

Abstract
Site-specific proteolysis is an important biological mechanism for the regulation of cellular processes such as gene expression, cell signaling, development, and apoptosis. In transcriptional regulation, specific proteolysis regulates the localization and activity of many regulatory factors. The C1 factor (HCF), a cellular transcription factor and coactivator, undergoes site-specific proteolytic processing at a series of unusual amino acid reiterations to generate a family of amino- and carboxyl-terminal polypeptides that remain tightly associated. Expression and purification of bacterially expressed domains of the C1 factor identifies an autocatalytic activity that is responsible for the specific cleavage of the reiterations. In addition, coexpression of the autocatalytic domain with a heterologous protein containing a target cleavage site demonstrates that the C1 protease may also function in trans.
Keywords: protease, enhancer, trans-cleavage, transport
Site-specific proteolysis plays a significant role in the regulation of many basic cellular processes. The control of cholesterol metabolism by proteolytic regulation of transcription factor SREBP (1, 2), the cleavage of amyloid precursor protein that may affect the development of Alzheimer's disease (3–6), the proteolytic activation of Notch during development (7–12), the intricate pathway of proteolytic targeting resulting in cell apoptosis (13–16), and the assembly of viral infectious particles (17) illustrate the importance and diversity of site-specific proteolysis in the regulation of both normal cellular as well as disease processes.
The C1 factor, a unique transcription factor, was identified as a required component of the herpes simplex virus (HSV) immediate early gene enhancer complex (18–24). A cellular protein, the factor functions as the coordinator of the enhancer assembly by direct interactions with the cellular POU-homeodomain protein Oct-1 (25) and the viral encoded transactivator α-trans-induction factor [αTIF (VP16)] (24, 26, 27). The C1 factor also interacts with GA-binding protein (GABP) and Sp1 (28), cellular transcription factors whose binding sites are adjacent to the core and contribute to the enhancer regulation (29–31). In addition, the factor also functions as a transcription coactivator for proteins such as GABP (28). Most significantly, the C1 factor may play a critical role in the regulation of HSV lytic-latent cycle, as the protein is uniquely sequestered in the cytoplasm of sensory neurons and is relocalized to the nucleus upon signals that reactivate latent virus in these cells (32).
As illustrated in Fig. 1, the C1 factor is expressed as a 230-kDa precursor containing a series of unique 20-amino acid reiterations (23, 33). Amino-terminal sequencing of the products isolated from mammalian cells demonstrated these repeats (VCSNPPCETHETGTTNTATT) and an additional sequence (102 cleavage site, SNMSSNQDPPPAASDQGEVE), located directly carboxyl terminal to the reiterations, are the sites of specific proteolytic processing that generates a family of amino- and carboxyl-terminal polypeptides ranging from 180 kDa to 102 kDa (33). Interestingly, the processing of the C1 factor results in polypeptides that do not segregate but rather remain tightly associated (23, 24), suggesting that processing of C1 may be a mechanism for regulating the activity of the protein.
Figure 1.
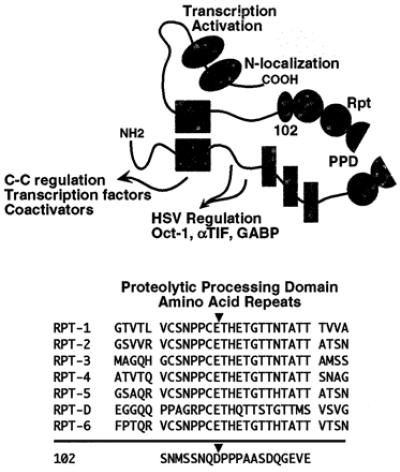
Functional domains and proteolytic processing of the C1 factor. A schematic representation of the C1 factor is shown with the functional domains indicated. The amino terminus (NH2) contains domains involved in interaction with transcription factors such as Oct-1, αTIF, GABP, and LZIP, as well as other coactivator proteins. In addition, a mutation in the NH2 results in a temperature sensitive cell-cycle G0-G1 block (C-C). The central proteolytic processing domain (PPD) contains the C1 factor reiterations (VCSNPPCETH… ) and the 102 cleavage site (SNQDPPPAAS… ). The carboxyl terminus encodes the transactivation domain and nuclear localization signal (N-localization). The sequence of the C1 factor 20 amino acid reiterations and 102 cleavage site are aligned and the cleavage positions are indicated.
Strikingly, the amino- and carboxyl-terminal domains of the C1 factor are both functionally distinct and functionally interdependent. As schematically illustrated in Fig. 1, the amino terminus of the protein contains sites for interaction of the factor with the HSV enhancer components Oct-1, αTIF, and GABP as well as additional cellular transcription factors (LZIP) and coactivators (28, 34–36). The amino terminus has also been implicated in the control of cell cycle as a mutation (P134S) results in a temperature-sensitive G0-G1 block (37). In contrast, the carboxyl terminus contains the sole nuclear localization sequence (38) as well as the protein's transactivation domain (data not shown).
To characterize this unusual processing, the mechanism of proteolysis was investigated. The results demonstrate that the C1 factor repeats are cleaved by an autocatalytic activity defining an additional functional domain of the protein. The catalysis suggests that the processing is involved in the regulation of the protein activity.
Materials and Methods
Polyclonals, Monoclonals, Western Blots, and Immunofluorescence.
The characterization of antigen affinity-purified anti-C1 polyclonal antibodies (Ab2125, Ab2159, Ab2131, and Ab2126) has been described (33, 39). Immunoprecipitation and Western blot analyses using anti-C1 affinity purified antibodies were as described (33, 39). Monoclonal antibodies (mAbAE1, mAbJA5, and mAbKG2) were produced against SDS/PAGE-purified fusion proteins expressed in Escherichia coli according to standard procedures and were screened by ELISA, Western blot, and immunoprecipitation assays for specific reactivity to the C1-derived antigen. Purified mAbs from ascites fluids were produced with selected monoclonal cell lines according to standard procedures. Immunofluorescent localization of the C1 factor in NIH 3T3 cells was done essentially as described (33).
Expression and Purification of Fusion Proteins.
Oligonucleotides encoding a six-histidine tag were inserted in the EcoRI site of pGEX1λ (Amersham Pharmacia) to generate pGEX1λhis. DNAs encoding domains of the C1 factor were ligated in-frame to generate amino-terminal glutathione S-transferase (GST) carboxyl-terminal histidine tag fusion proteins pGST5cahis (C1 amino acids 1177–1787), pGST5caΔ1 (amino acids 1177–1525), and pGST5caΔ2 (amino acids 1270–1525). Control plasmids contained the C1 DNA inserts in the opposite noncoding orientation (pGST5cainv). pRpt1-6ppa and pRpt4-6ppa contained C1 DNA sequences coding for amino acids 1003–1463 and 1278–1463, respectively, in pET21d (Novagen). The expression and purifications of these proteins by Ni-affinity chromatography (Novagen) were as recommended by the manufacturer in the presence of 6 M GuHCl. Proteins eluted from the resin were dialyzed to 50 mM Tris, 10% glycerol, 150 mM NaCl, 0.1 mM EDTA, and 5 mM DTT.
Denaturation-Renaturation Assays.
Aliquots of Ni-affinity purified proteins were resolved by SDS/PAGE and visualized by KCl staining. Full-length fusion proteins and control proteolytic products were eluted in buffer A (50 mM Tris, pH 7.9/0.1% SDS/200 mM NaCl/0.1 mM EDTA/5 mM DTT), precipitated with acetone, solubilized in buffer B (50 mM Tris, pH 7.9/150 mM NaCl/0.1 mM EDTA/1 mM DTT/20% glycerol) with 6 M guanidine HCl, and dialyzed to buffer B. Full-length GST5cahis was isolated, denatured, and renatured as described above in the presence of various protease inhibitors or a protease inhibitor mixture (Complete, Boehringer Mannheim). Equivalent aliquots of the reactions were resolved by SDS/PAGE, transferred to immobilon, and visualized by Western blot analysis with anti-C1 Ab2125 and Ab2131. Precursor and products were quantitated by chemifluorescence (Molecular Dynamics Storm). For analysis of the effect of denaturants on the autoproteolysis activity, aliquots of purified GST5cahis protein were treated with guanidine HCl, urea, deoxycholate, or were heat-treated at 65°C in buffer C (20 mM Tris, pH 7.4/100 mM NaCl/5% glycerol/0.05% Nonidet P-40/10 mM BME) for 30 min before dialysis to buffer C. Aliquots of reactions were resolved by SDS/PAGE, transferred to immobilon, and stained with Coomassie brilliant blue. The blots were subsequently developed for chemiluminescence or quantitative chemifluorescence (Molecular Dynamics Storm).
Amino-Terminal Sequencing.
Aliquots of bacterial protein preparations or reaction mixtures were resolved by SDS/PAGE, transferred to immobilon, and stained with Coomassie brilliant blue. Polypeptides selected for amino-terminal sequencing were based on Western blot analysis using anti-C1, anti-GST, and anti-histag antibodies. The amino-terminal sequences of excised bands were determined by standard procedures (Mark Garfield, Twinbrook II, Research Technologies Branch, Structural Biology Section, National Institutes of Health).
Transcleavage System.
Oligonucleotides encoding C1 cleavage sites 2–3 (amino acids 1074–1126) were inserted in-frame in the linker domain of the bacteriophage λ cI repressor cassette pcI.Bss-pAlterEX2 as described (40) to generate pcI.rpt2-3. JM109 E. coli cells were cotransformed with pcI.Bss-pAlterEX2 or pcI.rpt2-3 and pGST5cahis1λ or pGST5cahisinv as described (40). The expression of GST5cahis was induced in cultures of the cotransformed strains with 1.5 mM IPTG for 1 h. The cultures were pelleted by centrifugation and resuspended to an equivalent OD600. Equivalent aliquots of the cultures were resolved by SDS/PAGE, transferred to immobilon, and stained with Coomassie brilliant blue to ensure equivalent extract protein extract loading. The blots were subsequently probed with anti-λ cI sera (gift of J. Roberts, Cornell University, Ithaca, NY) and developed for chemiluminescence or quantitative chemifluorescence (Molecular Dynamics Storm).
Results
Proteolytic Processing of C1 Fusion Proteins Expressed in E. coli.
In the course of functional analysis of the C1 factor, a number of C1 domains were expressed in E. coli. As shown in Fig. 2, GST-5ca-his fusion protein was produced encoding C1 amino acids 1177–1787. This fusion contained C1 reiterations 4–6, the 102 cleavage site, and a portion of the carboxyl terminus. Upon expression in E. coli, the resulting purified protein preparation contained a series of polypeptides ranging from the 95-kDa full-length fusion to 27-kDa polypeptides. The pattern of polypeptides remained consistent in several preparations and purification procedures. As shown in Fig. 2, a representative preparation was subjected to Western blot analysis with anti-C1 sera Ab2125 and anti-GST. Ab2125 detected the full-length polypeptide as well as proteins from 87 to 45 kDa. In contrast, anti-GST reacted with the full-length protein and polypeptides primarily in the 50- to 27-kDa range.
Figure 2.
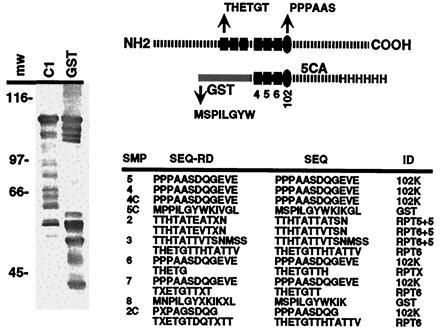
Processing of GST-5ca-his in E. coli. The structure of GST-5ca-his is illustrated relative to the WT C1 protein. The C1 factor reiterations and 102 cleavage site are represented by the filled rectangles and oval, respectively. The appropriate amino terminus derived by cleavage at the C1 repeat (THETG) or 102 site (PPPAAS) is shown as well as the amino terminus of the GST protein (MSPIL). A representative affinity-purified preparation of GST-5ca-his was resolved by SDS/PAGE and was subjected to Western blot analysis with C1- and GST-specific antisera. The amino-terminal sequences of isolated polypeptides from several preparations are shown: SMP, sample; SEQ-RD, NH2 sequence read; SEQ, deduced NH2 sequence; and ID, identified cleavage. Because of the complexity of the protein profile, numerous products from several independent protein preparations were sequenced, and the compiled nonredundant product sequences are listed. Multiple sequences indicate that the sample contained major and minor polypeptides.
To determine whether these polypeptides resulted from the appropriate proteolysis of the C1 factor fusion protein in E. coli, selected proteins were subjected to amino-terminal sequencing. As shown in Fig. 2, the amino terminus of several polypeptides corresponded to the correct proteolytic cleavage of the C1 factor repeats (THETG… ). Furthermore, similar to mammalian cells, the most abundant polypeptide was derived by processing at the 102-kDa site (PPPAASDQ… ). Interestingly, in addition to the correct cleavage species, the preparation also contained several polypeptides whose amino termini were exactly 5 amino acids carboxyl terminal to the natural mammalian cleavage position in repeat 5 and repeat 6 (TTHTATTATSN vs. THETGTTHTATT). As discussed below, these products may reflect an altered recognition of the cleavage site.
Autocatalysis of the C1 Factor in Vitro.
The appropriate proteolysis of the GST-5ca-his in E. coli suggested that this portion of the protein might encode an autocatalytic activity. Therefore, the full-length 95-kDa fusion protein as well as control proteolytic products were purified to homogeneity by SDS/PAGE, isolated, and renatured. As shown in Fig. 3, renaturation of purified eluted full-length protein (FL) resulted in a polypeptide pattern that was nearly identical to the initial GST-5ca-his preparation (Prep). In contrast, the proteolytic products representing the amino-terminal GST (band 5) and the 102 cleavage product (band 4) did not exhibit any additional degradation. The amino-terminal sequences of the polypeptides derived from the renaturation reaction demonstrated that these proteins resulted from appropriate proteolytic processing at rpt 5, rpt 6, and the 102 sites [THETG.. and PPPAAS (Fig. 3)]. Furthermore, in contrast to the products detected in the directly purified bacterial preparation (refer to Fig. 2), no products were identified with altered cleavage sites. These results indicated that the C1 factor contained an autocatalytic activity responsible for the proteolysis of the C1 factor to generate the family of NH2- and COOH- polypeptides. However, although the full-length protein was purified by SDS/PAGE, a slight possibility remained that the C1 reiterations were the target of a bacterial protease. Therefore, Rpt4-6-his, containing the C1 reiterations 4, 5, and 6, was expressed and purified in a similar manner. As shown in Fig. 3, no cleavage products of Rpt4-6-his were detected either in the initial expression preparation or after purification and renaturation of the full-length protein. The results indicated that the C1 factor reiterations are not proteolytically processed by an endogenous bacterial protease. Furthermore, these cleavage site repeats are not sufficient to enact autocatalytic cleavage.
Figure 3.
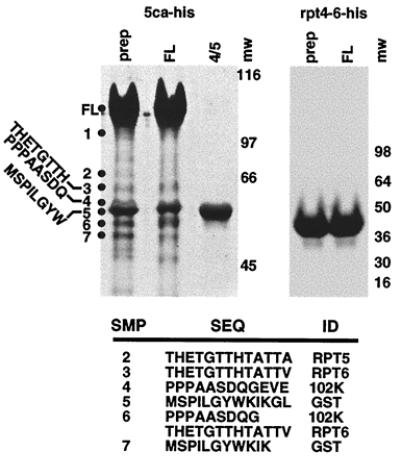
Autocatalytic processing of GST-5ca. Aliquots of affinity-purified preparations (Prep) of GST-5ca-his and Rpt4-6-his were resolved along with gel-purified, renatured full-length (FL) protein and cleavage products 4/5 (4/5). The amino-terminal sequences of isolated polypeptides from the GST-5ca-his denaturation-renaturation reaction are shown, along with the identification of the cleavage site.
Characterization of the C1 Factor Autoprocessing Activity.
In using the purification-renaturation assay of the full-length GST-5ca-his fusion protein as described, the C1 autoproteolytic activity was classified by inhibitor analysis. As shown in Fig. 4 Upper, a protease inhibitor mixture significantly suppressed the generation of the major 102 proteolytic product. The inhibition of product formation in the presence of protease inhibitors clearly indicates that the cleavage products are not polypeptides that copurify with the full-length GST-5ca-his but are, rather, autocatalytic products. Therefore, renaturation of GST-5ca-his was done in the presence of various serine, aspartate, and cystein protease inhibitors. After quantitation of the precursor and the 102 product, only the irreversible serine protease inhibitor pefabloc was highly effective in suppressing the autocatalysis.
Figure 4.
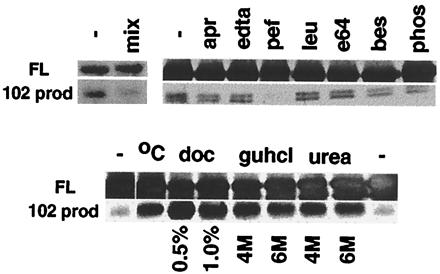
Characterization of the C1 factor autoproteolytic activity. (Upper) Full-length GST-5ca-his (FL) was isolated by SDS/PAGE and was incubated in the absence (−) or presence of the indicated protease inhibitor: mix, inhibitor mixture; apr, aprotinin, 5 μg⋅ml−1; EDTA, 1 mg⋅ml−1; pef, pefablock, 0.5 mg⋅ml−1; leu, leupeptin, 2 μg⋅ml−1; bes, bestatin, 40 μg⋅ml−1; e64, 10 μg⋅ml−1; phos, phosphoramidon, 200 μg⋅ml−1. (Lower) Aliquots of purified GST-5ca-his were subjected to denaturation procedures as follows: (−), no treatment; °C, 65°C; doc, deoxycholate; guhcl, guanidine HCl; and urea. Quantitation of the precursor and 102 site cleavage products (102 prod) was as described.
Although processing of the C1 factor in mammalian cells is efficient, processing of the GST-5ca-his in E. coli or in vitro was relatively inefficient, suggesting that appropriate protein folding, cofactors, modification, or accessory proteins may play a role in regulation of the catalysis. To address the effects of folding of the fusion protein, the full-length protein was subjected to various denaturing agents. Aliquots of the purified preparation were heated to 65°C or were incubated with deoxycholate, guanidine HCl, or urea. After dialysis, the reactions were resolved by SDS/PAGE, and the precursor and 102 product were quantitated. As illustrated (Fig. 4 Lower), each treatment stimulated the generation of the major proteolytic product, although treatment with deoxycholate was most effective. In contrast, neither the addition of Ca2+, Zn2+, Mn2+, or Mg2+ nor prolonged incubation periods had any significant effect upon the activity (data not shown).
Previously, insertion of C1 processing sites in a donor protein (Oct-1) resulted in inefficient processing of the target in mammalian cells (41), suggesting that the C1 factor proteolytic activity may also function in trans. Therefore, this was investigated by using an E. coli coexpression assay as illustrated in Fig. 5 (40). The C1 cleavage sites 2–3 (amino acids 1074–1122) were inserted into the linker domain of the bacteriophage λ cI repressor cassette. The cI.rpt2-3 target protein or the control cI.Bss were coexpressed with GST-5ca or GST-5cainv containing the C1 sequences inserted in the noncoding orientation. After induction of the expression of the C1 construct, the cleavage of the targets were monitored by quantitative chemifluroescent analysis using anti-cI sera. The induction of GST-5ca resulted in a significant reduction in the steady-state levels of cI.rpt2-3, indicating that the C1 factor protease could effectively function in trans. In contrast, no change was evident in the absence of induction or after induction of the control. Furthermore, the specificity of this trans-cleavage reaction for the C1 factor repeats is illustrated by the lack of processing of the control parental cI.Bss repressor.
Figure 5.
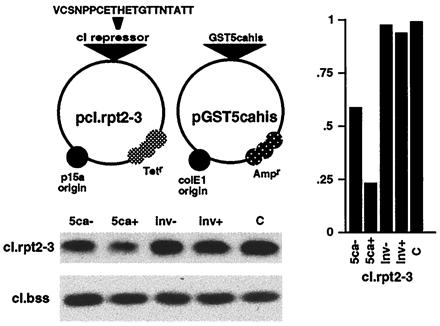
Transcleavage of a heterologous target protein. The structures of the transcleavage assay coexpression plasmids are schematically illustrated. pcI.rpt2-3 expresses the cI repressor containing C1 repeats 2–3 whereas pGST5cahis expresses the indicted C1 fusion protein. The steady state levels of the target cI repressor are shown in the cI Western blot for the coexpression cultures containing pcI.rpt2-3 or the control pcI.Bss and either GST-5ca (5ca), GST-5cainv (inv), or pgst-1λ control vector (C). (−) and (+) indicate uninduced and induced cultures, respectively. Chemifluorescent quantitation of the precursor cI.rpt2-3 is graphically illustrated relative to the control culture. Only the precursor protein is monitored as the cleavage products are rapidly degraded (H. J. Sices and T.M.K., unpublished observations).
Localization of C1 Processing.
Previous studies had suggested that the processing of the C1 factor occurred in the nucleus of mammalian tissue culture cells (41). However, these studies used constructs encoding the C1 protein in transfection assays and did not address the processing of the endogenous protein. Therefore, monoclonal antibodies were developed to distinct domains of the C1 protein including sequences between repeats 3 and 4 (interrepeat domain). The determinants and specificity of these antibodies were determined by ELISA assays, Western blots, and/or immunoprecipitations (data not shown). mAbJA5 and mAbKG2 were produced against the proteolytic processing domain whereas mAbAE1 was derived against carboxyl-terminal sequences of the C1 factor. As shown by the Western blot in Fig. 6 Upper, mAbJA5 reacts with a determinant located between C1 processing sites 3 and 4 (interrepeat domain) whereas the determinant for mAbKG2 is located within the domain encoding processing sites 4 through 6 (Fig. 6). Furthermore, in Western blots of mammalian cell extracts, mAbKG2 clearly reacted with polypeptides that coreact with previously defined anti-C1 seras (data not shown). These monoclonals and previously defined polyclonal seras (Ab2131 and Ab2125) were used to detect the endogenous C1 protein by immunofluorescent staining (Fig. 6 Lower). Ab2125, Ab2131, and mAbAE1 detect primarily nuclear C1 protein, although cytoplasmic staining is evident above the control background. In contrast, mAbJA5 detects punctate cytoplasmic protein whereas the mAbKG2 pattern is a compilation of that exhibited with mAbAE1 (primarily nuclear) and mAbJA5 (punctate cytoplasmic). The distinct pattern of immunofluorescence suggests that the JA5 epitope (interrepeat domain) is proteolytically removed before transport of the C1 factor to the nucleus.
Figure 6.
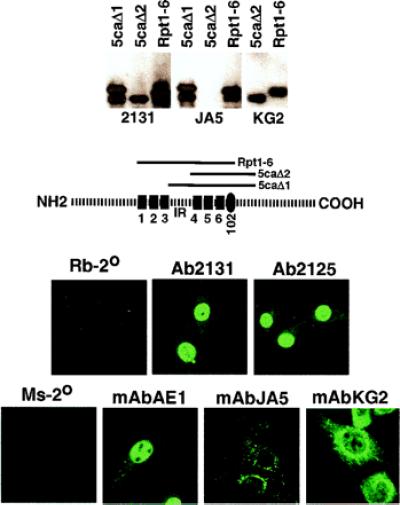
Immunofluorescent localization of C1 factor processing. (Upper) Aliquots of the illustrated bacterially expressed C1 proteins were resolved by SDS/PAGE, and transferred blots were probed with the indicated polyclonal or monoclonal sera. (Lower) 3T3 cells were stained with the indicated anti-C1 polyclonal or monoclonal sera. Rb-2° and Ms-2° are controls stained only with secondary antibodies.
Discussion
Site-specific proteolysis is a significant biological mechanism for the regulation of a diverse set of basic cellular processes, including gene expression. Studies on the processing of transcription factors such as NFκB and SREBP have illustrated how proteolysis may regulate transcription pathways by redirecting the localization of these proteins. Similarly, the regulation of developmental determinants such as Notch and growth factor receptors by proteolytic processing controls important cell signaling pathways.
An unusual multifunctional protein, the C1 factor (HCF) undergoes a series of site-specific proteolytic events. The protein is expressed as a large 230-kDa precursor that is processed primarily at a series of 20-amino acid repeats present in the central domain of the protein. However, in contrast to the proteolytic regulation of other transcription factors such as SREBP in which proteolysis results in release of a functional domain of the protein, the C1 factor undergoes a series of processing events that results in polypeptides that remain tightly associated. The resulting family of amino- and carboxyl-terminal polypeptides function as (i) a coordinator of the assembly of the HSV immediate early multiprotein enhancer complexes; (ii) a coactivator for some transcription factors such as GABP by direct interaction with the protein's transactivation domain; and (iii) a mediator of cell cycle progression. Most significantly, the C1 factor has been implicated in the regulation of the HSV lytic-latent cycle. In sensory neurons, the site of latent HSV, the C1 factor is uniquely sequestered in the cytoplasm and is rapidly relocalized to the nucleus upon signals that reactivate virus from latency.
Demonstrated by the activity of the GST-C1 fusion proteins, the C1 factor contains an autocatalytic activity that is responsible for the specific cleavage at the C1 amino acid repeats as well as the predominant 102 cleavage site. Interestingly, the 102 site (SNMSSNQDPPPAASDQGEVE) bears little primary sequence resemblance to the repeats (VCSNPPCETHETGTTNTATT). This site is located directly adjacent to the carboxyl-terminal sixth repeat and suggests the possibility that recognition of the C1 factor repeat may align cleavage at a distinct spatial site. The aberrant cleavage of the GST-5ca-his fusion protein in E. coli, where some cleavages occurred exactly 5 amino acids carboxyl terminal to the natural cleavage position, may reflect a distinct folding and site recognition. This hypothesis is strengthened by the absence of these altered cleavage products in the reactions using renatured full-length GST-5ca.
The autocatalytic domain of the C1 factor encoded by GST-5ca exhibits a low efficiency of cleavage in E. coli relative to mammalian cells. Denaturation-renaturation studies indicate that the folded structure of the expressed protein may account for the inefficiency. However, the activity also may be regulated by protein interactions or modification in its natural environment. In addition, although GST-5ca-his contains a domain that is sufficient for accurate autocatalysis, additional domains or other protein cofactors may play a significant role in determining the activity. Interestingly, GST-5ca contains the interrepeat domain between repeats 3 and 4, repeats 4–5-6, the 102 cleavage site, and a portion of the carboxyl terminus of the protein. Therefore, it is possible that the unusual C1 factor reiterations themselves play a significant role in the recognition and cleavage process. However, as shown by the lack of processing of the Rpt4-6-his protein (containing repeats 4–5-6), this region is not sufficient to direct autocatalysis.
The reiterations/cleavage sites of the C1 factor are, to date, unique. Although the catalytic domain can function in trans, it is not likely to be a primary function, because similar sequences have not been identified in other proteins. In fact, although homologs of the C1 factor have been found in many species [Caenorhabditis elegans U61948, zebrafish AI626241, mouse (39), and hamster D45419], the C. elegans homolog does not even contain the processing domain, suggesting that it has evolved in more complex organisms in concert with other protein functions.
Although the unique reiterations and processing of the C1 may impact on the protein's functions, the actual role of the processing remains unclear. As illustrated in Fig. 7, the processing may be important for transport-localization, interaction with other cofactors, or regulation of the C1 factor's conformation and activity. As demonstrated, some processing of the C1 factor does occur in the cytoplasm as antiseras recognizing the interrepeat domain localize only cytoplasmic protein whereas others localize primarily nuclear protein. It is possible, therefore, that initial cytoplasmic cleavage results in release of the protein for transport to the nucleus, where additional processing continues, affecting the interaction of the C1 factor with other components. Such a mechanism may be important in the signaling transport of the C1 factor that is uniquely exhibited in sensory neurons. Although cytoplasmic processing of the C1 factor is consistent with the observed accumulation of the interrepeat domain in the cytoplasm and the processed amino and carboxyl termini in the nucleus, it remains formally possible that the interrepeat domain is specifically exported from the nucleus after cleavage or that this localization may reflect an alternate, nonautocatalytic pathway of C1 factor proteolysis.
Figure 7.
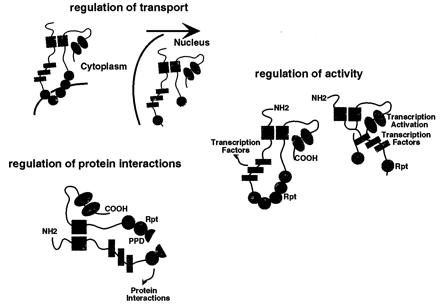
Potential roles of proteolytic processing of the C1 factor. Several potential roles for the proteolytic processing of the C1 factor are schematically illustrated: the initial proteolysis results in release and transport to the nucleus; the regulation of the interaction of proteins with the C1 repeat domain by selective proteolysis; and the regulation of the C1 factor activity by proteolytic alteration in C1 factor conformation allowing alternate interactions between the amino and carboxyl-terminal domains.
It is important to note that, although the NH2- and COOH-terminal polypeptides of the C1 factor can be coprecipitated and copurified, processing may allow segregation of the components under some conditions (42). Segregation would dissociate important interdependent domains of the C1 factor (protein interaction domains, transcription activation domain, nuclear localization signals), thereby also affecting its function. As the nuclear localization signal resides within the extreme carboxyl terminus of the C1 factor (38), processing results in a dependence on the carboxyl terminus for nuclear import. The autocatalytic function of the C1 factor defines an additional activity of this unusual transcriptional coactivator and suggests a role for proteolytic processing in the regulation of this protein.
Acknowledgments
We thank J. Roberts for λ-cI antisera; B. Moss, P. Sharp, J. Yewdell, and members of the Laboratory of Viral Diseases for helpful discussions; M. Garfield for amino-terminal protein sequencing; A. Pacheco and M. Leusink for technical assistance, J. Yewdell for microscopy assistance; R. Scarr, M. Smith, M. Beddall, and P. Sharp for communicating data before publication; and B. Moss, P. Sharp, A. McBride, and J. Yewdell for critical reading of this manuscript. These studies were supported by the Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health (T.M.K.).
Abbreviations
- HSV
herpes simplex virus
- GABP
GA-binding protein
- GST
glutathione S-transferase
- αTIF
α-trans-induction factor
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160266697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160266697
References
- 1.Brown M S, Goldstein J L. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 2.Brown M S, Goldstein J L. Proc Natl Acad Sci USA. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassar R, Bennett B D, Babu-Khan S, Kahn S, Mendiaz E A, Denis P, Teplow D B, Ross S, Amarante P, Loeloff R, et al. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 4.Sinha S, Lieberburg I. Proc Natl Acad Sci USA. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younkin S G. J Physiol (Paris) 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- 6.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 7.Artavanis-Tsakonas S, Rand M D, Lake R J. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 8.Kimble J, Simpson P. Annu Rev Cell Dev Biol. 1997;13:333–361. doi: 10.1146/annurev.cellbio.13.1.333. [DOI] [PubMed] [Google Scholar]
- 9.Struhl G, Adachi A. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- 10.Struhl G, Greenwald I. Nature (London) 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 11.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm J S, Schroeter E H, Schrijvers V, Wolfe M S, Ray W J, Goate A, Kopan R. Nature (London) 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 12.Schroeter E H, Kisslinger J A, Kopan R. Nature (London) 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 13.Salvesen G S, Dixit V M. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 14.Muzio M. Int J Clin Lab Res. 1998;28:141–147. doi: 10.1007/s005990050035. [DOI] [PubMed] [Google Scholar]
- 15.Nunez G, Benedict M A, Hu Y, Inohara N. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 16.Patel T, Gores G J, Kaufmann S H. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- 17.Fields B N, Knipe D M, Howley P M. Fields Virology. New York: Lippincott–Raven; 1996. [Google Scholar]
- 18.Gerster T, Roeder R G. Proc Natl Acad Sci USA. 1988;85:6347–6351. doi: 10.1073/pnas.85.17.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katan M, Haigh A, Verrijzer C P, van der Vliet P C, O'Hare P. Nucleic Acids Res. 1990;18:6871–6880. doi: 10.1093/nar/18.23.6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kristie T M, LeBowitz J H, Sharp P A. EMBO J. 1989;8:4229–4238. doi: 10.1002/j.1460-2075.1989.tb08608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristie T M, Sharp P A. Genes Dev. 1990;4:2383–2396. doi: 10.1101/gad.4.12b.2383. [DOI] [PubMed] [Google Scholar]
- 22.Xiao P, Capone J P. Mol Cell Biol. 1990;10:4974–4977. doi: 10.1128/mcb.10.9.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson A C, LaMarco K, Peterson M G, Herr W. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 24.Kristie T M, Sharp P A. J Biol Chem. 1993;268:6525–6534. [PubMed] [Google Scholar]
- 25.Mahajan S S, Wilson A C. Mol Cell Biol. 2000;20:919–928. doi: 10.1128/mcb.20.3.919-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayes S, O'Hare P. J Virol. 1993;67:852–862. doi: 10.1128/jvi.67.2.852-862.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson A C, Cleary M A, Lai J S, LaMarco K, Peterson M G, Herr W. Cold Spring Harbor Symp Quant Biol. 1993;58:167–178. doi: 10.1101/sqb.1993.058.01.021. [DOI] [PubMed] [Google Scholar]
- 28.Vogel J L, Kristie T M. EMBO J. 2000;19:683–690. doi: 10.1093/emboj/19.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones K A, Tjian R. Nature (London) 1985;317:179–182. doi: 10.1038/317179a0. [DOI] [PubMed] [Google Scholar]
- 30.LaMarco K L, McKnight S L. Genes Dev. 1989;3:1372–1383. doi: 10.1101/gad.3.9.1372. [DOI] [PubMed] [Google Scholar]
- 31.Triezenberg S J, LaMarco K L, McKnight S L. Genes Dev. 1988;2:730–742. doi: 10.1101/gad.2.6.730. [DOI] [PubMed] [Google Scholar]
- 32.Kristie T M, Vogel J L, Sears A E. Proc Natl Acad Sci USA. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kristie T M, Pomerantz J L, Twomey T C, Parent S A, Sharp P A. J Biol Chem. 1995;270:4387–4394. doi: 10.1074/jbc.270.9.4387. [DOI] [PubMed] [Google Scholar]
- 34.Freiman R N, Herr W. Genes Dev. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes T A, La Boissiere S, O'Hare P. J Biol Chem. 1999;274:16437–16443. doi: 10.1074/jbc.274.23.16437. [DOI] [PubMed] [Google Scholar]
- 36.Lu R, Yang P, O'Hare P, Misra V. Mol Cell Biol. 1997;17:5117–5126. doi: 10.1128/mcb.17.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goto H, Motomura S, Wilson A C, Freiman R N, Nakabeppu Y, Fukushima K, Fujishima M, Herr W, Nishimoto T. Genes Dev. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 38.LaBoissiere S L, Hughes T, O'Hare P. EMBO J. 1999;18:480–489. doi: 10.1093/emboj/18.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kristie T M. J Biol Chem. 1997;272:26749–26755. doi: 10.1074/jbc.272.42.26749. [DOI] [PubMed] [Google Scholar]
- 40.Sices H J, Kristie T M. Proc Natl Acad Sci USA. 1998;95:2828–2833. doi: 10.1073/pnas.95.6.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson A C, Peterson M G, Herr W. Genes Dev. 1995;9:2445–2458. doi: 10.1101/gad.9.20.2445. [DOI] [PubMed] [Google Scholar]
- 42.Scarr R B, Smith M R, Beddall M, Sharp P A. Mol Cell Biol. 2000;20:3568–3575. doi: 10.1128/mcb.20.10.3568-3575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]