaud C., Lyonnet S., Abadie V., Blandin-Savoja F., Feingold J., Saudubray J. M., Frézal J., Munnich A. Clinical and molecular heterogeneity of phenylalanine hydroxylase deficiencies in France. Am J Hum Genet. 1988 Dec;43(6):914–921. [PMC free article] [PubMed] [Google Scholar]
- Riess O., Michel A., Speer A., Meiske W., Cobet G., Coutelle C. Linkage disequilibrium between RFLP haplotype 2 and the affected PAH allele in PKU families from the Berlin area of the German Democratic Republic. Hum Genet. 1988 Apr;78(4):343–346. doi: 10.1007/BF00291732. [DOI] [PubMed] [Google Scholar]
- Sullivan S. E., Moore S. D., Connor J. M., King M., Cockburn F., Steinmann B., Gitzelmann R., Daiger S. P., Woo S. L. Haplotype distribution of the human phenylalanine hydroxylase locus in Scotland and Switzerland. Am J Hum Genet. 1989 May;44(5):652–659. [PMC free article] [PubMed] [Google Scholar]
- Woo S. L. Collation of RFLP haplotypes at the human phenylalanine hydroxylase (PAH) locus. Am J Hum Genet. 1988 Nov;43(5):781–783. [PMC free article] [PubMed] [Google Scholar]
- Woo S. L., Lidsky A. S., Güttler F., Chandra T., Robson K. J. Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. Nature. 1983 Nov 10;306(5939):151–155. doi: 10.1038/306151a0. [DOI] [PubMed] [Google Scholar]