

Abstract
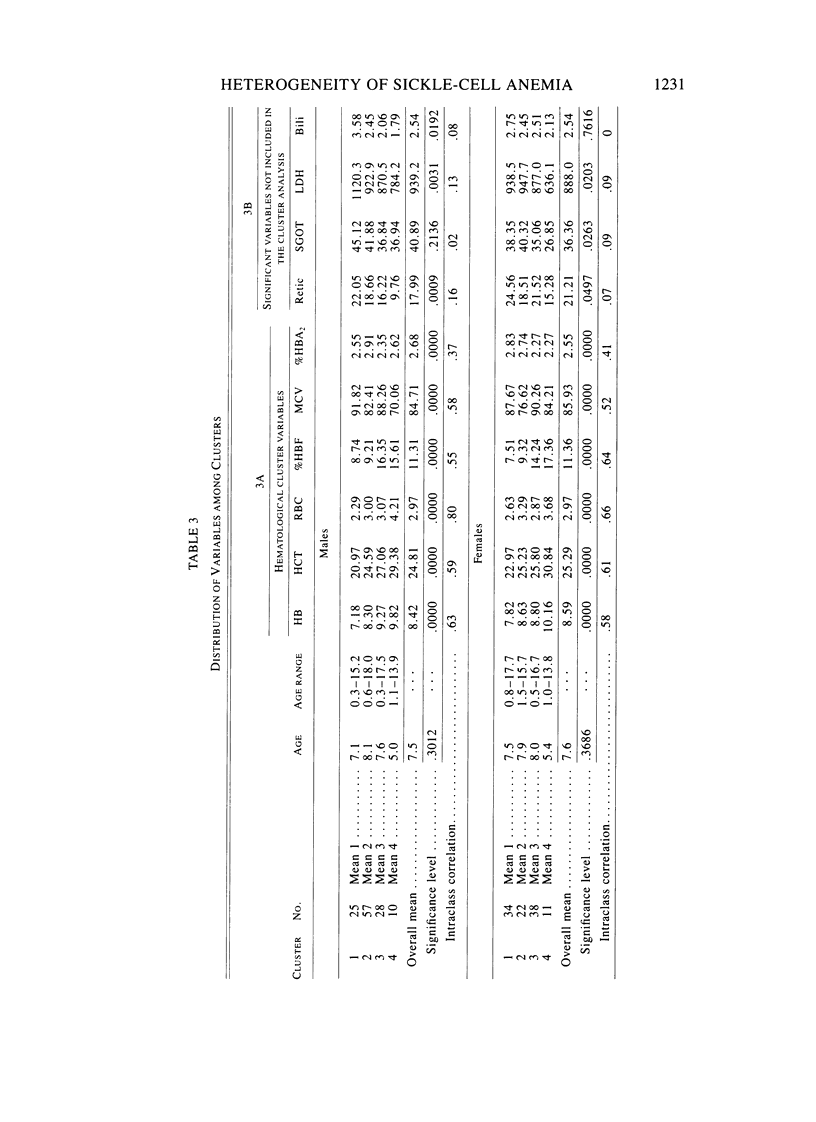
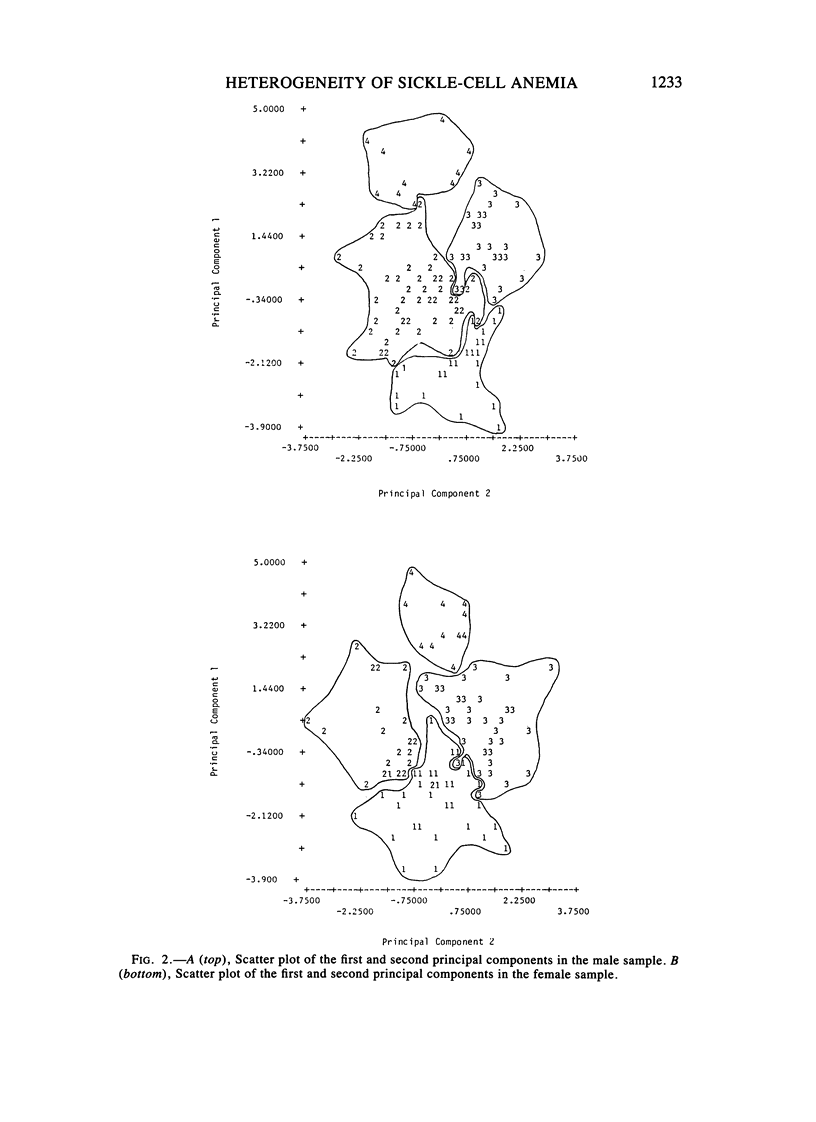
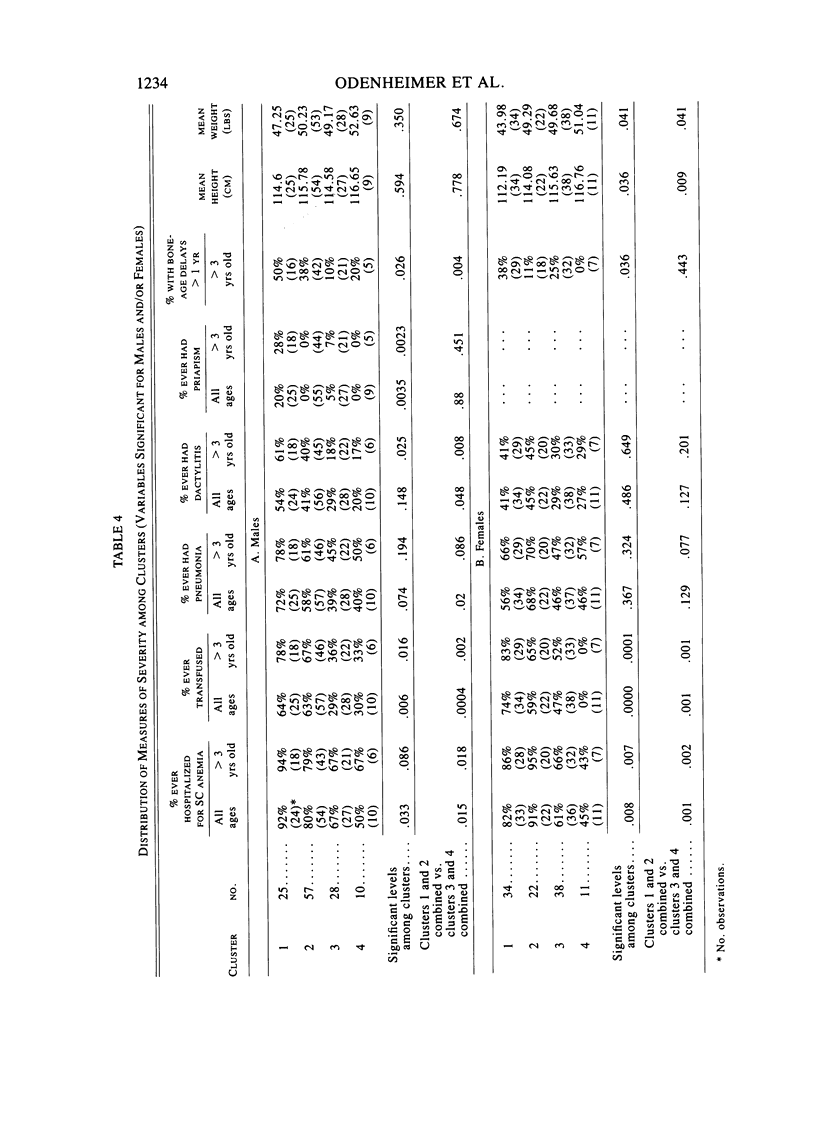
Factors that influence the heterogeneity of the disease expression of sickle-cell anemia are not well understood. This study examines the ability of a profile of six hematological variables (HB, HCT, RBC, %Hb F, MCV, and %HBA2) to predict the severity of disease measured on 225 patients ranging from 0.2 to 18 years of age. Four subgroups of patients were identified separately in each sex using cluster analysis techniques. In each sex, mean hemoglobin concentration and percent Hb F increased across the four clusters from 7 to 10 gm/dl and from 7% to 16%, respectively. Mean cell volumes were approximately 90, 80, 90, and 75 in groups 1, 2, 3, and 4, respectively; thus MCV did not increase in an orderly progression along with HB and %Hb F. We studied the distribution of four anthropometric, five physical examination, and seven clinical measures of disease severity among clusters. In each sex, subgroups differed significantly (P less than .05) for percent ever hospitalized for sickle-cell anemia, percent ever transfused, and percent with bone-age delays greater than 1 year. In addition, male clusters differed significantly for percent ever having had pneumonia, priapism, or dactylitis, and females differed significantly for height and weight. %Hb F and its inverse relationship with %HBA2 was more highly associated with the measures of severity than the degree of anemia or MCV. This study establishes the utility of a vector of hematological variables as a predictor of heterogeneity of measures of clinical manifestations among young patients with sickle-cell anemia. The role of alpha-thalassemia and genetic factors that affect Hb F levels were considered as possible explanations for the observed heterogeneity.
Full text
PDF










Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- ALLISON A. C. Properties of sickle-cell haemoglobin. Biochem J. 1957 Feb;65(2):212–219. doi: 10.1042/bj0650212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S. A. Milder variant of sickle-cell disease in Arabs in Kuwait associated with unusually high level of foetal haemoglobin. Br J Haematol. 1970 Nov;19(5):613–619. doi: 10.1111/j.1365-2141.1970.tb01645.x. [DOI] [PubMed] [Google Scholar]
- Bookchin R. M., Nagel R. L. Ligand-induced conformational dependence of hemoglobin in sickling interactios. J Mol Biol. 1971 Sep 14;60(2):263–270. doi: 10.1016/0022-2836(71)90292-0. [DOI] [PubMed] [Google Scholar]
- Davis L. R. Changing blood picture in sickle-cell anaemia from shortly after birth to adolescence. J Clin Pathol. 1976 Oct;29(10):898–901. doi: 10.1136/jcp.29.10.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover G. J., Boyer S. H., Charache S., Heintzelman K. Individual variation in the production and survival of F cells in sickle-cell disease. N Engl J Med. 1978 Dec 28;299(26):1428–1435. doi: 10.1056/NEJM197812282992603. [DOI] [PubMed] [Google Scholar]
- Dover G. J., Boyer S. H., Pembrey M. E. F-cell production in sickle cell anemia: regulation by genes linked to beta-hemoglobin locus. Science. 1981 Mar 27;211(4489):1441–1444. doi: 10.1126/science.6162200. [DOI] [PubMed] [Google Scholar]
- ERSLEV A. J., ATWATER J. EFFECT OF MEAN CORPUSCULAR HEMOGLOBIN CONCENTRATION ON VISCOSITY. J Lab Clin Med. 1963 Sep;62:401–406. [PubMed] [Google Scholar]
- Embury S. H., Dozy A. M., Miller J., Davis J. R., Jr, Kleman K. M., Preisler H., Vichinsky E., Lande W. N., Lubin B. H., Kan Y. W. Concurrent sickle-cell anemia and alpha-thalassemia: effect on severity of anemia. N Engl J Med. 1982 Feb 4;306(5):270–274. doi: 10.1056/NEJM198202043060504. [DOI] [PubMed] [Google Scholar]
- Felice A. E., Webber B., Miller A., Mayson S. M., Harris H. F., Henson J. B., Gravely M. E., Huisman T. H. The association of sickle cell anemia with heterozygous and homozygous alpha-thalassemia-2: in vitro HB chain synthesis. Am J Hematol. 1979;6(2):91–106. doi: 10.1002/ajh.2830060202. [DOI] [PubMed] [Google Scholar]
- Haghshenass M., Ismail-Beigi F., Clegg J. B., Weatherall D. J. Mild sickle-cell anaemia in Iran associated with high levels of fetal haemoglobin. J Med Genet. 1977 Jun;14(3):168–171. doi: 10.1136/jmg.14.3.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkness D. R. Hematological and clinical features of sickle cell diseases: a review. Hemoglobin. 1980;4(3-4):313–334. doi: 10.3109/03630268008996214. [DOI] [PubMed] [Google Scholar]
- Higgs D. R., Aldridge B. E., Lamb J., Clegg J. B., Weatherall D. J., Hayes R. J., Grandison Y., Lowrie Y., Mason K. P., Serjeant B. E. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med. 1982 Jun 17;306(24):1441–1446. doi: 10.1056/NEJM198206173062402. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Gunay U., Mason R. G., Vida L. N., Ferenc C. Sickle cell syndromes. I. Hemoglobin SC-alpha-thalassemia. Pediatr Res. 1976 Jun;10(6):613–620. doi: 10.1203/00006450-197606000-00010. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Koshy M., Mason R. G., Vida L. N. Sickle cell syndromes. II. The sickle cell anemia-alpha-thalassemia syndrome. J Pediatr. 1978 Apr;92(4):556–561. doi: 10.1016/s0022-3476(78)80287-x. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Mason R. G., Tremaine L. M., Vida L. N. Sickle cell syndromes. III. Silent-carrier alpha-thalassemia in combination with hemoglobin S and hemoglobin C. Pediatr Res. 1979 Oct;13(10):1109–1111. doi: 10.1203/00006450-197910000-00005. [DOI] [PubMed] [Google Scholar]
- Huisman T. H., Miller A., Schroeder W. A. A G gamma type of the hereditary persistence of fetal hemoglobin with beta chain production in cis. Am J Hum Genet. 1975 Nov;27(6):765–777. [PMC free article] [PubMed] [Google Scholar]
- Maniatis A., Frieman B., Bertles J. F. Increased expression in erythrocytic Ii antigens in sickle cell disease and sickle cell trait. Vox Sang. 1977 Jul;33(1):29–36. doi: 10.1111/j.1423-0410.1977.tb02233.x. [DOI] [PubMed] [Google Scholar]
- Martinez G., Colombo B. A new type of hereditary persistence of foetal haemoglobin: is a diffusible factor regulating gamma-chain synthesis? Nature. 1974 Dec 20;252(5485):735–736. doi: 10.1038/252735a0. [DOI] [PubMed] [Google Scholar]
- May A., Huehns E. R. The mechanism of the low oxygen affinity of red cells in sickle cell disease. Hamatol Bluttransfus. 1972;10:279–283. [PubMed] [Google Scholar]
- Papayannopoulou T., Chen P., Maniatis A., Stamatoyannopoulos G. Simultaneous assessment of i-antigenic expression and fetal hemoglobin in single red cells by immunofluorescence. Blood. 1980 Feb;55(2):221–232. [PubMed] [Google Scholar]
- Pembrey M. E., Wood W. G., Weatherall D. J., Perrine R. P. Fetal haemoglobin production and the sickle gene in the oases of Eastern Saudi Arabia. Br J Haematol. 1978 Nov;40(3):415–429. doi: 10.1111/j.1365-2141.1978.tb05813.x. [DOI] [PubMed] [Google Scholar]
- Perrine R. P., Brown M. J., Clegg J. B., Weatherall D. J., May A. Benign sickle-cell anaemia. Lancet. 1972 Dec 2;2(7788):1163–1167. doi: 10.1016/s0140-6736(72)92592-5. [DOI] [PubMed] [Google Scholar]
- Powars D. R., Schroeder W. A., Weiss J. N., Chan L. S., Azen S. P. Lack of influence of fetal hemoglobin levels or erythrocyte indices on the severity of sickle cell anemia. J Clin Invest. 1980 Mar;65(3):732–740. doi: 10.1172/JCI109720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON A. R., ROBSON M., HARRISON A. P., ZUELZER W. W. A new technique for differentiation of hemoglobin. J Lab Clin Med. 1957 Nov;50(5):745–752. [PubMed] [Google Scholar]
- SINGER K., CHERNOFF A. I., SINGER L. Studies on abnormal hemoglobins. II. Their identification by means of the method of fractional denaturation. Blood. 1951 May;6(5):429–435. [PubMed] [Google Scholar]
- Seakins M., Gibbs W. N., Milner P. F., Bertles J. F. Erythrocyte Hb-S concentration. An important factor in the low oxygen affinity of blood in sickle cell anemia. J Clin Invest. 1973 Feb;52(2):422–432. doi: 10.1172/JCI107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serjeant G. R., Ashcroft M. T. Delayed skeletal maturation in sickle cell anemia in Jamaica. Johns Hopkins Med J. 1973 Feb;132(2):95–102. [PubMed] [Google Scholar]
- Serjeant G. R. Fetal haemoglobin in homozygous sickle cell disease. Clin Haematol. 1975 Feb;4(1):109–122. [PubMed] [Google Scholar]
- Serjeant G. R., Richards R., Barbor P. R., Milner P. F. Relatively benign sickle-cell anaemia in 60 patients aged over 30 in the West Indies. Br Med J. 1968 Jul 13;3(5610):86–91. doi: 10.1136/bmj.3.5610.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serjeant G. R., Serjeant B. E., Mason K. Heterocellular hereditary persistence of fetal haemoglobin and homozygous sickle-cell disease. Lancet. 1977 Apr 9;1(8015):795–796. doi: 10.1016/s0140-6736(77)92976-2. [DOI] [PubMed] [Google Scholar]
- Sing C. F., Risser D. R., Howatt W. F., Erickson R. P. Phenotypic heterogeneity in cystic fibrosis. Am J Med Genet. 1982 Oct;13(2):179–195. doi: 10.1002/ajmg.1320130209. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos G., Wood W. G., Papayannopoulou T., Nute P. E. A new form of hereditary persistence of fetal hemoglobin in blacks and its association with sickle cell trait. Blood. 1975 Nov;46(5):683–692. [PubMed] [Google Scholar]
- Steinberg M. H., Dreiling B. J., Morrison F. S., Necheles T. F. Mild sickle cell disease. Clinical and laboratory studies. JAMA. 1973 Apr 16;224(3):317–321. [PubMed] [Google Scholar]
- Stevens M. C., Hayes R. J., Vaidya S., Serjeant G. R. Fetal hemoglobin and clinical severity of homozygous sickle cell disease in early childhood. J Pediatr. 1981 Jan;98(1):37–41. doi: 10.1016/s0022-3476(81)80529-x. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., Cartner R., Clegg J. B., Wood W. G., Macrae I. A., Mackenzie A. A form of hereditary persistence of fetal haemoglobin characterized by uneven cellular distribution of haemoglobin F and the production of haemoglobins A and A2 in homozygotes. Br J Haematol. 1975 Feb;29(2):205–220. doi: 10.1111/j.1365-2141.1975.tb01815.x. [DOI] [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Blankson J., McNeil J. R. A new sickling disorder resulting from interaction of the genes for haemoglobin S and alpha-thalassaemia. Br J Haematol. 1969 Dec;17(6):517–526. doi: 10.1111/j.1365-2141.1969.tb01402.x. [DOI] [PubMed] [Google Scholar]
- Wood W. G., Weatherall D. J., Clegg J. B., Hamblin T. J., Edwards J. H., Barlow A. M. Heterocellular hereditary persistence of fetal haemoglobin (heterocellular HPFH) and its interaction with beta thalassaemia. Br J Haematol. 1977 Aug;36(4):461–473. doi: 10.1111/j.1365-2141.1977.tb00986.x. [DOI] [PubMed] [Google Scholar]
- van Enk A., Lang A., White J. M., Lehmann H. Benign obstetric history in women with sickle-cell anaemia associated with -thalassaemia. Br Med J. 1972 Dec 2;4(5839):524–526. doi: 10.1136/bmj.4.5839.524. [DOI] [PMC free article] [PubMed] [Google Scholar]