

Abstract
Yeast vacuoles undergo priming, docking, and homotypic fusion, although little has been known of the connections between these reactions. Vacuole-associated Vam2p and Vam6p (Vam2/6p) are components of a 65S complex containing SNARE proteins. Upon priming by Sec18p/NSF and ATP, Vam2/6p is released as a 38S subcomplex that binds Ypt7p to initiate docking. We now report that the 38S complex consists of both Vam2/6p and the class C Vps proteins [Reider, S. E. and Emr, S. D. (1997) Mol. Biol. Cell 8, 2307–2327]. This complex includes Vps33p, a member of the Sec1 family of proteins that bind t-SNAREs. We term this 38S complex HOPS, for homotypic fusion and vacuole protein sorting. This unexpected finding explains how Vam2/6p associates with SNAREs and provides a mechanistic link of the class C Vps proteins to Ypt/Rab action. HOPS initially associates with vacuole SNAREs in “cis” and, after release by priming, hops to Ypt7p, activating this Ypt/Rab switch to initiate docking.
The compartmentation of eukaryotic cells requires passage of proteins between organelles by membrane vesicle budding and fusion. Proteins synthesized at the endoplasmic reticulum are partitioned into budding vesicles that fuse to the Golgi. The process of budding, transport, and fusion continues to other subcellular destinations including the plasma membrane and the lysosome.
The yeast counterpart of the mammalian lysosome is the vacuole. Several mutant screens have identified genes (VPS) that function in vacuole protein sorting. One type of screen (vps) exploited the aberrant secretion of vacuolar proteases (1–3). Together with screens for altered vacuolar peptidase activity (pep), vacuole inheritance (vac), and vacuole morphology (vam), more than 40 complementation groups have been identified (4–6). The VPS genes have been grouped into six classes based on vacuole morphology, ranging from normal vacuole appearance (class A) to multiple vacuoles (class B) to cells containing highly fragmented vacuoles or no recognizable vacuolar structures (class C) (7, 8). Many of the proteins encoded by the VPS genes have been studied, and their functions in vacuole transport pathways are being elucidated (9).
We have studied the homotypic fusion of yeast vacuoles, the last step in their inheritance to the bud during cell division. This process is studied in vitro with a biochemical assay in which vacuoles from a proteinase A-deficient strain (BJ3505, pep4Δ) are mixed with vacuoles from a strain deleted in the gene encoding the membrane-associated alkaline phosphatase (DKY6281, pho8Δ) (10). Vacuole fusion allows Pep4p to gain access to pro-Pho8p and process it to its catalytically active form that can be assayed spectrophotometrically. Homotypic vacuole fusion occurs in the obligately ordered subreactions of priming, docking, and fusion. Priming is driven by hydrolysis of ATP by Sec18p (yeast NSF) (11, 12). This triggers the release of Sec17p (yeast α-SNAP) from the vacuole membrane and the disassembly of a large cis-SNARE complex, which includes a t-SNARE (Vam3p), three v-SNAREs (Nyv1p, Vti1p, and Ykt6p), and a homolog of neuronal SNAP-25 (Vam7p) (12–15). This SNARE disassembly is accompanied by the activation of Vam3p which, in turn, is stabilized by the chaperone LMA1 (16–18). Priming is followed by a two-stage docking event that brings the vacuoles into physical contact (19). The first stage of docking is reversible and SNARE-independent, requiring instead a small, Rab-like GTPase called Ypt7p (20). The second stage, which is irreversible, involves trans-SNARE complex formation between apposed vacuoles (19). Recently, it was shown that phosphatidylinositol 4,5-biphosphate is needed for both the priming and docking subreactions (21). The final subreaction, that of vacuole fusion, depends on calcium/calmodulin and protein phosphatase 1, leading to the release of LMA1 (18, 22, 23).
We recently have studied two regulators of vacuole docking, Vam2p (Vps41p) and Vam6p (Vps39p) (24, 25). vam2 and vam6 mutants have fragmented vacuoles like the ypt7 (vam4) mutant. VAM2 and VAM6 are classified as class B VPS genes (6–8). Approximately half of Vam2p and Vam6p is associated with vacuoles, based on in vivo analysis of green fluorescent protein fusion constructs and in vitro analysis of both fractionated cell extracts and isolated, intact vacuoles (24, 26). Antibodies to either Vam2p or Vam6p block the docking stage of in vitro vacuole fusion, as seen for antibodies to Ypt7p (20). Vam2p and Vam6p (Vam2/6p) initially are part of a large 65S protein complex on the vacuole that includes Vam3p and Nyv1p. Priming by Sec18p and ATP triggers complex disassembly. This priming-dependent disassembly leads to a smaller, 38S Vam2/6p complex that can associate with Ypt7p, thereby establishing Vam2/6p as an essential physical and functional link between priming and docking (24, 25).
We now report that the Vam2/6p complex also includes all four proteins encoded by class C VPS genes: Vps11p (Vam1p), Vps16p (Vam9p), Vps18p (Vam8p), and the Sec1p homolog Vps33p (Vam5p). We term the complex that contains these six proteins HOPS for homotypic vacuole fusion and vacuole protein sorting and to reflect the fact that it hops from one set of associations to another. The HOPS complex initially is found in a 65S complex with SNAREs before priming, is released without SNAREs after priming, and thereby gains the capacity to associate specifically with the GTP-bound form of Ypt7p. We also show that antibodies to all four class C Vps proteins block vacuole fusion, as seen previously for antibodies to Vam2p, Vam6p, Ypt7p, and the SNAREs. The discovery that this HOPS Ypt/Rab effector complex contains Vps33p, a member of the Sec1p family of proteins that bind t-SNAREs (27–29), provides clear, physical evidence for the long-sought link between Ypt/Rab function and SNAREs. Surprisingly, whereas most Rab effectors are thought to bind to Rab proteins before SNAREs, HOPS is transferred from cis-associated SNAREs to the Ypt/Rab by the priming action of Sec18p/NSF.
Materials and Methods
Vacuole Isolation.
Vacuoles were isolated from yeast strains BJ3505 and DKY6281 as described (30). Large-scale frozen vacuole preparations of BJ3505 vacuoles were used for studies of glutathione S-transferase (GST)-Ypt7p association and for immunoprecipitation experiments. Briefly, a 1-liter shaker culture was grown to OD600 = 1.0 in 1% yeast extract, 2% Bacto-peptone, and 2% dextrose (1× YPD) at 30°C and 225 rpm, added to 5 liters of 1× YPD in a fermentor (New Brunswick Scientific), and grown to an OD600 of ≈7.0 at 30°C and 800-rpm agitation with periodic supplementation of 5× YP, 50% dextrose, and 1 M NaOH to maintain adequate nutrient levels and the pH at 6.0. Cells were harvested [4,400 × g, 10 min, 4°C; JA-10 rotor (Beckman)] and spheroplasted (30) except that postlysis centrifugation was at 1,400 × g for 5 min at 4°C (JA-14 rotor; Beckman). Spheroplast pellets were resuspended in an equal volume of PS buffer (10 mM Pipes, pH 6.8/200 mM sorbitol) containing 8% (wt/vol) Ficoll and then lysed by dextran treatment as described (30). Lysates were diluted with an equal volume of PS buffer containing 4% (wt/vol) Ficoll and centrifuged [300,000 × g, 1 h, 4°C; Type 60 Ti rotor (Beckman)]. Vacuoles were harvested from the top of the 4% Ficoll buffer, diluted 5-fold with PS buffer, and centrifuged (15,000 × g, 10 min, 4°C; JA-14 rotor). Vacuoles were resuspended at 0.3 mg/ml in PS buffer with 10% glycerol and frozen dropwise in liquid nitrogen for long-term storage at −85°C.
GST-Tagged Class C Vps Protein Preparation.
Polyclonal antibodies were raised in rabbits against GST-tagged fusion proteins. DNA fragments corresponding to the carboxyl-terminal portions of Vps5p (289 aa), Vps11p (354 aa), Vsp16p (348 aa), Vps17p (552 aa), Vps18p (342 aa), Vps33p (225 aa), and Vps45p (578 aa) were amplified by PCR from yeast genomic DNA by using the following primers: VPS5 5′ primer-CGC GGA TCC ACA AGT GAT GAC TTT AGT TC, VPS5 3′ primer-CGC GGA TCC TAT TCA TTT TAC TTA CTG T; VPS11 5′ primer-CGC GGA TCC TTA GAA GCC TGT TTG GCA T, VPS11 3′ primer-CGC GGA TCC AAC ATG CGC TGT TTC AG; VPS16 5′ primer-CGC GGA TCC GGT AGT CCT GAC ATG GAA G, VPS16 3′ primer-CGC GGA TCC TTC GCG TTC CAT GGG T; VPS17 5′ primer-CGC GGA TCC ATG ACT TCG GCT GTA CCT, VPS17 3′ primer-CGC GGA TCC GCA ACC ATT TTT ATC TGT TAT; VPS18 5′ primer-CGC GGA TCC AAT AAG TTA ATT CCA ACA ATT TT, VPS18 3′ primer-CGC GGA TCC TAT TTA GGT AAG TAA ATA CTC; VPS33 5′ primer-CGC GGA TCC ATC GAC TCT TGG GGC ATT, VPS33 3′ primer-CGC GGA TCC TCT AGA AAA TTA AAG GAT ACA; and VPS45 5′ primer-CGC GGA TCC ATG AAC CTT TTT GAT GTG G, VPS45 3′ primer-CGC GGA TCC ACG CAT GCA CAT AGA AGG. DNA fragments were digested with BamHI, purified and cloned into pGEX-4T-1 (Amersham Pharmacia), and transformed into Escherichia coli BL21 (Stratagene). Transformants were examined for correct gene orientation by diagnostic restriction enzyme digests and expression of the fusion proteins. E. coli strains harboring fusion protein constructs were grown at 37°C to an OD600 of 0.5. Protein expression was induced by the addition of isopropyl β-d-thiogalactoside to 1 mM, and cultures were incubated for 3 h. Cells from a 3-liter culture were harvested at 700 × g for 5 min at 4°C (JA-10 rotor; Beckman), suspended in 120 ml of PBS (31), centrifuged as above, and suspended in 120 ml of PBS. Cells then were lysed by two freeze–thaw cycles, incubation with 0.1 mg/ml lysozyme for 10 min at room temperature, sonication (two times, 30 s each), and incubation with 1% Triton X-100 for 10 min at 4°C. Lysates were centrifuged at 10,000 × g for 10 min at 4°C (JA-20 rotor; Beckman); fusion proteins were found in the pellet. Pellets were suspended in 120 ml of 50 mM citrate, adjusted to pH 6.0 with solid Na2HPO4, for 30 min at room temperature and centrifuged at 18,000 × g for 10 min at 4°C (JA-20 rotor). Pellets were suspended in 30 ml of 50 mM citrate buffer, pH 6.0, containing 1.5% N-laroylsarcosine for 30 min at room temperature and centrifuged at 48,000 × g for 10 min at 4°C (JA-20 rotor). The Sarkosyl extracts, which contained the GST-tagged class C Vps proteins, were dialyzed at 4°C for 36 h with three changes of PBS containing 1.5% N-laroylsarcosine.
Antibody Purification.
Polyclonal antibodies were raised in rabbits to the GST-tagged class C Vps proteins or to peptides (CLE DTE QWQ KDG FDL NSK KT and CIE DEH AAD KIT NEN DDF SEA) from the Vps33p sequence. For purification, GST-tagged class C Vps proteins or peptides were cross-linked to Sulfolink Coupling Gel (Pierce) according to the manufacturer's instructions, except that 1.5% N-laroylsarcosine was included during the postreduction gel-filtration step for the fusion proteins. Antibodies were affinity-purified on the cognate antigen- or peptide-Sulfolink column (32). Acid eluates were neutralized with 100 mM Tris⋅Cl, pH 9.4, and dialyzed into PS buffer as above.
Results
The Class C Vps Proteins Associate with Ypt7p.
Membrane trafficking depends on small GTPases of the Ypt/Rab family (33, 34). For homotypic vacuole fusion in yeast, Ypt7p mediates an early, reversible stage of vacuole docking that occurs before the pairing in trans of v- and t-SNAREs (19, 20). Ypt7p effectors for vacuole fusion have remained elusive until the recent finding that Vam2p/Vps41p and Vam6p/Vps39p, two vacuole-associated proteins, associate in vitro with recombinant Ypt7p in a GTP-dependent manner (25).
To further characterize the proteins that associate with Ypt7p, vacuole detergent extracts were incubated with glutathione-immobilized GST-Ypt7p that had been preloaded with either no nucleotide, guanosine 5′-[γ-thio]triphosphate (GTP[γS]; a nonhydrolyzable analog of GTP), or GDP (Fig. 1). When analyzed by silver-staining, several polypeptides selectively associated with GST-Ypt7p:GTP[γS]. Less association was seen with the GDP or nucleotide-free forms of the fusion protein. Additionally, these polypeptides did not associate with GST-Ypt1p:GTP[γS], the Rab-like GTPase that functions in vesicle transport from the endoplasmic reticulum to the Golgi and in an early intra-Golgi transport step (37). Each of these Ypt7p-associated polypeptides was excised from preparative gels and submitted for identification by mass spectroscopy. The largest of these polypeptides was Vam6p, consistent with previous immunoblot analysis (25). Surprisingly, the other four polypeptides were the class C Vps proteins: Vps11p, Vps16p, Vps18p, and Vps33p. These four class C Vps proteins were already known to interact physically as a high-molecular-weight complex and to have a functional role in multiple vacuole-targeting pathways (38), but their exact mode of action was not clear. Further, they had not been implicated in homotypic vacuole fusion, nor were they known to be Ypt7p effectors. Vps11p and Vps18p are hydrophilic proteins with C-terminal RING finger zinc-binding domains that are thought to mediate protein/protein interactions (39–41). Vps16p is also a hydrophilic protein, but lacks known protein motifs or homologies with characterized proteins (42). Vps33p is a member of the Sec1p family of proteins (43, 44), which bind and modulate the activity of t-SNAREs (27–29). Because these proteins associate specifically with the GTP form of Ypt7 (Fig. 1), they are part of an effector complex for this Ypt/Rab protein. Although we have observed Vam2p in this complex by immunoblot analysis (data not shown), this protein was not detected here, perhaps because it is more susceptible to degradation and/or is less abundant. Lower-molecular-weight proteins submitted for sequencing were breakdown products of GST-Ypt7p (data not shown).
Figure 1.
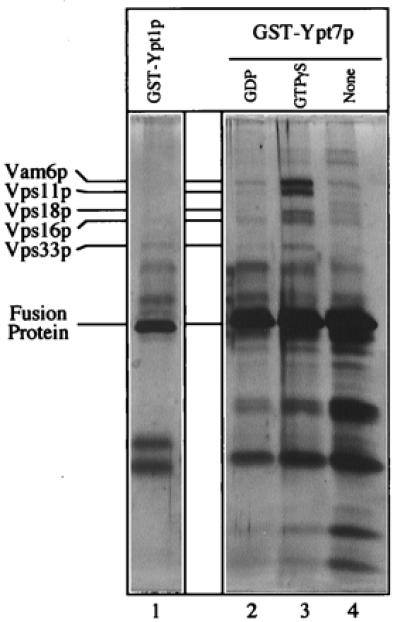
The class C Vps proteins associate with glutathione-immobilized GST-Ypt7p:GTP[γS]. Frozen BJ3505 vacuoles (300 μg) were diluted to 1.5 ml with PS buffer (see Materials and Methods) and sedimented (13,000 × g, 10 min, 4°C). Vacuole pellets were suspended in 500 μl of IP solubilization buffer [20 mM Hepes, pH 7.4/150 mM NaCl/10% glycerol/1% Triton X-100/1× protease inhibitor mixture (PIC; ref. 30)] and incubated on ice for 10 min. After sedimentation (13,000 × g, 10 min, 4°C), the solubilized vacuolar supernatant was transferred to fresh tubes containing GST-Ypt1p (lane 1) or GST-Ypt7p (lanes 2–4) that had been prebound to 40 μl (packed volume) of glutathione-Sepharose beads (Amersham Pharmacia) and preloaded with nucleotides (see below). [Fusion proteins underwent nucleotide exchange before incubation with the solubilized vacuolar supernatants. Each fusion protein (120 μg) was mixed by nutation with 40 μl of glutathione-Sepharose beads in 50 mM Tris·Cl, pH 8.0, for 20 min at room temperature. Bound material was sedimented (10,000 × g, 1 min, 4°C), and the supernatant was decanted. Beads were incubated (20 min, room temperature) in 500 μl of PS elution buffer (PS buffer with 2 M NaCl, 20 mM EDTA, 5 mM GDP, 1 mM DTT, and 1× PIC) and then sedimented and washed four times by resuspension in 1 ml of PS buffer and sedimentation (10,000 × g, 1 min, 4°C). Beads with fusion proteins were incubated (20 min, room temperature) with either 4 mM GTP[γS] (lanes 1 and 3), 4 mM GDP (lane 2), or no nucleotide (lane 4) in 500 μl of PS loading buffer (PS buffer containing 150 mM KCl, 4.5 mM MgCl2, 0.5 mM MnCl2, 1 mM DTT, and 1× PIC) and collected (10,000 × g, 1 min, 4°C).] Detergent extracts of vacuoles were mixed with beads by nutation for 2 h at 4°C. Unbound material was decanted from the beads after centrifugation (10,000 × g, 1 min, 4°C). Beads were resuspended twice in 1 ml of IP solubilization buffer and sedimented as above, and then bound proteins were eluted with 500 μl of PS elution buffer. Elution was by nutation for 15 min at room temperature followed by centrifugation. The eluate was precipitated by 12.5% trichloroacetic acid for 15 min on ice followed by centrifugation (13,000 × g, 15 min, 4°C). The pellet was resuspended in 1 ml of 80% acetone and sedimented as above. Protein pellets were dried and resuspended in 50 μl SDS/PAGE loading buffer, and polypeptides were separated on 10% acrylamide gels (35). Visualization was by silver staining (36); mass spectroscopy analysis (at the Howard Hughes Medical Institute/Keck Lab, Yale University) was performed on polypeptides extracted from a similar preparative gel stained with Coomassie brilliant blue.
Vam2/6p and the Class C Vps Proteins Are Part of a Large Complex on the Vacuole Membrane.
To determine whether the class C Vps proteins are part of the 65S complex that includes Vam2/6p (26) and the Vam3p and Nyv1p SNAREs (25), vacuole detergent extracts were fractionated on sucrose velocity gradients and analyzed by immunoblotting (Fig. 2). Under the conditions of this gradient analysis, Vam2p and Vam6p (Fig. 2A) cosediment with each other, with the 65S marker ferritin (25), and with each of the class C Vps proteins (Fig. 2B). Priming by ATP and Sec18p disassembles the 65S Vam2/6p complex to a 38S complex in parallel with the disassembly of the cis-SNARE complex (13) and the release of Sec17p (12). As with Vam2/6p (Fig. 2A), the four class C Vps proteins shift from 65S to 38S when the vacuoles are primed (Fig. 2B).
Figure 2.
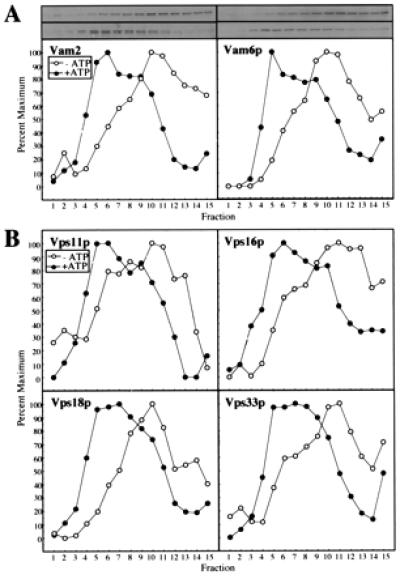
Cofractionation on sucrose velocity gradients of HOPS complex proteins. Freshly isolated BJ3505 vacuoles (400 μg) were diluted to 1.5 ml with PS buffer and sedimented (13,000 × g, 10 min, 4°C). Pellets were resuspended in 2 ml of either priming mixture [5 mM ATP, 5 mM MgCl2, 1 mg/ml creatine kinase, 100 mM creatine phosphate, 0.1× PIC, and 25 ng of his6-Sec18p/μg vacuolar protein (11)] or priming inhibition mixture (priming mixture, but with 0.16 units of apyrase/μg vacuolar protein substituting for ATP) and incubated for 10 min at 27°C. Vacuoles then were sedimented as above, resuspended in 1 ml of PS buffer, and sedimented. Vacuole pellets were suspended in 400 μl of PS solubilization buffer (PS buffer plus 1% Triton X-100 and 1× PIC) and incubated on ice for 10 min. After centrifugation (13,000 × g, 10 min, 4°C), the solubilized vacuolar supernatants were loaded on 11.6-ml continuous (40–10%, wt/vol) sucrose gradients in PS buffer plus 1% Triton X-100. Samples were centrifuged at 250,000 × g for 6 h at 4°C (SW 41 rotor; Beckman). Fractions of 800 μl were collected from the top of the gradient, and aliquots were separated by SDS/PAGE (10% acrylamide; ref. 35), analyzed by immunoblotting to the indicated proteins, and quantitated by scanning densitometry. ○, −ATP; ●, +ATP. (A) Vam2p and Vam6p. (B) Vps11p, Vps16p, Vps18p, and Vps33p.
Vam2/6p and the Class C Vps Proteins Are in the Same Complex.
Vam2/6p and the class C Vps proteins are at least partially localized to vacuoles (25, 26, 36), cofractionate on sucrose gradients at a high molecular weight (Fig. 2), and are effectors of Ypt7p (Fig. 1). To test whether they are directly associated with each other, vacuole detergent extracts were incubated with protein A-Sepharose-immobilized antibodies to Vam2/6p, to each of the four class C Vps proteins, or to a control antibody to the E. coli SecE protein. Immunoprecipitated proteins then were analyzed by immunoblotting (Fig. 3). In each case, antibodies to Vam2/6p and the class C Vps proteins cross-immunoprecipitated all six proteins (Fig. 3A, lanes 2–7). The efficiency of the cross-immunoprecipitations was remarkably similar to the efficiency with which each antibody immunoprecipitated its cognate protein, suggesting that the majority of each of these proteins exist in a complexed state. The specificity of the immunoprecipitations is highlighted by the lack of cross-immunoprecipitation with antibodies to SecE (lane 8) and by the fact that none of the antibodies immunoprecipitated the vacuole membrane protein Pho8p. We also addressed the issue of specificity by using affinity-purified antibodies that had been prepared to a peptide from the Vps33p sequence. Immunoprecipitations were performed in the presence of nonspecific or specific peptide inhibitors (Fig. 3B). The Vps33p peptide antibody was as efficient at immunoprecipitating Vps33p (lanes 1 and 2) as the Vps33p fusion protein antibodies (Fig. 3A, lanes 1 and 7), and the addition of a 3:1 molar excess of SecE peptide before the addition of the vacuole detergent extract had no effect (Fig. 3B, lane 2). In contrast, Vps33p peptide efficiently blocked the immunoprecipitation of both Vps33p and Vam6p (lane 3). Thus, Vam2/6p and the class C Vps proteins are all part of the same large, 65S complex on the vacuole membrane (Fig. 3), which can be disassembled to a 38S subcomplex by priming (Fig. 2), and the proteins thereby can be activated to bind to Ypt7p (Fig. 1 and ref. 25).
Figure 3.
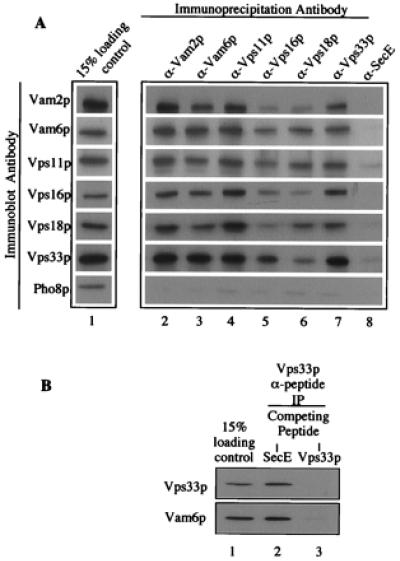
Coimmunoprecipitation of HOPS complex subunits. (A) Detergent-solubilized vacuolar supernatants (500 μl) were prepared from 200 μg of frozen BJ3505 vacuoles as described in Fig. 1 (lane 1). Each vacuole extract was mixed by nutation for 2 h at 4°C with 20 μl (packed volume) of protein A-Sepharose CL-4B beads (Amersham Pharmacia), which had been cross-linked to affinity-purified antibodies to Vam2p (lane 2), Vam6p (lane 3), Vps11p (lane 4), Vps16p (lane 5), Vps18p (lane 6), Vps33p (lane 7), or IgG antibodies to E. coli SecE (lane 8). [Before mixing, beads were mock-eluted with 200 μl of 1% (wt/vol) SDS at 37°C for 10 min followed by sedimentation (10,000 × g, 1 min, 4°C). Beads were washed three times in 500 μl of IP solubilization buffer and then preblocked by a 15-min nutation in 500 μl of IP solubilization buffer containing 2% (wt/vol) BSA (Fraction V; Sigma). Beads were sedimented (10,000 × g, 1 min, 4°C) and then suspended twice in 500 μl of IP solubilization buffer and sedimented.] Beads were collected by sedimentation (10,000 × g, 1 min, 4°C) and washed two times by resuspension in 1 ml of IP solubilization buffer and sedimentation. Bound proteins were eluted with 200 μl of 1% (wt/vol) SDS as above. After sedimentation (10,000 × g, 1 min, 4°C) to remove beads, 180 μl of the eluate was supplemented with 45 μl of 5× SDS/PAGE loading buffer (45). Proteins were analyzed by SDS/PAGE (10% acrylamide) and immunoblotting. (B) Immunoprecipitations were as described in A, except that the antibodies were to Vps33p peptides (see Materials and Methods). After incubating the beads in IP solubilization buffer plus 2% (wt/vol) BSA and before mixing with vacuole detergent extracts, the beads were mixed with a 3:1 molar excess of either SecE peptide (ATV AFA REA ATE VRK VIW PTR QET C; SecE, lane 2) or Vps33 peptides (Vps33p, lane 3) for 1 h at room temperature.
The Class C Vps Proteins Are Needed for Vacuole Fusion.
Homotypic vacuole fusion can be measured in vitro by mixing populations of purified vacuoles from two yeast strains, one with a deletion of PHO8 and the other with deletions in vacuole proteases. Vacuoles from the first strain have normal proteases but no Pho8p, whereas vacuoles from the second strain lack proteases and, therefore, accumulate catalytically inactive pro-Pho8p. Upon incubation with ATP, these vacuoles undergo priming, docking, and fusion, allowing the proteases to gain access to the proenzyme and process it to its active form, which can be assayed colorimetrically (30). Using this assay, we found (Fig. 4) that the reaction is blocked by antibody to each of the four class C Vps proteins, as shown previously for antibody to Vam2p or Vam6p (24), but is not blocked by antibody to other Vps proteins such as Vps45p, Vps17p, and Vps5p. This is consistent with studies showing that Vps33p is required for in vitro trafficking from the endosome to the vacuole (46). The sensitivity of the reaction to antibody to each class C Vps protein as well as to antibody to Vam2p and Vam6p is in accord with the physical evidence that they are part of the same complex.
Figure 4.
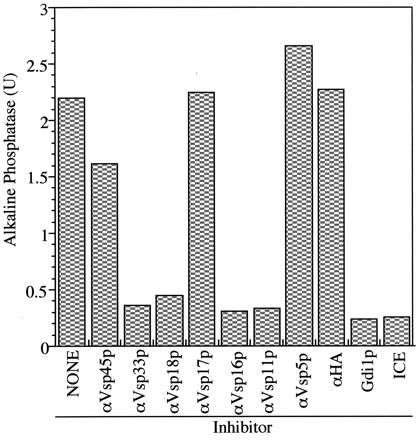
Antibody to each of the class C Vps proteins inhibits homotypic vacuole fusion. Homotypic vacuole fusion was assayed as described (30) but without cytosol. Before the preparation of the fusion assay mix, each vacuole population was incubated with 0.05 mg/ml affinity-purified antibodies either to the class C Vps proteins (Vps11p, Vps16p, Vps18p, and Vps33p), other Vps proteins (Vps5p, Vps17p, and Vps45p), or hemagglutinin (HA; epitope 12CA5). A separate reaction was treated with 60 μg/ml Gdi1p as a control for complete inhibition of fusion. One unit (U) of alkaline phosphatase activity produces 1 μmol of p-nitrophenol/min per μg BJ3505 vacuolar protein.
Discussion
The class C vps mutants, selected by their defects in protein delivery to the vacuole, contain very small, highly fragmented vacuoles or vacuole-related organelles (7, 8). A visual screen for mutants with such an altered vacuole morphology yielded the same four genes: VAM1/VPS11, VAM5/VPS33, VAM8/VPS18, and VAM9/VPS16 (6). We now report that these four encoded proteins, together with those encoded by VAM2/VPS41 and VAM6/VPS39, are part of a large HOPS complex that is essential for homotypic vacuole fusion and vacuole protein sorting. The HOPS complex initially is part of a larger 65S complex that includes the vacuolar SNAREs Vam3p and Nyv1p, thereby providing a biochemical explanation for the known genetic interactions between VPS33 and VAM3 (47). This 65S complex is disassembled by the action of Sec18p and Sec17p, and the released 38S HOPS subcomplex binds specifically to the active GTP-bound form of Ypt7p to initiate docking.
The Sec1p family of proteins are known to associate specifically with t-SNAREs and to modulate their conformations and associations with other SNAREs (27–29). Our current findings demonstrate that a Sec1p family member, Vps33p, is a subunit of a Ypt/Rab effector complex, i.e., the HOPS complex. These findings cast the studies of Price et al. (25) in a new light, because the presence of Vps33p offers a plausible mechanism for how this complex can be associated in a 65S complex with SNAREs and why priming is needed to release HOPS from the 65S complex and allow association with the GTP-bound form of Ypt7p.
How can the HOPS complex be needed for both homotypic vacuole fusion and for protein delivery to the vacuole? In addition to its function on the vacuole, Vam2/Vps41p also functions on the Golgi in the formation of transport vesicles destined for the endosome (48). It is possible that the other members of HOPS also have a function at the Golgi in initiating trafficking to the endosome. Our current functional biochemical studies of vacuole fusion by using purified wild-type vacuoles (Fig. 4), as well as the sequential physical association of the HOPS complex with vacuolar SNAREs (24, 25) and Ypt7p (Fig. 1), establish that the HOPS complex has a direct role in homotypic vacuole fusion.
No specific function has been attributed to the HOPS complex, but it is needed until the vacuoles dock. For example, Vam2p and Vam6p antibodies inhibit vacuole fusion at the same stage as Gdi1p, a protein that extracts the GDP form of Ypt7p from the vacuole membrane (25). Antibodies to the class C Vps proteins also inhibit at this stage (data not shown). The HOPS complex binds to GST-Ypt7p:GTP[γS] (Fig. 1 and ref. 25) and thus serves as a Ypt7p effector. The interactions of HOPS complex proteins and Ypt7p may occur during the early “tethering” step of docking, when vacuoles become loosely and reversibly associated, before the trans-pairing of SNARE proteins across apposed vacuoles. This model is in accord with the proposed function of other large complexes that function with Rab/Ypt proteins to tether vesicles in other intracellular transport pathways (49). These tethering factors, which may include the Exocyst for Golgi-to-plasma membrane traffic (50), Uso1p and TRAPP for endoplasmic reticulum-to-Golgi traffic (51, 52), EEA1 and Rabaptin-5 for endosome homotypic fusion (53), p115, giantin, and GM130 for intra-Golgi traffic (54), and Vac1p for Golgi-to-endosome traffic (55, 56), exhibit some notable relationships with the HOPS complex. Vam2p and Vam6p are high-molecular-weight proteins and, like p115 and Uso1p, could extend out from membrane surfaces to aid in vesicle/target tethering. Localization studies of green fluorescent protein-Vam6p (26) and green fluorescent protein-Vps33p (57) suggest positioning at a few discrete sites on the vacuole membrane, which may dictate, like the Exocyst, a locus for docking and fusion. The HOPS complex also contains Vps33p, one of several Sec1p homologs that are known to modulate the activity of t-SNAREs. Like the HOPS complex, Vac1p also interacts with both a Ypt/Rab protein, Vps21p, and a Sec1p homolog, Vps45p (55, 56). Together, these complexes may provide an important functional link between activated Rab proteins and SNAREs, although in no case other than the HOPS complex has the Sec1p homolog been shown to be part of a complex that binds directly to the Ypt/Rab protein. Additionally, in the case of homotypic vacuole fusion, the order of associations and their causal relationships now have been delineated. This novel order of interactions, proceeding from (cis) SNAREs to the Rab/Ypt protein, raises the intriguing question of whether Ypt7:GTP:HOPS then interacts directly with SNAREs to catalyze trans-assembly of SNARE pairs (19).
Although the function of each HOPS complex subunit in homotypic vacuole fusion is unknown, the functions of several class C Vps proteins have been studied relative to other vacuole-sorting pathways. Both Vps11p and Vps18p contain C-terminal RING finger zinc-binding domains with important roles in biosynthetic, endocytic, and autophagic vacuole-targeting pathways (38). It will be important to determine whether these HOPS complex subunits perform similar functions to support homotypic fusion as they do for heterotypic-trafficking reactions. There are four Sec1p homologs in yeast (Sec1p, Sly1p, Vps33p, and Vps45p), and each may regulate the docking of vesicles to their respective targets through interactions with t-SNAREs. Vps33p, however, is unusual among the Sec1p-like proteins in that it contains an ATP-binding cassette. This cassette appears to be indispensable for Vps33p function because a point mutation in this region leads to a class C vps phenotype (43) and a partial deletion of the cassette impairs the proper targeting and processing of carboxypeptidase Y (57). Both the integrity of this ATP-binding cassette as well as cellular ATP levels govern the distribution of Vps33p between the cytosol and perivacuolar spots (57). These perivacuolar spots may be the same as those visualized for Vam6p (26) and may represent the HOPS complex on the vacuole surface. Previous studies have shown that the release of Vam2p and Vam6p from primed vacuoles depends on ATP after the priming step (25). Perhaps ATP binding by Vps33p governs the localization of the entire complex. It is noteworthy that Vps11p also contains an ATP-binding cassette, although its functionality has not been fully tested. Finally, there is a dynamic interaction among the class C Vps proteins by which Vps16p links the association of Vps33p to Vps11p and Vps18p (38). These interactions and their relationship to Vam2/6p may be important to the function of the complex as a whole.
It appears that the activity of HOPS complex proteins could be conserved in similar vesicle-trafficking pathways of other organisms. Indeed, homologs of VAM2 and VPS33 can be identified from plants to mammals (58, 59). VAM2, VPS18, and VPS33 also have been identified in Drosophila as the eye pigmentation mutants light (lt), deep orange (dor), and carnation (car), respectively (45, 60, 61). Genetic studies have revealed an association of lt and dor (61) whereas a high-molecular-weight complex containing Dor and Car has been shown to associate with endosomal membranes (45), as in yeast (ref. 38; this study). Further molecular characterization of these mutants indicates defects in the biogenesis of the pigmentation granule, a compartment related to the vacuole/lysosome (45). It will be of interest to determine whether these proteins function specifically in the docking and fusion of vesicles to this lysosomal compartment, as in yeast homotypic vacuole fusion.
Further progress in understanding the functions of the HOPS complex in yeast will require its purification to homogeneity and the identification of each of its subunit polypeptides. In addition, it will be vital to establish a functional assay for the HOPS complex; the demonstration that it can bind GST-Ypt7p:GTP[γS] may be a first step toward such a reconstitution of function.
Acknowledgments
We thank Naomi Thorngren for expert technical assistance and Susan Oldfield for assistance with the preparation of this manuscript. This work was supported by a grant from the National Institute of General Medical Sciences. D.F.S. is supported by National Institutes of Health Postdoctoral Fellowship GM220025-02 and G.E. is supported by the Human Frontiers of Science Program Organization.
Abbreviations
- HOPS
homotypic fusion and vacuole protein sorting
- VPS
vacuole protein sorting
- VAM
vacuole morphology
- GTP[γS]
guanosine 5′-[γ-thio]triphosphate
References
- 1.Rothman J H, Stevens T. Cell. 1986;47:1041–1051. doi: 10.1016/0092-8674(86)90819-6. [DOI] [PubMed] [Google Scholar]
- 2.Bankiatis V A, Johnson L M, Emr S D. Proc Natl Acad Sci USA. 1986;83:9075–9079. doi: 10.1073/pnas.83.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson J R, Klionsky D J, Banta L M, Emr S D. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones E W. Genetics. 1977;85:23–33. doi: 10.1093/genetics/85.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisman L S, Emr S D, Wickner W T. Proc Natl Acad Sci USA. 1990;87:1076–1080. doi: 10.1073/pnas.87.3.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wada Y, Ohsumi Y, Anraku Y. J Biol Chem. 1992;267:18665–18670. [PubMed] [Google Scholar]
- 7.Banta L M, Robinson J S, Klionsky D J, Emr S D. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raymond C K, Howald-Stevenson I, Vater C A, Stevens T H. Mol Biol Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant N J, Stevens T H. Micro Mol Biol Rev. 1998;62:230–247. doi: 10.1128/mmbr.62.1.230-247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Condradt B, Haas A, Wickner W. J Cell Biol. 1994;126:99–110. doi: 10.1083/jcb.126.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas A, Wickner W. EMBO J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer A, Wickner W, Haas A. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 13.Ungermann C, Nichols B J, Pelham H R B, Wickner W. J Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ungermann C, Wickner W. EMBO J. 1998;17:3269–3276. doi: 10.1093/emboj/17.12.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ungermann C, von Mollard G F, Jensen O N, Margolis N, Wickner W. J Cell Biol. 1999;145:1435–1442. doi: 10.1083/jcb.145.7.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Z, Wickner W. J Cell Biol. 1996;132:787–794. doi: 10.1083/jcb.132.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Mayer A, Muller E, Wickner W. J Cell Biol. 1997;136:299–306. doi: 10.1083/jcb.136.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Z, Sato K, Wickner W. Cell. 1998;93:1125–1134. doi: 10.1016/s0092-8674(00)81457-9. [DOI] [PubMed] [Google Scholar]
- 19.Ungermann C, Sato K, Wickner W. Nature (London) 1998;396:543–548. doi: 10.1038/25069. [DOI] [PubMed] [Google Scholar]
- 20.Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. EMBO J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A. Mol Biol Cell. 2000;11:807–817. doi: 10.1091/mbc.11.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters C, Mayer A. Nature (London) 1998;396:575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 23.Peters C, Andrews P D, Stark M J R, Cesaro-Tadic S, Glatz A, Podtelejnikov A, Mann M, Mayer A. Science. 1999;285:1084–1087. doi: 10.1126/science.285.5430.1084. [DOI] [PubMed] [Google Scholar]
- 24.Price A, Wickner W, Ungermann C. J Cell Biol. 2000;148:1223–1230. doi: 10.1083/jcb.148.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price A, Seals D, Wickner W, Ungermann C. J Cell Biol. 2000;148:1231–1238. doi: 10.1083/jcb.148.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura N, Hirata A, Ohsumi Y, Wada Y. J Biol Chem. 1997;272:11344–11349. doi: 10.1074/jbc.272.17.11344. [DOI] [PubMed] [Google Scholar]
- 27.Carr C M, Grote E, Munson M, Hughson F M, Novick P J. J Cell Biol. 1999;146:333–344. doi: 10.1083/jcb.146.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misura K M S, Scheller R H, Wels W I. Nature (London) 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 29.Yang B, Steegmaier M, Gonzalez L C, Jr, Scheller R H. J Cell Biol. 2000;148:247–252. doi: 10.1083/jcb.148.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haas A. Methods Cell Sci. 1995;17:283–294. [Google Scholar]
- 31.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 32.Harlow E, Lane O. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 33.Lazar T, Gotte M, Gallwitz D. Trends Biochem Sci. 1997;22:468–472. doi: 10.1016/s0968-0004(97)01150-x. [DOI] [PubMed] [Google Scholar]
- 34.Novick P, Zerial M. Curr Opin Cell Biol. 1997;9:496–504. doi: 10.1016/s0955-0674(97)80025-7. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Blum H, Beier H, Gross H J. Electrophoresis. 1987;8:93–99. [Google Scholar]
- 37.Jedd G, Richardson C, Litt R, Segev N. J Cell Biol. 1995;131:583–590. doi: 10.1083/jcb.131.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reider S E, Emr S D. Mol Biol Cell. 1997;8:2307–2327. doi: 10.1091/mbc.8.11.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preston R A, Manolson M F, Becherer K, Weidenhammer E, Kirkpatrick D, Wright R, Jones E W. Mol Cell Biol. 1991;11:5801–5812. doi: 10.1128/mcb.11.12.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson J R, Graham T R, Emr S D. Mol Cell Biol. 1991;12:5813–5824. doi: 10.1128/mcb.11.12.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saurin A J, Borden K L B, Boddy M N, Freemont P S. Trends Biochem Sci. 1996;21:208–214. [PubMed] [Google Scholar]
- 42.Horazdovsky B F, Emr S D. J Biol Chem. 1993;268:4953–5962. [PubMed] [Google Scholar]
- 43.Banta L M, Vida T A, Herman P K, Emr S D. Mol Cell Biol. 1990;10:4638–4649. doi: 10.1128/mcb.10.9.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wada Y, Kitamoto K, Kanbe T, Tanaka K, Anraku Y. Mol Cell Biol. 1990;10:2214–2223. doi: 10.1128/mcb.10.5.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sevrioukov E A, He J, Moghrabi N, Sunio A, Krämer H. Mol Cell. 1999;4:479–486. doi: 10.1016/s1097-2765(00)80199-9. [DOI] [PubMed] [Google Scholar]
- 46.Vida T, Gerhardt B. J Cell Biol. 1999;146:85–97. doi: 10.1083/jcb.146.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darsow T, Reider S E, Emr S D. J Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rehling P, Darsow T, Katzmann D J, Emr S D. Nat Cell Biol. 1999;1:346–353. doi: 10.1038/14037. [DOI] [PubMed] [Google Scholar]
- 49.Pfeffer S R. Nat Cell Biol. 1999;1:E17–E22. doi: 10.1038/8967. [DOI] [PubMed] [Google Scholar]
- 50.Guo W, Roth D, Walch-Solimena C, Novick P. EMBO J. 1999;18:1071–1080. doi: 10.1093/emboj/18.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao X, Ballew N, Barlowe C. EMBO J. 1998;17:2165–2165. doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates J R, Abeliovich H, Ferro-Novick S. EMBO J. 1998;17:2494–2503. doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christoforidis S, McBride H M, Burgoyne R D, Zerial M. Nature (London) 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura N, Lowe M, Levine T P, Rabouille C, Warren G. Cell. 1997;89:445–455. doi: 10.1016/s0092-8674(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 55.Peterson M R, Burd C G, Emr S D. Curr Biol. 1999;9:159–162. doi: 10.1016/s0960-9822(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 56.Tall G G, Hama H, DeWald D B, Horazdovsky B F. Mol Biol Cell. 1999;10:1873–1889. doi: 10.1091/mbc.10.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerhardt B, Kordas T J, Thompson C M, Patel P, Vida T. J. Biol. Chem. 1998. 15818–15829. [DOI] [PubMed] [Google Scholar]
- 58.Pevsner J, Hsu S, Hyde P S, Scheller R H. Gene. 1996;183:7–14. doi: 10.1016/s0378-1119(96)00367-8. [DOI] [PubMed] [Google Scholar]
- 59.Radisky, D. C., Snyder, W. B., Emr, S. D. & Kaplan, J. (1997) 94, 5662–5666. [DOI] [PMC free article] [PubMed]
- 60.Shestopal S A, Makunin I V, Belyaeva E S, Ashburner M, Zhimulev I F. Mol Gen Genet. 1997;253:642–648. doi: 10.1007/s004380050367. [DOI] [PubMed] [Google Scholar]
- 61.Warner T S, Sinclair D A R, Fitzpatrick K A, Singh M, Devlin R H, Honda B M. Genome. 1998;41:236–243. [PubMed] [Google Scholar]