

Abstract
Proteome analysis is most commonly accomplished by a combination of two-dimensional gel electrophoresis (2DE) to separate and visualize proteins and mass spectrometry (MS) for protein identification. Although this technique is powerful, mature, and sensitive, questions remain concerning its ability to characterize all of the elements of a proteome. In the current study, more than 1,500 features were visualized by silver staining a narrow pH range (4.9–5.7) 2D gel in which 0.5 mg of total soluble yeast protein was separated. Fifty spots migrating to a region of 4 cm2 were subjected to MS protein identification. Despite the high sample load and extended electrophoretic separation, proteins from genes with codon bias values of <0.1 (lower abundance proteins) were not found, even though fully one-half of all yeast genes fall into that range. Proteins from genes with codon bias values of <0.1 were found, however, if protein amounts exceeding the capacity of 2DE were fractionated and analyzed. We conclude that the large range of protein expression levels limits the ability of the 2DE-MS approach to analyze proteins of medium to low abundance, and thus the potential of this technique for proteome analysis is likewise limited.
The genomics revolution has changed the paradigm for the comprehensive analysis of biological processes and systems. It is now hypothesized that biological processes and systems can be described based on the comparison of global, quantitative gene expression patterns from cells or tissues representing different states. To test this hypothesis, it is essential that methods for the precise measurement of gene expression be developed and applied.
Several methods, including serial analysis of gene expression, oligonucleotide and cDNA microarrays, and large-scale sequencing of expressed sequence tags have been developed to globally and quantitatively measure gene expression at the mRNA level (1, 2). The discovery of posttranscriptional mechanisms that control rate of synthesis and half-life of proteins (3) and the ensuing nonpredictive correlation between mRNA and protein levels expressed by a particular gene (4, 5) indicate that direct measurement of protein expression also is essential for the analysis of biological processes and systems.
Global analysis of gene expression at the protein level is now also termed proteomics. The standard method for quantitative proteome analysis combines protein separation by high-resolution (isoelectric focusing/SDS-PAGE) two-dimensional gel electrophoresis (2DE) with mass spectrometric (MS) or tandem MS (MS/MS) identification of selected protein spots. Important technical advances related to 2DE and protein MS have increased sensitivity, reproducibility, and throughput of proteome analysis while creating an integrated technology.
By using 2DE with extended pH range and high-sensitivity protein identification by electrospray ionization and MS/MS, we have evaluated the potential of the 2DE-MS strategy to serve as the technology base for comprehensive and quantitative proteome analysis.
Materials and Methods
Yeast Strain and Growth Conditions.
The source of protein for all experiments was yeast strain YPH499 (MATa ura3-52 lys2-801 ade2-101 leu2-1 his3-200 trp1-63) (6). Cells were grown to log phase (2 × 107 cells/ml) in yeast extract/peptone rich medium with 2% galactose at 30°C. Protein was harvested as described by Garrels and coworkers (7). Harvested protein was lyophilized, resuspended in isoelectric focusing gel rehydration solution, and stored at −80°C.
Preparation of Narrow-Range Immobilized pH Gradient (nrIPG) Strips.
nrIPGs were cast in a U-frame on a GelBond PAG film according to nomograms (application note 324, Amersham Pharmacia Biotech). The pH gradient was poured in a 125 × 260 × 0.5-mm frame resulting in gradient pH strips of approximately 25 cm in length. For casting a 4.5–5.5 pH gradient, the following acidic and basic solutions were mixed. Acidic solution: 1 ml of acrylamide (30% T/3% C), 0.358 ml of Immobiline pK 4.6, 0.163 ml of Immobiline pK 9.3, 2.15 ml of 87% glycerol, 7.5 μl of N,N,N′,N′-tetramethylethylenediamine (TEMED), and 7.5 μl of ammonium persulfate (equilibrated to 7.5 ml of total volume). Basic solution: 1 ml of acrylamide (30% T, 3% C), 0.657 ml of Immobiline pK 4.6, 0.604 ml of Immobiline pK 9.3, 0.8 ml of 87% glycerol, 7.5 μl of TEMED, and 7.5 μl of ammonium persulfate (equilibrated to 7.5 ml of total volume). The pH of the basic solution was neutralized with 4 M HCl before polymerization. After a 16-h polymerization at 37°C, and drying, approximately 0.3 mm wide nrIPG strips were cut.
2DE and Protein Identification.
Protein (500 μg) was mixed with IPG rehydration buffer (8 M urea/2% NP-40/10 mM DTT; final volume = 360 μl). The strips were allowed to rehydrate overnight, focused, equilibrated, apposed to the second dimension (10%) gels, and run as described (8). Gels were silver stained (9) and dried between two sheets of cellophane. A 4-cm2 region of the gel was arbitrarily selected, and 50 protein spots were excised and subjected to in-gel tryptic digestion (9). Extracted peptides were stored at −20°C until analysis by microcapillary liquid chromatography (LC)-MS/MS as described (5, 10, 11). MS/MS spectra were searched automatically against the yeast protein database by using sequest software (12). Multiple peptides from each protein generally were detected, adding confidence to the protein identifications.
Identification of Low Abundance Proteins.
Soluble yeast protein was harvested as described above. Fifty milligrams of protein in loading buffer (75 mM Tris buffer, pH 6.8/10% glycerol/0.5% SDS/0.01% bromophenol blue) was loaded into a single large well (10 cm in length) of a 10% polyacrylamide gel slab (150 × 120 × 15 mm). The gel was run at 40 mA constant and stained with colloidal Coomassie blue. A strip (3 mm × 100 mm) representing proteins with molecular masses of ≈68–85 kDa was cut from the gel. The gel strip was cut into 1-mm3 pieces, which were subjected to in-gel digestion with trypsin (9). Extracted peptides were lyophilized, resolubilized in buffer A (5 mM KH2PO4/25% acetonitrile, pH 3.0), loaded onto a polysulfoethyl A column (2.1 × 250 mm; PolyLC, Columbia, MD), and eluted with buffer B (same as buffer A with 250 mM KCl added) in a 60-min linear gradient from 0 to 100% B at a flow rate of 200 μl/min. Fractions were collected every minute. Five fractions (numbers 10, 15, 20, 25, and 30) were reduced in volume from 200 μl to 50 μl, and 5 μl of each sample was independently analyzed by microcapillary LC-MS/MS. Gradients for on-line LC-MS were extended to 2 h, and one MS scan was followed by four MS/MS scans on the four most-intense peptide ions. More than 4,000 MS/MS spectra were collected in each 2-h run. sequest analysis was performed against the yeast database without the tryptic constraint, and a peptide sequence was considered to be a match if (i) the cross-correlation score was greater than 2.0; (ii) the ends were tryptic, and (iii) the molecular mass of the matched protein was between 68 and 85 kDa.
Results
Analysis of Yeast Proteins by nrIPG-2DE.
A standard 2D gel with an isoelectric focusing range of pH 3–10 and a protein load of 50 μg results in a gel with more than 1,000 features visualized by silver staining (5). By narrowing the pH range from 7 to 1 unit/20 cm gel and by increasing the protein load to 500 μg, more than 1,500 spots could be visualized on a single silver-stained gel (Fig. 1A). By extrapolation, if seven 2D gels were run, each with a connective 1 pH unit range, more than 10,000 features might be visualized from a yeast cell lysate. This number of features surpasses the number of proteins predicted to be potentially expressed in yeast (6,139 proteins) (13). To characterize the proteins detected by nrIPG-2DE, an arbitrary region of 4 cm2 was chosen from the gel (Fig. 1B), and the proteins migrating to 50 spots in the selected area were identified by MS techniques (11, 14) (Table 1).
Figure 1.
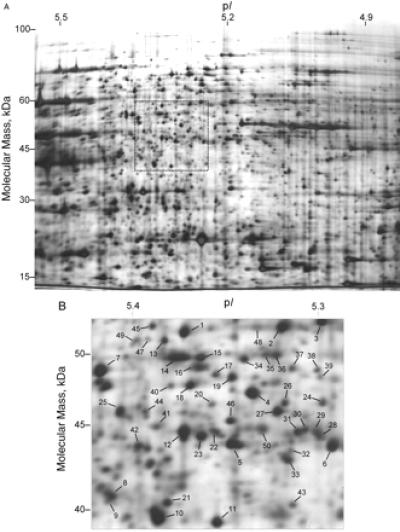
(A) Narrow pH range isoelectric focusing 2D gel (pH 4.9–5.5). Soluble yeast protein (500 μg) was loaded onto the gel. More than 1,500 features were visible by silver staining. (B) An arbitrary 4-cm2 region of the gel was selected for analysis. Numbers show the 50 spots that were identified by the MS techniques described in the text.
Table 1.
Proteins identified in selected area from Fig. 1B
| Spot no. | Molecular mass, kDa | pI* | Codon bias* | Gene name* |
|---|---|---|---|---|
| 1 | 53.6 | 5.26 | 0.61 | HXK1 |
| 2 | 53.6 | 5.26 | 0.61 | HXK1 |
| 3 | 53.8 | 5.13 | 0.76 | HXK2 |
| 4 | 69.6 | 4.96 | 0.83 | SSA1 |
| 5 | 66.4 | 5.23 | 0.91 | SSB1 |
| 66.4 | 5.29 | 0.88 | SSB2 | |
| 6 | 66.4 | 5.23 | 0.91 | SSB1 |
| 7 | 52.7 | 5.45 | 0.44 | YFR044C |
| 48.3 | 7.24 | 0.33 | KGD2 | |
| 8 | 69.6 | 4.96 | 0.83 | SSA1 |
| 62.0 | 5.22 | 0.27 | DAK1 | |
| 41.9 | 5.28 | 0.38 | TOM40 | |
| 46.7 | 6.27 | 0.93 | ENO1 | |
| 9 | 69.6 | 4.96 | 0.83 | SSA1 |
| 10 | 93.1 | 6.03 | 0.89 | EFT1 |
| 35.6 | 5.37 | 0.34 | CAR1 | |
| 11 | 38.6 | 5.15 | 0.41 | THR1 |
| 12 | 42.7 | 5.18 | 0.62 | GPD1 |
| 13 | 59.2 | 5.91 | 0.2 | AIP2 |
| 14 | 49.4 | 5.37 | 0.75 | GDH1 |
| 52.7 | 5.45 | 0.44 | YFR044C | |
| 15 | 50.1 | 5.29 | 0.45 | SUB2 |
| 16 | 50.1 | 5.29 | 0.45 | SUB2 |
| 48.3 | 7.24 | 0.33 | KGD2 | |
| 49.4 | 5.37 | 0.75 | GDH1 | |
| 17 | 57.8 | 4.82 | 0.55 | HSP60 |
| 18 | 57.8 | 4.82 | 0.55 | HSP60 |
| 49.6 | 5.39 | 0.30 | PRO2 | |
| 19 | 54.4 | 5.38 | 0.66 | ALD6 |
| 20 | 69.6 | 4.96 | 0.83 | SSA1 |
| 66.4 | 5.23 | 0.91 | SSB1 | |
| 54.4 | 7.55 | 0.97 | PYK1 | |
| 21 | 41.7 | 5.39 | 0.83 | ACT1 |
| 22 | 66.4 | 5.32 | 0.91 | SSB1 |
| 66.4 | 5.37 | 0.88 | SSB2 | |
| 23 | 66.4 | 5.32 | 0.91 | SSB1 |
| 24 | 52.7 | 5.18 | 0.57 | ATP2 |
| 25 | 69.6 | 4.96 | 0.83 | SSA1 |
| 69.3 | 4.90 | 0.89 | SSA2 | |
| 46.7 | 6.27 | 0.93 | ENO1 | |
| 26 | 49.4 | 5.30 | 0.19 | GDH3 |
| 69.7 | 4.72 | 0.59 | KAR2 | |
| 27 | 66.4 | 5.23 | 0.91 | SSB1 |
| 49.4 | 5.30 | 0.19 | GDH3 | |
| 28 | 66.4 | 5.29 | 0.88 | SSB2 |
| 29 | 66.4 | 5.29 | 0.88 | SSB2 |
| 81.2 | 4.69 | 0.66 | HSP82 | |
| 30 | 42.7 | 5.18 | 0.62 | GPD1 |
| 31 | 42.7 | 5.18 | 0.62 | GPD1 |
| 66.4 | 5.23 | 0.91 | SSB1 | |
| 69.6 | 4.96 | 0.83 | SSA1 | |
| 32 | 69.3 | 4.90 | 0.89 | SSA2 |
| 33 | 69.3 | 4.90 | 0.89 | SSA2 |
| 49.4 | 5.30 | 0.19 | GDH3 | |
| 75.2 | 5.74 | 0.50 | GRS1 | |
| 34 | 47.9 | 5.32 | 0.27 | RPT3 |
| 35 | 49.4 | 5.37 | 0.75 | GDH1 |
| 47.9 | 5.38 | 0.27 | RPT3 | |
| 50.1 | 5.29 | 0.45 | SUB2 | |
| 48.4 | 5.12 | 0.52 | TIF3 | |
| 67.7 | 5.09 | 0.55 | VMA1 | |
| 52.7 | 5.45 | 0.44 | YFR044C | |
| 36 | 48.4 | 5.17 | 0.52 | TIF3 |
| 52.7 | 5.45 | 0.44 | YFR044C | |
| 37 | 54.5 | 5.38 | 0.66 | ALD6 |
| 38 | 69.5 | 4.97 | 0.24 | SSA4 |
| 39 | 57.8 | 4.82 | 0.55 | HSP60 |
| 40 | 66.4 | 5.29 | 0.88 | SSB2 |
| 41 | 46.8 | 5.70 | 0.96 | ENO2 |
| 42 | 49.4 | 5.37 | 0.75 | GDH1 |
| 43 | 42.1 | 5.21 | 0.64 | SAM2 |
| 44 | 48.1 | 4.90 | 0.25 | RPT5 |
| 45 | 60.9 | 5.34 | 0.17 | HIS7 |
| 46 | 45.2 | 5.25 | 0.55 | NPL3 |
| 47 | 51.0 | 5.39 | 0.42 | YER081W |
| 48 | 53.6 | 5.26 | 0.61 | HXK1 |
| 49 | 91.8 | 4.80 | 0.41 | CDC48 |
| 50 | 42.7 | 5.18 | 0.62 | GPD1 |
Gene names, predicted molecular masses, pl values, and codon bias values are from the Yeast Protein Database (YPD) (13).
Evaluation of Protein Abundance Based on Codon Bias Value.
The codon bias value for a gene is its propensity to use only one of several codons to incorporate a specific amino acid into the polypeptide chain (5, 15, 16). It is known that more highly expressed proteins have large codon bias values (>0.2). The proteins identified in the selected area were examined with respect to the codon bias values of their respective genes. Fig. 2A shows the distribution of codon bias values (codon bias distribution; CBD) for the entire yeast genome with the largest number of genes falling in the 0.0–0.1 category. Fig. 2B shows the CBD for genes predicted from the yeast genome to be present in the selected gel region based on theoretical pI and predicted molecular mass values. There are 58 proteins predicted to run within the selected area, and their CBD is similar to the distribution of the entire genome. Fig. 2C shows the CBD for the genes identified from the selected gel area. No protein with a codon bias value of <0.1 was found, even though more than one-half of yeast genes fall into this range. Fig. 2D shows the CBD of the genes both predicted and found in the gel region. Only 14 of the 58 proteins were found within the area. Additionally, no protein was found that had a codon bias value <0.1 even though 10 of these proteins (i.e., YPL053C, YLL006W, YNL330C, YMR270C, YGL100W, YGL213C, YMR028W, YDR400W, YBR246W, and YDL024C) are likely expressed because mRNA expression has been reported (6). These data indicate that proteins of lesser abundance were not detected and that a larger number of proteins migrated to the selected region of the gel than were predicted.
Figure 2.
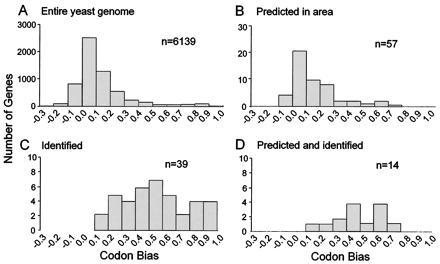
Codon bias value distributions in the yeast genome and on a narrow-range isoelectric focusing gel. (A) The entire yeast genome. (B) Genes predicted to run within selected area based on pI (5.25–5.45) and molecular mass (38–54 kDa). (C) Unique genes detected in selected area. (D) Genes both predicted and found. Codon bias value is a predictor of protein abundance with lower abundance proteins generally arising from genes with codon bias values <0.2. No low abundance proteins were detected with codon bias values <0.1 even though more than one-half of the proteins in yeast fall into this category.
Evaluation of Comigrating Proteins and Differential Migration Products.
Products expressed from a single gene can migrate to multiple spots on 2D gels for a variety of reasons, including differential protein processing and posttranslational or artifactual modifications. In this study, we encountered numerous examples of this behavior (Table 1). The protein products from multiple genes also can run to the same coordinates on a gel. Surprisingly, several occurrences of comigrating proteins were found when using the nrIPGs for 2DE. For example, the protein products of six different genes all migrated to a faintly silver-stained spot (Table 2). Both differential migration and comigration of proteins complicate comparative, quantitative pattern analyses of 2D gel databases.
Table 2.
Proteins comigrating in a single silver-stained spot on a 2D gel (spot 35 from Fig. 1B)
| Gene name* | Peptide sequences identified† | pI* | Molecular mass, kDa |
|---|---|---|---|
| GDH1 | (K)VIELGGTVVSLSDSK | ||
| (K)FIAEGSNMGSTPEAIAVFETAR | |||
| (R)EIGYLFGAYR | 5.50 | 49.6 | |
| (K)VLPIVSVPER | |||
| RPT3 | (R)ENAPSIIFIDEVDSIATK | 5.32 | 47.9 |
| SUB2 | (R)DVQEIFR | ||
| (K)LTLHGLQQYYIK | |||
| (R)INLAINYDLTNEADQYLHR | 5.36 | 50.3 | |
| (K)NKDTAPHIVVATPGR | |||
| (R)FLQNPLEIFVDDEAK | |||
| TIF3 | (R)GSNFQGDGREDAPDLDWGAAR | ||
| (R)ADLVAVLK | |||
| (K)ITIPIETANANTIPLSELAHAK | 5.17 | 48.5 | |
| (R)EREEVDIDWTAAR | |||
| (R)EREEPDIDWSAAR | |||
| VMA1 | (K)VGHDNLVGEVIR | 5.09 | 67.7 |
| YFR044C | (R)YPSLSIHGVEGAFSAQGAK | ||
| (K)LVYGVDPDFTR | |||
| (K)FISEQLSQSGFHDIK | 5.54 | 52.9 | |
| (R)TELIHDGAYWVSDPFNAQFTAAK | |||
| (K)ILIDGIDEMVAPLTEK |
Large Starting Amounts Are Required to Analyze Low Abundance Proteins.
Because no proteins from genes with codon bias values of <0.1 were detected in this study, we attempted to detect low abundance proteins by increasing the amount of starting protein beyond the capacity of 2D gels. Fifty milligrams of protein was separated by one-dimensional SDS-PAGE, and a 3 mm × 10 cm strip was cut out of the Coomassie-stained gel. Peptides recovered after tryptic digestion of this sample were separated by strong cation exchange chromatography and selected fractions were analyzed by reverse-phase microcapillary LC-MS/MS. The number of proteins detected between 68 and 85 kDa was 193. This is more than one-third of the proteins predicted to migrate to the selected region of the gel (Fig. 3). The CBD of the genes detected was similar, but not identical, to the CBD for the entire genome, and many proteins with codon bias values <0.1 were detected.
Figure 3.
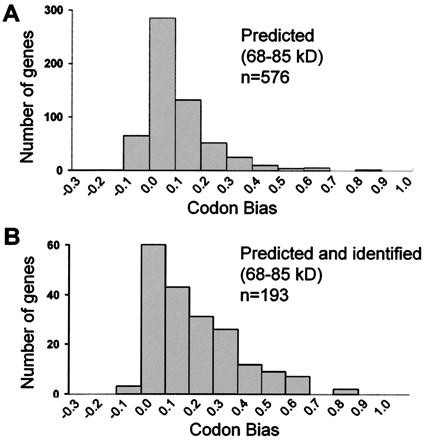
Low abundance proteins can be detected if the starting protein load is large. Fifty milligrams of soluble yeast protein was separated by SDS/PAGE in a single 10-cm wide lane. A band corresponding to a molecular mass range of 68 to 85 kDa was excised and in-gel trypsinized, and peptides were separated by strong cation exchange chromatography followed by on-line separation and analysis by LC-MS/MS techniques. Proteins in the gel band were identified from MS/MS spectra by the computer program sequest(12). (A) Distribution of codon bias for genes predicted to be between 68 and 85 kDa in molecular mass. This is a similar pattern as that shown in A, which shows the distribution for all yeast genes. (B) Distribution of codon bias for genes identified within the gel band. Many proteins from genes with low codon bias values (<0.1) were identified, including three protein kinases and a transcription factor.
These results indicate that low abundance proteins can be analyzed if larger starting amounts of proteins are used. Table 3 shows the calculated effect of larger starting loads on visualizing individual low abundance proteins. Calculated data for proteins present at 1,000, 100, and 10 copies/cell represent relatively low abundance proteins compared to proteins present at 104–106 copies/cell. Indeed, when previously using standard 2DE techniques and 40 μg of protein load of whole yeast lysate, the average protein abundance detected was 51,200 copies/cell and no proteins were detected with abundances <1,000 copies/cell (5). With 0.5 mg of starting protein, proteins present at 1,000 copies/cell could be visualized by silver staining, but those present at 100 and 10 copies/cell could not be visualized (Table 3). To achieve meaningful comparative and global protein expression profiles of different cell states by 2DE, prefractionation of milligram amounts of protein before 2DE is essential. However, it is likely that during preelectrophoretic protein fractionation, the ability for accurate protein quantification becomes compromised.
Table 3.
Theoretical required total starting protein amounts for individual protein visualization by staining
| Protein abundance, copies per cell | Silver staining*
|
Coomassie staining*
|
||
|---|---|---|---|---|
| Protein amount, mg† | Number of cells | Protein amount, mg† | Number of cells | |
| 10 | 20.073 | 1.20 × 109 | 2007.3 | 1.20 × 1011 |
| 100 | 2.007 | 1.20 × 108 | 200.7 | 1.20 × 1010 |
| 1,000 | 0.201 | 1.20 × 107 | 20.1 | 1.20 × 109 |
| 10,000 | 0.020 | 1.20 × 106 | 2.0 | 1.20 × 108 |
| 100,000 | 0.002 | 1.20 × 105 | 0.2 | 1.20 × 107 |
Protein detection limits for silver and Coomassie staining were 1 and 100 ng, respectively.
† Soluble yeast protein was calculated based on 1 mg of yeast protein being derived from harvesting 6 × 107 cells. Calculations are based on a protein molecular mass of 50 kDa and 100% efficiencies of the procedures used.
Discussion
The ability to identify proteins separated by 2DE has resulted in the analysis of many hundreds of spots. However, in studies in which total cell yeast lysates were separated by 2DE and the resulting spots were identified, only abundant proteins have been detected (5, 16–18). In this study, we have systematically examined the potential of the 2DE-MS/MS technology to provide global protein expression profiles from unseparated yeast cell lysates. The results indicate that the method is unsuited for the analysis low abundance proteins and that statements about the feasibility and straightforwardness of proteome analysis based on 2DE-MS should be rethought (17).
The focus of this study was a 2D gel with a 1 pH unit isoelectric focusing range. More than 1,500 spots could be visualized by silver staining. By extrapolation, a full complement of 1 pH unit gels might contain as many as 10,500 features (1,500 spots × 7 pH units). This greatly exceeds the 6,139 proteins predicted from the yeast genome (13). It might be concluded based on spot counting alone that 2D gels could be the basis for global proteome analysis. However, analysis of protein spots by MS identified generally abundant proteins (codon bias values >0.2). Clearly the number of spots on a 2D gel is not representative of the overall number or classes of expressed genes that can be analyzed.
Differential protein processing (producing more than one spot per protein) and comigrating spots present problems for both quantitative protein expression comparison and database matching. We detected as many as six proteins comigrating to the same faintly silver-stained spot (Table 2). In addition, many proteins were detected in multiple spots within the gel region selected for analysis (Table 1), and it is likely that other forms of the same proteins migrated to spots outside of the selected area. With protein levels in yeast cells being expressed over a range of at least 5 orders of magnitude, small populations of differentially processed or posttranslationally modified forms of abundant proteins compete for localization in the gel with lower abundance proteins and greatly increase the overall number of features visible on a 2D gel.
There are 61 possible codons that code for 20 amino acids. Codon bias is a measure of the propensity of an organism to selectively use certain codons that result in the incorporation of the same amino acid residue in a growing polypeptide chain. The larger the codon bias value, the fewer the number of codons that are used to encode the protein (15). It is thought that codon bias is a measure of protein abundance because highly expressed proteins generally have large codon bias values (16). We have shown previously that codon bias appears to be an excellent indicator of the boundaries of current 2D gel proteome analysis technology (5). There are thousands of yeast genes with expressed mRNA (6) and likely expressed protein with codon bias values <0.1 (Fig. 2A). In this study, we detected none of them by a 2D gel-based approach (Fig. 2C). Indeed, in every examined yeast proteome study (5, 16–18) in which the combined total number of identified proteins is >400, this same observation is true. It is expected that for the more complex cells of higher eukaryotic organisms the detection of low abundance proteins would be even more challenging than in yeast. Clearly, highly abundant proteins are overwhelmingly detected in proteome studies. If proteome analysis is to provide truly meaningful information about cellular and regulatory processes, it must be able to penetrate to the level of regulatory proteins, which are typically of low abundance.
In this study, we examined the use of 2D gels with extended separation range and increased sample load. The data indicated that there was a somewhat improved detection of proteins of moderate abundance but that low abundance proteins characterized by codon bias values <0.1 (more than one-half of the genes in the yeast genome) were still generally undetectable. We therefore conclude that the current proteome technology, used without sample preenrichment, is not suitable for the global detection of proteins expressed by the cell and that the construction of complete, quantitative proteome maps based on the 2DE-MS/MS approach will be very challenging, even for relatively simple, unicellular organisms.
Low abundance proteins were, however, detected if larger starting amounts were used. Beginning with 50 mg of total yeast protein and by using a strategy that included SDS/PAGE, in-gel digestion, strong cation exchange chromatography separation, and on-line microcapillary LC-MS/MS techniques, we successfully detected 193 proteins with more than 60 proteins from genes with codon bias values of <0.1 including three protein kinases (YNL020C, YLR248W, and YMR216C) and one transcription factor (YML007W). These data support the need to develop strategies that allow for large starting amounts of protein to provide at least femtomole amounts of each protein to be delivered to the MS (Table 3). The most favorable techniques currently use multiple chromatographic steps such as the pairing of strong cation exchange and reverse-phase chromatography in this study (19).
Conclusions
The 2D-gel-based proteome analysis has been successfully used to detect and characterize marker proteins that are idiotypic for a specific physiologic or pathologic state of a cell or tissue. However, it is now apparent that the 2DE-MS/MS approach is unsuitable to detect, identify, and quantify every protein in a sample, a task that seems necessary for the comprehensive analysis and eventual mathematical description of biological processes and systems. For this reason, it is necessary to develop novel techniques that allow for much increased starting amounts while permitting large-scale quantitative comparison of protein expression.
Acknowledgments
We thank Amersham Pharmacia Biotech for technical support and Jimmy Eng for expert computer programming. This work was supported in part by grants from the National Science Foundation (Science and Technology Center for Molecular Biotechnology), National Institutes of Health (HG00041, RR11823, T32HG00035, and AI41109), and the Merck Genome Research Institute.
Abbreviations
- MS/MS
tandem MS
- LC
liquid chromatography
- 2DE
two-dimensional gel electrophoresis
- IPG
immobilized pH gradient
- nrIPG
narrow-range IPG
- CBD
codon bias distribution
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160270797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160270797
References
- 1.Lashkari D A, DeRisi J L, McCusker J H, Namath A F, Gentile C, Hwang S Y, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1997;94:13057–13062. doi: 10.1073/pnas.94.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velculescu V E, Zhang L, Vogelstein B, Kinzler K W. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 3.Varshavsky A. Proc Natl Acad Sci USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Futcher B, Latter G I, Monardo P, McLaughlin C S, Garrels J I. Mol Cell Biol. 1999;19:7357–7368. doi: 10.1128/mcb.19.11.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gygi S P, Rochon Y, Franza B R, Aebersold R. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Velculescu V E, Zhang L, Zhou W, Vogelstein J, Basrai M A, Bassett D E, Jr, Hieter P, Vogelstein B, Kinzler K W. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- 7.Garrels J I, Futcher B, Kobayashi R, Latter G I, Schwender B, Volpe T, Warner J R, McLaughlin C S. Electrophoresis. 1994;15:1466–1486. doi: 10.1002/elps.11501501210. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez J C, Hochstrasser D, Rabilloud T. Methods Mol Biol. 1999;112:221–225. doi: 10.1385/1-59259-584-7:221. [DOI] [PubMed] [Google Scholar]
- 9.Shevchenko A, Wilm M, Vorm O, Mann M. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 10.Gygi S P, Rist B, Gerber S A, Turecek F, Gelb M H, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 11.Corthals G L, Gygi S P, Aebersold R, Patterson S D. In: Proteome Research: 2D Gel Electrophoresis and Detection Methods. Rabilloud T, editor. New York: Springer; 1999. pp. 197–231. [Google Scholar]
- 12.Eng J, McCormack A L, Yates J R. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 13.Costanzo M C, Hogan J D, Cusick M E, Davis B P, Fancher A M, Hodges P E, Kondu P, Lengieza C, Lew-Smith J E, Lingner C, et al. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gygi S P, Han D K M, Gingras A C, Sonenberg N, Aebersold R. Electrophoresis. 1999;20:310–319. doi: 10.1002/(SICI)1522-2683(19990201)20:2<310::AID-ELPS310>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Kurland C G. FEBS Lett. 1991;285:165–169. doi: 10.1016/0014-5793(91)80797-7. [DOI] [PubMed] [Google Scholar]
- 16.Garrels J I, McLaughlin C S, Warner J R, Futcher B, Latter G I, Kobayashi R, Schwender B, Volpe T, Anderson D S, Mesquita F R, et al. Electrophoresis. 1997;18:1347–1360. doi: 10.1002/elps.1150180810. [DOI] [PubMed] [Google Scholar]
- 17.Shevchenko A, Jensen O N, Podtelejnikov A V, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M. Proc Natl Acad Sci USA. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrot M, Sagliocco F, Mini T, Monribot C, Schneider U, Shevchenko A, Mann M, Jeno P, Boucherie H. Electrophoresis. 1999;20:2280–2298. doi: 10.1002/(SICI)1522-2683(19990801)20:11<2280::AID-ELPS2280>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Link J, Eng J, Schieltz D M, Carmack E, Mize G J, Morris D R, Garvik B M, Yates J R. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]