

Abstract
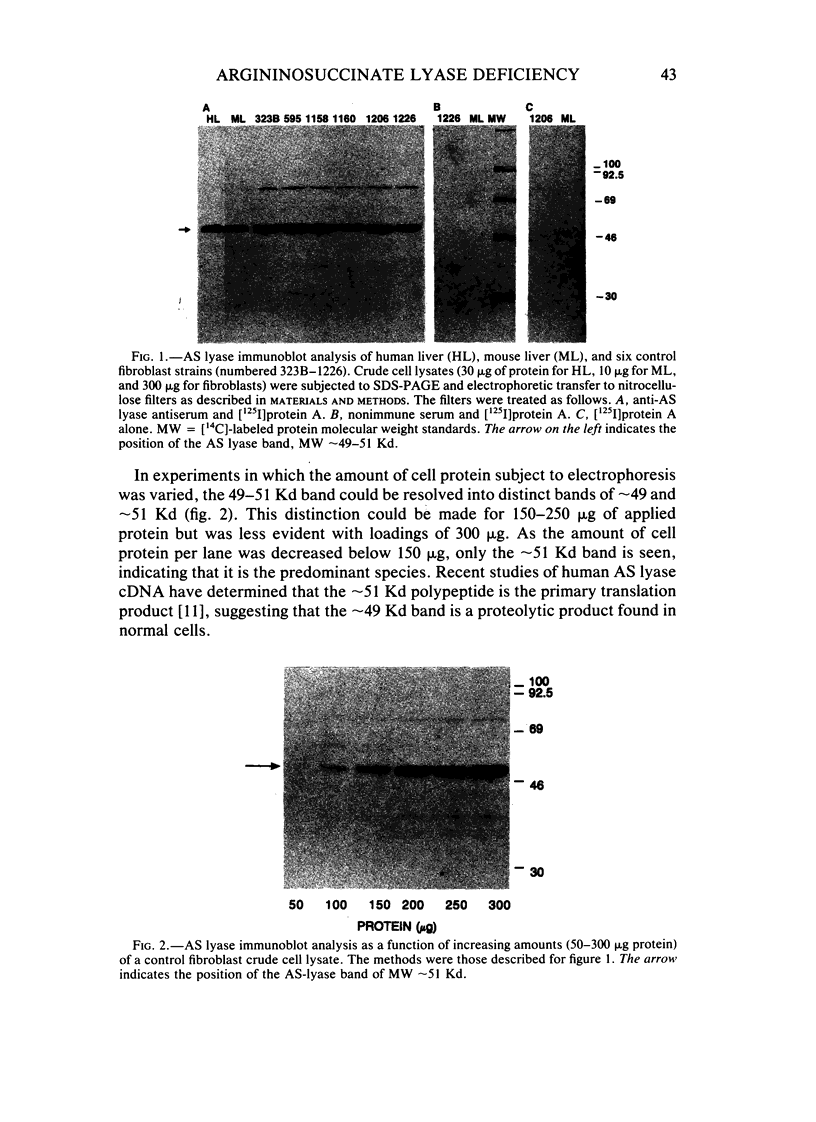
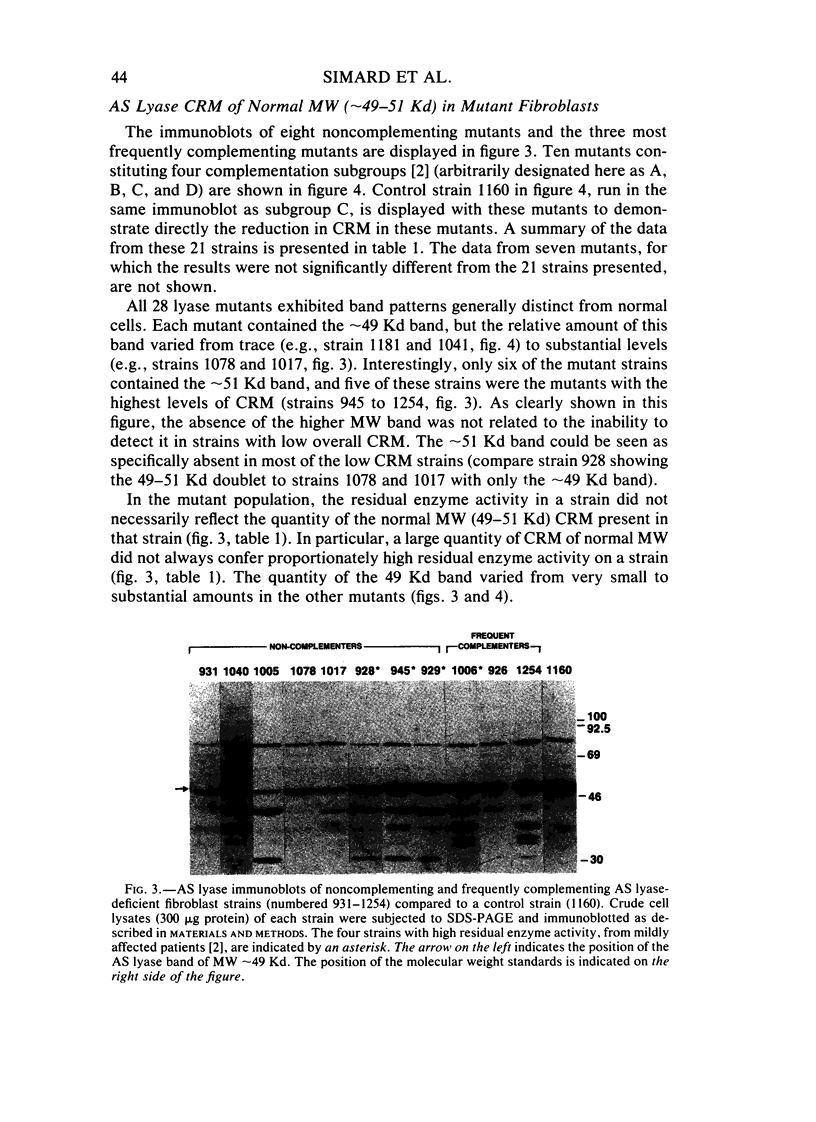
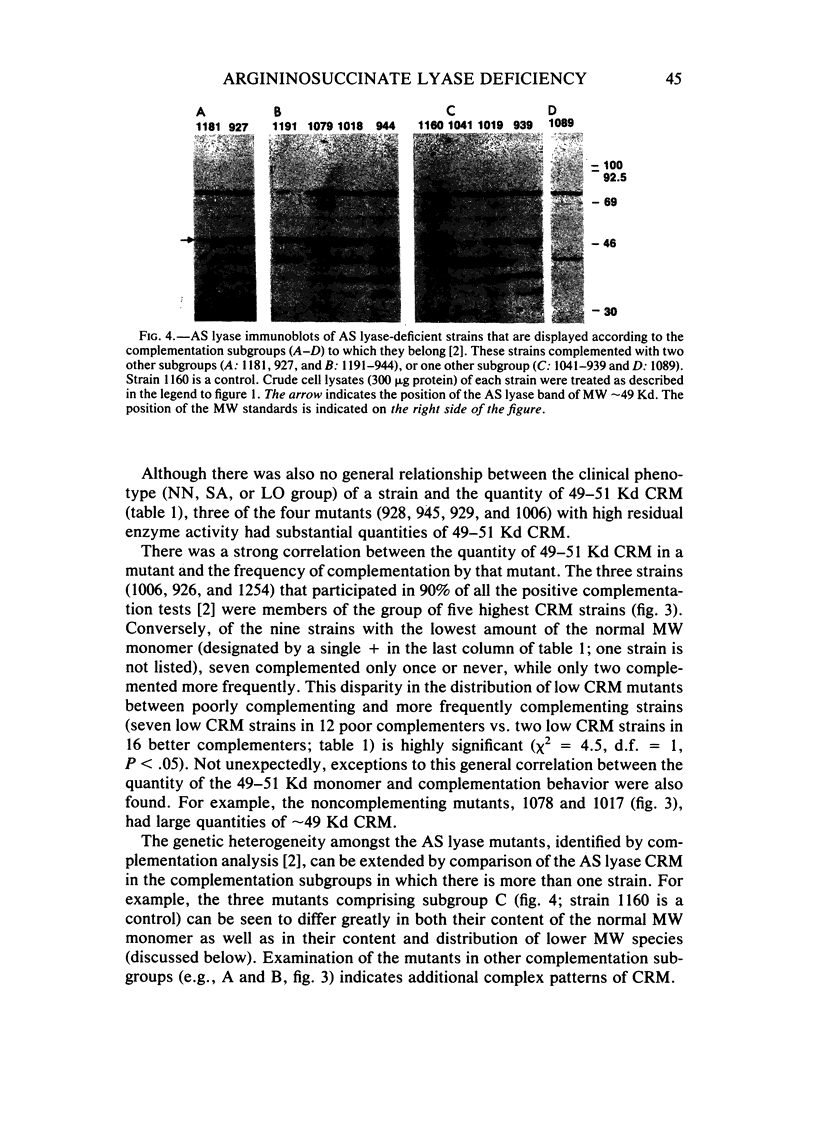
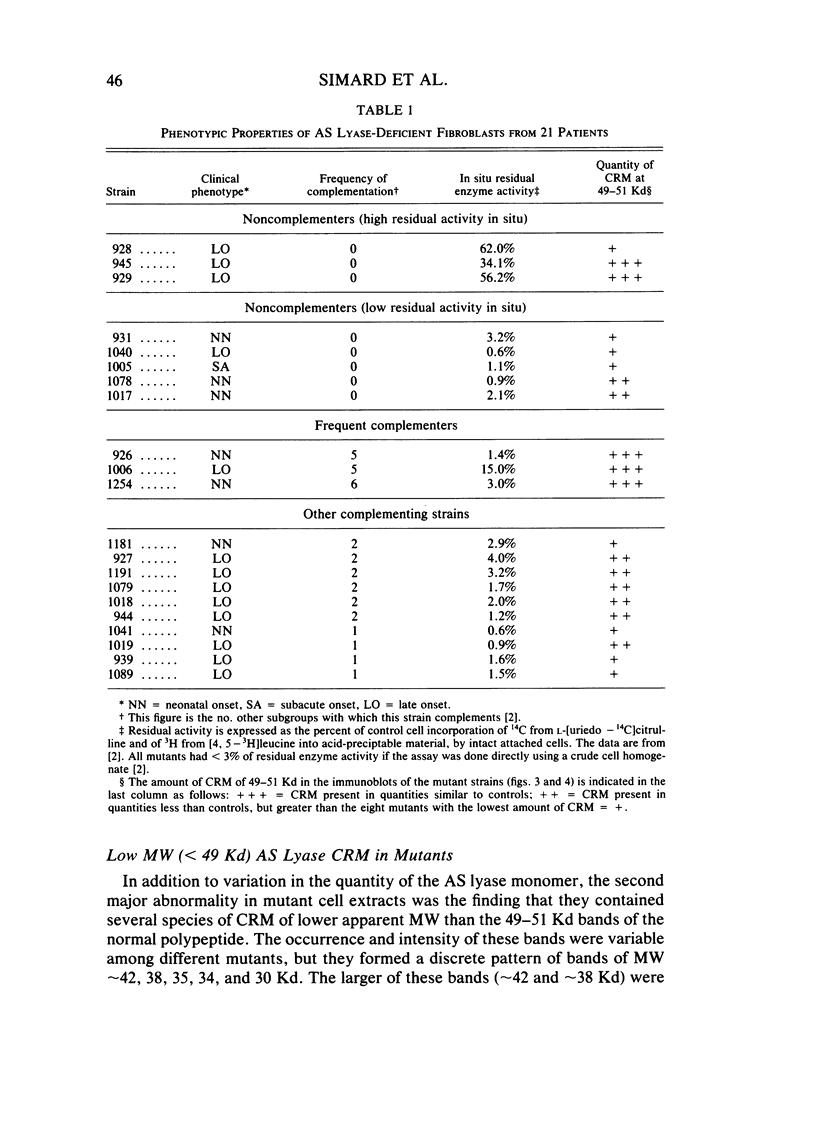
Argininosuccinate lyase (AS lyase) deficiency is an inborn error of the urea cycle with extensive clinical and genetic heterogeneity. We investigated the biochemical basis of the enzyme defect and the genetic heterogeneity in this disorder using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting of fibroblast extracts. The AS lyase monomer in control fibroblasts was present in two bands of approximately 51 and approximately 49 Kd. Each of 28 mutant strains had some cross-reactive material (CRM) of the lower (approximately 49 Kd) MW, in quantities ranging from trace to substantial levels. The approximately 51 Kd band was found in only six mutants with near-normal amounts of AS lyase CRM or high residual enzyme activity. The residual AS lyase enzyme activity in a mutant did not necessarily reflect the amount of the 49-51 Kd monomer in that strain. In contrast, there was a strong general correlation between the quantity of 49-51 Kd CRM in a mutant and the frequency of complementation by that mutant. In addition to the CRM of normal molecular weight (MW) (49-51 Kd), the majority of mutants (but not controls) had significant CRM present in one to five bands of MW less than 49 Kd. The immunoprecipitation of at least one of these low MW bands was inhibited by purified human AS lyase. Mutants indistinguishable by clinical, enzymatic, or complementation analysis have been shown to be heterogeneous in their content of AS lyase CRM, greatly extending the number of distinct mutant alleles identified at this locus. These data demonstrate that multiple unique mutations in the structural gene coding for the monomer cause AS lyase deficiency and that the AS lyase monomers made by these mutants may be unstable. Integration of these findings with enzymatic and complementation data has indicated the functional domain of the AS lyase monomer likely to be altered in certain mutants.
Full text
PDF








Images in this article



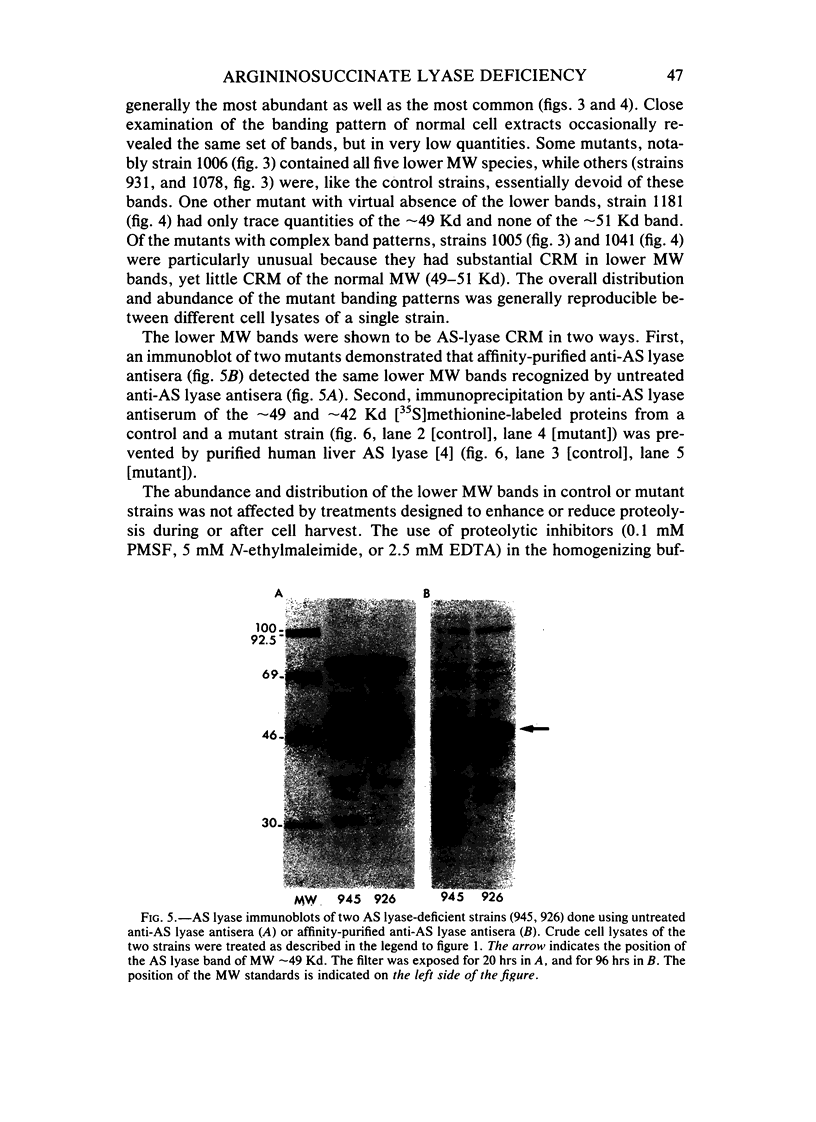
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Beacham I. R., Schweitzer B. W., Warrick H. M., Carbon J. The nucleotide sequence of the yeast ARG4 gene. Gene. 1984 Sep;29(3):271–279. doi: 10.1016/0378-1119(84)90056-8. [DOI] [PubMed] [Google Scholar]
- CRICK F. H., ORGEL L. E. THE THEORY OF INTER-ALLELIC COMPLEMENTATION. J Mol Biol. 1964 Jan;8:161–165. doi: 10.1016/s0022-2836(64)80156-x. [DOI] [PubMed] [Google Scholar]
- Capecchi M. R., Capecchi N. E., Hughes S. H., Wahl G. M. Selective degradation of abnormal proteins in mammalian tissue culture cells. Proc Natl Acad Sci U S A. 1974 Dec;71(12):4732–4736. doi: 10.1073/pnas.71.12.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. Mechanisms of intracellular protein breakdown. Annu Rev Biochem. 1982;51:335–364. doi: 10.1146/annurev.bi.51.070182.002003. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- McInnes R. R., Shih V., Chilton S. Interallelic complementation in an inborn error of metabolism: genetic heterogeneity in argininosuccinate lyase deficiency. Proc Natl Acad Sci U S A. 1984 Jul;81(14):4480–4484. doi: 10.1073/pnas.81.14.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien W. E., Barr R. H. Argininosuccinate lyase: purification and characterization from human liver. Biochemistry. 1981 Mar 31;20(7):2056–2060. doi: 10.1021/bi00510a049. [DOI] [PubMed] [Google Scholar]
- Palekar A. G., Mantagos S. Human liver arginiosuccinase purification and partial characterization. J Biol Chem. 1981 Sep 10;256(17):9192–9194. [PubMed] [Google Scholar]
- Robertson A., Hill W. G. Identity of different mutations for deleterious genes. Nature. 1983 Jan 13;301(5896):176–177. doi: 10.1038/301176a0. [DOI] [PubMed] [Google Scholar]
- Robinson B. H., Oei J., Sherwood W. G., Applegarth D., Wong L., Haworth J., Goodyer P., Casey R., Zaleski L. A. The molecular basis for the two different clinical presentations of classical pyruvate carboxylase deficiency. Am J Hum Genet. 1984 Mar;36(2):283–294. [PMC free article] [PubMed] [Google Scholar]
- Stanners C. P., Eliceiri G. L., Green H. Two types of ribosome in mouse-hamster hybrid cells. Nat New Biol. 1971 Mar 10;230(10):52–54. doi: 10.1038/newbio230052a0. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979 Sep;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]