

Abstract
Macrophages from Tpl2 knockout (Tpl2–/–) mice exhibit a defect in ERK activation by lipopolysaccharide (LPS). This impairs the nucleocytoplasmic transport of the tumor necrosis factor α (TNF-α) mRNA and prevents the induction of TNF-α by LPS. As a result, Tpl2–/– mice are resistant to LPS/d-galactosamine-induced shock. We demonstrate that Tpl2 is essential for ERK signals transduced by members of the TNF receptor superfamily, such as CD40 and the TNF receptor 1. Thus, ERK activation was impaired in Tpl2–/– B cells and macrophages stimulated with agonistic CD40 antibody or TNF-α, whereas the induction of other mitogen-activated protein kinases, such as JNK and p38, and the activation of NF-κB were unaffected. Tpl2 was recruited to a CD40/TRAF6 complex in response to CD40 stimulation. Moreover, TRAF6, which when overexpressed activates ERK, failed to do so in Tpl2–/– cells. The selective signaling defect resulting from the inactivation of Tpl2 allowed us to demonstrate that CD40-mediated ERK activation contributes to immunoglobulin production but is not essential for B-cell proliferation.
Keywords: CD40/ERK/signaling/TNF/Tpl2
Introduction
The Tpl2/Cot proto-oncogene encodes a serine/threonine protein kinase that is activated by provirus insertion in MoMuLV-induced T-cell lymphomas and MMTV- induced mammary carcinomas (Patriotis et al., 1993; Erny et al., 1996). Earlier studies had shown that overexpressed Tpl2 activates the ERK, JNK and p38 mitogen-activated protein kinase (MAPK) pathways, NFAT and NF-κB, induces IL-2 expression in EL4 and Jurkat T cells, promotes cell cycle progression and is highly oncogenic in mice (Patriotis et al., 1994; Salmeron et al., 1996; Ceci et al., 1997; Tsatsanis et al., 1998a,b; Lin et al., 1999). Despite the profound biological effects of overexpressed Tpl2, Tpl2 knockout (Tpl2–/–) mice are viable and have no obvious phenotypic defects (Dumitru et al., 2000). However, a detailed analysis of these mice revealed a critical physiological role for Tpl2 in endotoxin shock. Thus, Tpl2–/– mice exhibit a defect in tumor necrosis factor α (TNF-α) production following lipopolysaccharide (LPS) inoculation, and they are resistant to LPS/d-galactosamine-induced shock. This defect was found to be the result of impaired ERK activation by LPS, which in turn is responsible for inhibiting the nucleocytoplasmic transport of the TNF-α mRNA (Dumitru et al., 2000). These observations provided an explanation to the recently described protection of Tpl2–/– mice from TNF-α-induced inflammatory bowel disease (Kontoyiannis et al., 2002). In other studies, we found that Tpl2-transduced ERK activation signals initiated by LPS in macrophages regulate the transcription of the cyclooxygenase-2 (COX-2) gene and the stability of its message. As a result, LPS-stimulated Tpl2–/– macrophages exhibit a defect in prostaglandin synthesis (Eliopoulos et al., 2002a). Recent work using NF-κB1–/– macrophages has shown that p105 NF-κB1 controls the stability and function of Tpl2, an observation that may explain the inability of these cells to activate ERK and its downstream target COX-2 in response to LPS stimulation (Waterfield et al., 2003). Taken together, these data demonstrate that Tpl2 plays a critical role in the regulation of LPS signaling and function. In this report, we present evidence that B cells and macrophages from Tpl2–/– mice are also defective in their ability to respond to members of the TNF-α cytokine family, such as CD40 ligand (CD40L) and TNF-α itself.
CD40, the receptor for CD40L, is a member of the TNF receptor (TNFR) superfamily. It is expressed in a plethora of cell types, including B cells, macrophages and dendritic, endothelial and epithelial cells (van Kooten and Banchereau, 2000). Increasing in vitro and in vivo evidence suggests that CD40 regulates growth and survival in a cell type-dependent manner. Thus, whereas CD40 transduces potent proliferative and anti-apoptotic signals in normal B cells, the treatment of lymphoma and carcinoma cell lines with CD40L may result in growth inhibition and apoptosis, an effect mediated in part through the upregulation of cytotoxic ligands of the TNF family (Funakoshi et al., 1994; Eliopoulos et al., 1996; Afford et al., 1999; Grell et al., 1999; Eliopoulos et al., 2000; Challa et al., 2002). Following stimulation with CD40L, the cytoplasmic C-terminus of CD40 recruits several members of the family of TNFR-associated factors (TRAFs). One of these proteins, TRAF6, has been reported to play an important role in ERK activation by CD40L (Kashiwada et al., 1998). Data shown in the present study demonstrate that the activation of ERK by TRAF6-transduced signals requires Tpl2.
The CD40/CD40L interactions play a central role in orchestrating the immune response. CD40L is produced by both Th1 and Th2 helper T cells, as well as by mast cells, basophils and eosinophils (van Kooten and Banchereau, 2000). Signals induced by CD40L expressed in Th1 and Th2 cells are required for macrophage and B-cell activation, respectively. Of particular interest is the ability of CD40L to cooperate with IL-4, which is also produced by Th2 cells, to regulate immunoglobulin G1 (IgG1) and E (IgE) isotype switching. CD40L and IL-4 are also produced by IgE-stimulated mast cells, basophils and eosinophils and further stimulate B cells to secrete IgE (Gauchat et al., 1993). We show that CD40-induced ERK activation signals, transduced via Tpl2, contribute to IgE production by B cells.
Results
B cells and macrophages from Tpl2–/– mice exhibit a defect in ERK activation following CD40 stimulation
Primary B-cell and macrophage cultures were generated from spleen and bone marrow of Tpl2+/+ and Tpl2–/– mice, as described in Materials and methods. Cell cultures were stimulated with agonistic anti-CD40 monoclonal antibody for various time intervals, and lysates were analyzed for MAPK activation by immunoblotting. Blots were probed with antibodies against the phosphorylated, active forms of ERK1, ERK2, JNK1, JNK2 and p38 MAPKs. Expression of total (phosphorylated and non-phosphorylated) ERK, JNK and p38 MAPKs was determined by probing the same western blots with antibodies against these proteins. The results show that ERK is activated by anti-CD40 in both B cells and macrophages derived from the Tpl2+/+ mice. However, the activation of ERK was severely impaired in B cells and moderately impaired in macrophages from Tpl2–/– mice. JNK and p38 MAPKs were equally phosphorylated in B cells and macrophages derived from Tpl2+/+ and Tpl2–/– mice (Figure 1).

Fig. 1. ERK activation by CD40 in B cells and macrophages depends on signals transduced via Tpl2. (A) Splenic B cells from Tpl2+/+ and Tpl2–/– mice were stimulated with anti-CD40. A western blot of cell lysates harvested at the indicated time points before and after anti-CD40 stimulation was probed with an antibody that recognizes phosphorylated ERK (upper) or total ERK (lower). (B and C) Western blots of the same cell lysates were probed with antibodies against phosphorylated or total JNK (B) and phosphorylated or total p38 MAPKs (C). (D–F) Western blots of cell lysates derived from unstimulated and anti-CD40-stimulated bone marrow-derived macrophages were probed with antibodies against phosphorylated or total ERK (D), phosphorylated or total JNK (E) and phosphorylated or total p38 MAPKs (F). At least four independent experiments were performed and gave similar results.
To determine whether Tpl2 is required for the transduction of CD40 signals that activate NF-κB, we performed electrophoretic mobility shift assays using nuclear proteins isolated from Tpl2+/+ and Tpl2–/– B cells before and 15 and 30 min after stimulation with anti-CD40 monoclonal antibody (mAb). The engagement of CD40 induced a significant increase in nuclear protein binding to a 32P-labeled oligonucleotide probe containing the HIV long terminal repeat (LTR) NF-κB binding site, but there was no difference between the Tpl2+/+ and Tpl2–/– cells with regard to the levels of activation (Figure 2, lanes 1–6). The nature and specificity of the NF-κB probe-bound proteins were assessed by incubating nuclear extracts isolated from anti-CD40-stimulated B cells with 50× excess unlabeled probe containing either the HIV LTR NF-κB consensus or the CREB binding site from the COX-2 promoter. Whereas the cold NF-κB probe abolished the interaction of B-cell nuclear proteins with the 32P-labeled HIV LTR probe, excess CREB-binding oligonucleotides had no effect (Figure 2, lanes 7 and 8). Furthermore, the incubation of CD40-stimulated B-cell nuclear extracts with antibodies against the p65 or p50 NF-κB subunits specifically induced a supershift in the DNA/protein complexes (Figure 2, lanes 9–11). Thus, Tpl2 does not appear to be essential for CD40-induced NF-κB activation in B cells.

Fig. 2. CD40 stimulation promotes comparable levels of NF-κB activation in B cells derived from Tpl2+/+ and Tpl2–/– mice, as determined by electrophoretic mobility shift assay (lanes 1–6). The specificity of the NF-κB binding was confirmed by the effects of an excess of cold NF-κB versus CREB-oligo probe (lanes 7 and 8), and the composition of the DNA probe/protein complexes was assessed by supershift analysis using antibodies against the p65 or p50 subunits of NF-κB or the unrelated transcription factor ATF2 (asterisks; lanes 9–11).
The activation of ERK in anti-CD40-stimulated B cells is MEK dependent
In an effort to determine the pathway through which Tpl2 regulates CD40-transduced ERK activation signals, we first examined the phosphorylation of MEK, the ERK kinase, in anti-CD40-stimulated B cells from Tpl2+/+ and Tpl2–/– mice. The results show that MEK phosphorylation is impaired in anti-CD40-stimulated Tpl2–/– cells (Figure 3A), suggesting that Tpl2 functions upstream of MEK. To determine whether CD40 signals that activate ERK are transduced via MEK, we examined the effects of the MEK inhibitor PD98059 on ERK activity in CD40-stimulated B cells. To this end, endogenous ERK1 and ERK2 were immunoprecipitated from Tpl2+/+ and Tpl2–/– B cells both before and 15 and 30 min after stimulation with anti-CD40. In parallel, ERK1 and ERK2 were immunoprecipitated from anti-CD40-stimulated wild-type B cells that were pretreated with 20 µM PD98059. In vitro kinase assays were carried out on the immunoprecipitates using GST–Elk1 as the substrate. The results showed that PD98059 blocks ERK activation by anti-CD40 (Figure 3B) and confirmed that ERK kinase activity is impaired in the absence of Tpl2 in CD40-stimulated B cells. We conclude that MEK phosphorylation is required for ERK activation following anti-CD40 stimulation and that Tpl2 functions upstream of MEK in this cascade of events.
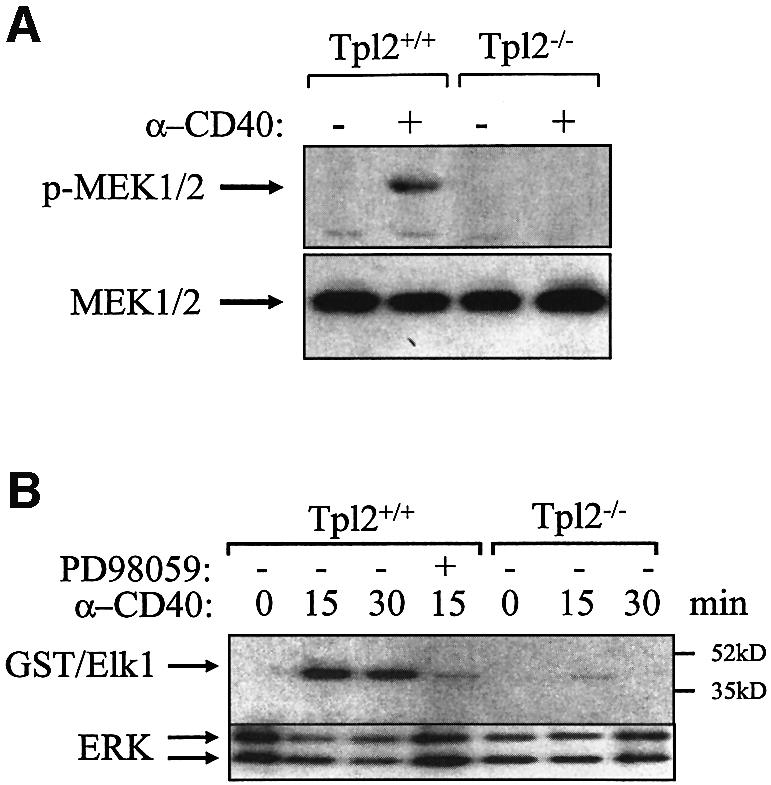
Fig. 3. ERK activation by Tpl2-transduced CD40 signals is MEK dependent. (A) A representative western blot of Tpl2+/+ and Tpl2–/– B-cell lysates harvested before and after anti-CD40 stimulation was probed with an antibody against the phosphorylated form of MEK (upper) or with an antibody that recognizes total MEK (lower). (B) In vitro kinase assays were carried out on anti-ERK immunoprecipitates derived from unstimulated and anti-CD40-stimulated Tpl2+/+ and Tpl2–/– B cells using GST–Elk1 as a substrate. Tpl2+/+ B-cell cultures were also pretreated for 45 min with 20 µM MEK inhibitor PD98059 before being stimulated with the anti-CD40 antibody for 15 min.
Tpl2 transduces TNF-α signals that activate ERK in macrophages
CD40 is a member of the TNFR family. Molecules that belong to this large family elicit diverse biological effects upon stimulation, including mitogenesis, cell cycle arrest, apoptosis or differentiation. In intact animals, several of these molecules function as immunomodulators and as mediators of cytotoxicity and inflammation. Therefore, we asked whether the CD40 signaling defect we observed in the Tpl2–/– mice is limited to CD40 or whether it also applies to other members of this family of receptors, such as the TNFRs. The important role of Tpl2 in the phenotypic effects of TNF-α is highlighted by the recently reported protection of Tpl2–/– mice from TNF-α-induced inflammatory bowel disease (Kontoyiannis et al., 2002). To address the contribution of Tpl2 to TNFR signaling, Tpl2+/+ and Tpl2–/– macrophages were stimulated with mouse TNF-α (mTNF-α). Western blots of cell lysates harvested at sequential time points were probed for ERK phosphorylation using antibodies that recognize phosphorylated and total ERK. The results demonstrate that Tpl2 is also required for ERK activation by TNF-α (Figure 4A). In agreement with our earlier data (Dumitru et al., 2000), PMA induced ERK activation with similar potency and kinetics in both Tpl2–/– and wild-type macrophage cultures (Figure 4A). To determine which of the two known TNFRs transduces Tpl2 signals that regulate ERK, RT–PCR was performed in RNA isolated from Tpl2+/+ and Tpl2–/– macrophages using primers specific for TNFR1 or TNFR2. In addition, Tpl2+/+ macrophages were pretreated with the blocking anti-TNFR1 mAb 55R170 or the anti-TNFR2 mAb TR75-54 and then stimulated with mTNF-α for 15 min before immunoblot analysis for phosphorylated and total ERK was performed. We found that, although both receptors are expressed in mouse macrophages, the TNFR1, but not TNFR2, transduces ERK signals (Figure 4B and C). This finding is consistent with a previous report demonstrating that the engagement of TNFR2 does not induce the activation of ERK in other cell types (Jupp et al., 2001). To confirm our observation, we stimulated Tpl2+/+ and Tpl2–/– macrophages with human TNF-α, which activates the TNFR1 but not TNFR2. Lysates were examined for ERK phosphorylation by immunoblot. It was found that ERK activation following TNFR1 stimulation is severely impaired in Tpl2–/– macrophages (Figure 4D). Therefore, Tpl2 transduces TNFR1 signals that activate ERK in macrophages.
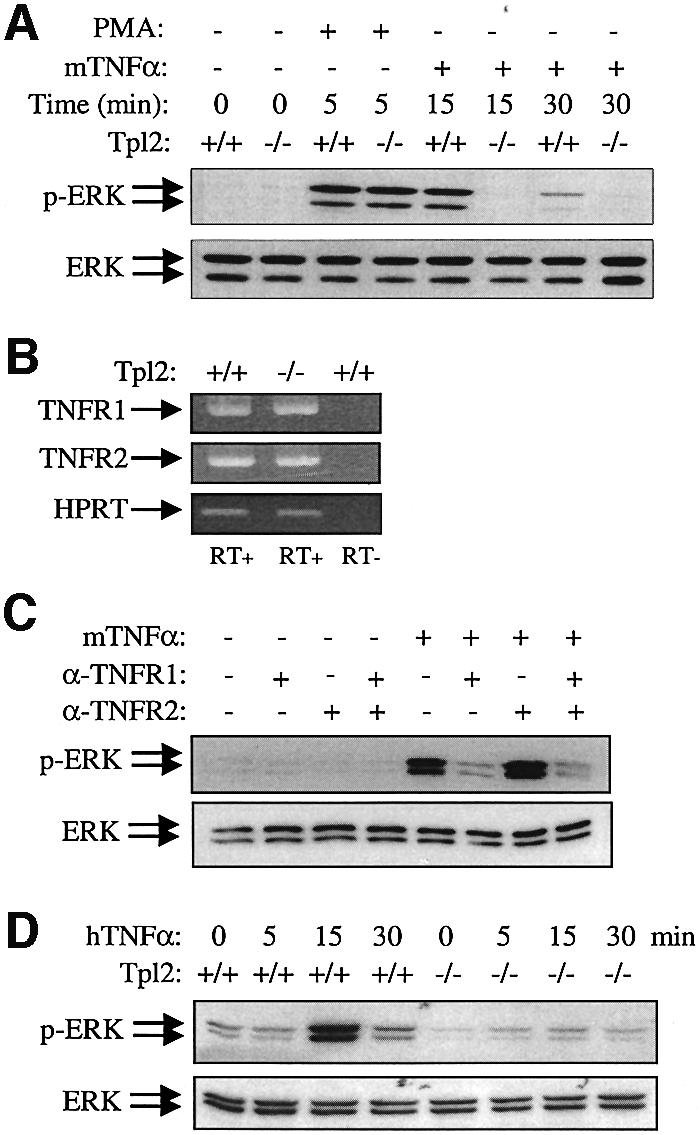
Fig. 4. ERK activation by TNF-α in macrophages depends on signals transduced via Tpl2. (A) Representative western blots of Tpl2+/+ and Tpl2–/– bone marrow-derived macrophage (BMDM) cell lysates, harvested at the indicated time points before and after PMA or murine TNF-α (mTNFα) stimulation, was probed with antibodies to phosphorylated ERK (upper) or total ERK (lower). (B) Tpl2+/+ and Tpl2–/– BMDMs express both TNFR1 and TNFR2, as determined by RT–PCR. Amplification of the housekeeping gene HPRT serves as a cDNA synthesis and amplification control, and the lack of DNA contamination is confirmed by the absence of amplification in the RT– sample. (C) TNFR1 transduces ERK signaling in mTNF-α-treated mouse BMDMs. BMDMs from Tpl2+/+ mice were pretreated for 30 min with 2 µg/ml blocking TNFR1, TNFR2 mAbs or a combination of these reagents and then stimulated for 15 min with 50 ng/ml mTNF-α. Lysates were examined for phosphorylated or total ERK. (D) The effects of human TNF-α (hTNFα), which exclusively stimulates the mTNFR1, on ERK activation in Tpl2+/+ and Tpl2–/– BMDMs.
Tpl2 is required for TRAF6-mediated ERK activation
Following CD40 stimulation, TRAF2, TRAF3 and TRAF6 are directly recruited to the cytoplasmic C-terminus of CD40 and transduce signals that regulate the MAPK pathways and NF-κB (Inoue et al., 2000; Chung et al., 2002). The CD40 domain that binds TRAF6 has been shown to contribute to the induction of Ig germline transcription in vitro (Leo et al., 1999) and the generation of long-lived plasma cells in vivo (Ahonen et al., 2002). TRAF6 has also been proposed to be unique among the TRAF molecules in that it activates the ERK pathway in human embryonic kidney (HEK) 293 and COS-1 cells (Reinhard et al., 1997; Kashiwada et al., 1998).
To assess the role of Tpl2 in TRAF6-induced ERK activation, we used SV40-transformed mouse embryo fibroblasts (MEFs) derived from Tpl2+/+ and Tpl2–/– mice. Transfection of MEFs with a CD40 expression construct, and subsequent stimulation with trimeric soluble CD40L, resulted in the phosphorylation of ERK, an effect that was significantly reduced in the knockout cells (Figure 5A). Therefore, like B cells and macrophages, CD40-stimulated fibroblasts also require Tpl2 for the efficient activation of ERK. We then examined the ability of exogenously expressed FLAG-tagged TRAF6 or TRAF2 to induce phosphorylation of cotransfected HA-ERK1 in Tpl2+/+ and Tpl2–/– MEFs. The results show that the absence of Tpl2 impairs the ability of TRAF6 to transduce ERK activation signals (Figure 5B). Consistent with previous reports in other cell types (Reinhard et al., 1997; Kashiwada et al., 1998), TRAF2 failed to activate this signaling pathway in either Tpl2+/+ or Tpl2–/– cells. The critical role of Tpl2 in TRAF6-transduced ERK activation was confirmed in SV40-transformed keratinocytes derived from Tpl2+/+ and Tpl2–/– mice. Following cotransfection with HA-ERK1 and increasing amounts of TRAF6, cells were lysed, and immunoprecipitated HA-ERK1 was subjected to in vitro kinase assays using GST–Elk1 as the substrate. These experiments consistently showed that TRAF6 promotes ERK activation in Tpl2+/+, but not Tpl2–/–, keratinocytes (Figure 5C). Using luciferase reporter assays, we also showed that, unlike ERK, NF-κB activation was induced by TRAF6 in both Tpl2+/+ and Tpl2–/– MEFs (Figure 5D).

Fig. 5. Tpl2 functions downstream of TRAF6 in the transduction of ERK activation signals. (A) CD40-mediated ERK activation is significantly impaired in mouse embryo fibroblasts (MEFs) isolated from Tpl2–/– mice. Tpl2+/+ and Tpl2–/– cells were transiently transfected with CD40 or control vector in combination with HA-ERK1. The transfected cultures were serum starved and stimulated with recombinant soluble CD40L as indicated. Lysates were examined for expression of HA-ERK1 and phosphorylated ERK1 by immunoblotting. (B) Tpl2 is required for TRAF6-induced ERK activation. MEFs from Tpl2+/+ and Tpl2–/– mice were transiently transfected with HA-ERK1 together with FLAG-tagged expression vectors for TRAF2 or TRAF6. Lysates were examined for expression of FLAG-TRAF, HA-ERK1 and phosphorylated ERK1 by immunoblot. (C) Tpl2 is essential for TRAF6-induced ERK activation in mouse keratinocytes. SV40-transformed keratinocytes from Tpl2+/+ and Tpl2–/– mice were transfected with HA-ERK1 and increasing amounts of TRAF6. ERK activation was determined by anti-HA immune-complex in vitro kinase assay (IVK) using GST–Elk1 as a substrate. Data are representative of four independent experiments. (D) TRAF6-induced NF-κB transactivation, assessed by luciferasse reporter assays, is not impaired in Tpl2–/– MEFs. Relative luciferase value (RLV) is the ratio of luciferase versus β-galactosidase measurements from duplicate determinations. Data are means ± SD from three independent experiments. (E) Endogenous Tpl2 and TRAF6 coimmunoprecipitate with aggregated CD40. Lysates from BJAB lymphoma cells stimulated with CD40L for 10 min or from untreated cultures were immunoprecipitated with the anti-CD40 mAb S2C6 (lanes 1 and 2) and probed with antibodies against TRAF6, Tpl2 or CD40. Lane 3 represents 40 µg of whole BJAB cell lysates (WCL) for Tpl2 and CD40 and 50 µg for TRAF6 detection. (F) CD40 augments the association of coexpressed Tpl2 and TRAF6. 293T cells were cotransfected with FLAG-tagged TRAF6, myc-tagged Tpl2 and/or CD40 as indicated. Cell lysates were immunoprecipitated with a FLAG antibody immobilized on G Sepharose beads. Immunoprecipitates were resolved in 9% SDS/PAGE and probed with either anti-myc (upper) or anti-TRAF6 antibody (lower).
The preceding data demonstrate that Tpl2 functions downstream of TRAF6 to transduce CD40 signals that activate ERK. TRAF6 is known to be recruited to the CD40 signaling complex following CD40 stimulation (Ishida et al., 1996). To determine whether Tp12 is also recruited to the complex, CD40 was immunoprecipitated from lysates of CD40L-stimulated BJAB human lymphoma cells or untreated cultures, and the immunoprecipitates were probed for Tpl2, TRAF6 and CD40. The results show that TRAF6 and Tpl2 coimmunoprecipitate with CD40 following stimulation (Figure 5E). Attempts to coimmunoprecipitate endogenous TRAF6 and Tpl2 from these lysates using a TRAF6 antibody were unsuccessful. This may be the result of weak and transient interactions. Alternatively, the TRAF6 antibody may recognize an epitope critical for the recruitment of Tpl2 or of a protein that binds Tpl2. Therefore, we performed coimmunoprecipitation assays in HEK 293 cells transiently transfected with myc-tagged Tpl2 and FLAG-tagged TRAF6 expression constructs. Lysates from these transfectants were immunoprecipitated with anti-FLAG and analyzed by western blotting for the presence of myc-Tpl2. The results show that Tpl2 coimmunoprecipitates with TRAF6 (Figure 5F). Moreover, this interaction was augmented in the presence of coexpressed CD40 (Figure 5F). We conclude that, following stimulation, CD40 recruits both Tpl2 and TRAF6 and that the assembled CD40/ TRAF6/Tp12 complex is required for ERK activation.
Cellular proliferation and expression of activation markers in Tpl2+/+ and Tpl2–/– B cells following CD40 stimulation
CD40L expressed by Th2 and Th1 cells contributes to B-cell and macrophage activation, respectively (van Kooten and Banchereau, 2000). The effects of CD40 stimulation in both B cells and macrophages are complex. Dissecting these effects will be facilitated by mutations that selectively block some, but not all, of the signaling pathways that are activated by CD40. The mutation of Tpl2 selectively blocks the activation of ERK in B cells and therefore provides us with a unique opportunity to examine the biological output of this pathway following CD40 stimulation.
CD40 ligation promotes the expression of several B-cell activation markers and induces B-cell proliferation. In an effort to determine whether the failure of CD40-generated signals to activate ERK in Tpl2–/– B cells affects these processes, we stimulated Tpl2+/+ and Tpl2–/– B cells with anti-CD40 mAb, and 48 h later we examined the expression of the activation markers CD54 (ICAM1), CD69, CD44 and CD95 (Fas) by flow cytometry. Cellular proliferation was also measured at the 48 h time point by [3H]thymidine incorporation. The results reveal no significant differences between the Tpl2+/+ and Tpl2–/– B cells (Figure 6), suggesting that none of these CD40-mediated effects is ERK dependent. CD40-induced proliferation measured at a later time point (72 h) was also unaffected (data not shown).

Fig. 6. Cellular proliferation and expression of activation markers in Tpl2+/+ and Tpl2–/– B cells following CD40 stimulation. (A) Proliferation in unstimulated (NT) and anti-CD40-stimulated splenic B cells from three wild-type (WT) and three Tpl2–/– (KO) mice was assessed by [3H]thymidine incorporation. Mean values (±SD) of triplicate determinations from a representative experiment are shown. Four independent experiments were performed and yielded similar results. (B) Induction of CD54, CD69, CD44 and CD95 in anti-CD40-stimulated Tpl2+/+ and Tpl2–/– B cells was examined by flow cytometry. Mean fluorescence intensity (mfi, mean values ± SD from three independent experiments) is shown.
Tpl2 regulates IgE production in B cells stimulated with anti-CD40 and IL-4
CD40L, in combination with cytokines produced by helper T cells, promotes B-cell activation (van Kooten and Banchereau, 2000). Ig genes expressed in activated B cells undergo rearrangements that are responsible for switching the Ig isotypes produced by these cells. Isotype switching depends on dual signals triggered by CD40L and other cytokines produced by the helper T cells (Purkerson and Isakson, 1992). IL-4, for example, which is the primary cytokine produced by Th2 cells, promotes switching to IgE and IgG1 (Jabara et al., 1990; Armitage et al., 1993; Bacharier and Geha, 2000).
To determine whether CD40 signals that activate ERK via Tpl2 have a role in IgE and IgG1 production, we analyzed the Ig isotypes produced by Tpl2+/+ and Tpl2–/– B cells following stimulation with anti-CD40, IL-4 or the combination of anti-CD40 and IL-4. The results showed that the combination of anti-CD40 and IL-4 induced the secretion of Igs and that the Tpl2–/– B cells were consistently partially defective in their ability to switch to the production of IgE in response to these stimuli in vitro (Figure 7A). The defect in IgE production was observed not only in purified splenic B cells from naïve mice but also in B cells from ovalbumin-immunized Tpl2–/– mice (see Supplementary figure 1A available at The EMBO Journal Online). However, IgG1 production was unaffected (see Supplementary figure 1B). Tpl2+/+ B cells treated with the MEK inhibitors PD98059 or UO126 were also defective in IgE secretion in response to CD40 and IL-4 stimulation (Figure 7B; data not shown), suggesting that switching to the production of IgE is partly regulated by the ERK pathway.

Fig. 7. B cells from Tpl2–/– mice are partially impaired in IgE production following CD40 and IL-4 stimulation. (A) Secreted IgE levels (means ± SD from seven independent experimental B-cell cultures) are reduced in Tpl2–/– B cells stimulated with anti-CD40 plus IL-4. This difference is statistically significant (p = 0.016). (B) Secreted IgE levels (means ± SD from four independent experimental B-cell cultures) are reduced in Tpl2+/+ B cells treated with IL-4 and anti-CD40 in the presence of the MEK inhibitor UO126. The levels of IgE from vehicle control (DMSO)-treated cultures are also shown. (C) CD40 does not affect the levels of STAT6 phosphorylation induced by IL-4. B cells from Tpl2+/+ and Tpl2–/– mice were stimulated with anti-CD40, IL-4 or anti-CD40 plus IL-4 for 15 min, and cell lysates were analyzed for the phosphorylation status of STAT6 by immunoblot analysis (upper). Equal loading was confirmed by probing the same blot for β-tubulin (lower). (D) IL-4 does not modify the MAPKs and NF-κB activation by CD40. B cells from Tpl2+/+ and Tpl2–/– mice were stimulated with anti-CD40, IL-4 or anti-CD40 plus IL-4 for 15 min, and cell lysates were analyzed for MAPK activation using antibodies that recognize either the phosphorylated or total forms of ERK, JNK and p38. IκBα degradation was assessed by immunoblot using an anti-IκBα polyclonal antibody. The experiments in (C) and (B) were carried out on the same cell lysates. This confirmed that both the anti-CD40 antibody and the IL-4 were functionally active when used alone.
These data raised the question of the relative contribution of IL-4 to CD40 signals. As IL-4-transduced signals leading to STAT6 activation have been shown to be critical for Ig class switching (Shimoda et al., 1996; Takeda et al., 1996), we examined the possibility that Tpl2–/– B cells may be defective in STAT6 phosphorylation. For this purpose, Tpl2+/+ and Tpl2–/– B cells were treated for 15 min with IL-4, anti-CD40 or both. Lysates of unstimulated and stimulated B cells were analyzed for STAT6 activation, using an antibody that detects the activated form of STAT6, which is phosphorylated on Tyr641. The results show that STAT6 phosphorylation is not impaired in IL-4-treated Tpl2–/– B cells. Therefore, CD40 stimulation does not affect the ability of IL-4 to transduce STAT6 activation signals (Figure 7C). To evaluate the possibility that IL-4 modulates CD40-induced signals, we examined whether Tpl2+/+ and Tpl2–/– B cells differ in their response to IL-4, anti-CD40 or IL-4 plus anti-CD40. To this end, the cell lysates used for the experiments shown in Figure 7B were analyzed for ERK, p38 and JNK phosphorylation. The results show that IL-4 does not modulate the ability of CD40 to engage the MAPK pathways (Figure 7D). Furthermore, IL-4 does not influence the ability of CD40 to transduce NF-κB signals, as determined by the effects of CD40 stimulation on the degradation of IκBα, a prerequisite for NF-κB activation (Figure 7D).
Discussion
Results presented in this report demonstrate that Tpl2 is required for ERK activation by CD40 and TNFR engagement in B cells and macrophages. Moreover, they demonstrate that Tpl2 is recruited to a CD40/TRAF6 signaling complex in response to CD40 ligation and that it plays an obligatory role in TRAF6-induced ERK activation. The failure of CD40 to activate ERK in B cells from Tpl2–/– mice allowed us to demonstrate that the Tpl2/ERK pathway influences IgE secretion by B cells stimulated with anti-CD40 and IL-4.
CD40 ligation activates all the MAPK pathways in macrophages and B cells. The data presented here demonstrate that Tpl2 is selectively required for the transduction of CD40 and TNF signals that activate the ERK MAPK pathway. Thus, the activation of ERK was abolished in CD40-stimulated Tpl2–/– B cells. Intriguingly, macrophages and CD40-transfected MEFs isolated from Tpl2–/– mice retain some ability to activate ERK in response to CD40 ligation, pointing to the complementary role of Tpl2 and a kinase other than Tpl2 in the regulation of CD40-mediated ERK signaling in these cell types. Earlier studies had shown that Tpl2 is also essential for LPS-induced ERK activation in macrophages (Dumitru et al., 2000). Taken together, the above findings suggest that the pathways of ERK activation by members of the TNF-α cytokine family and LPS converge and that Tpl2 functions downstream from the point of convergence of these pathways.
A critical role for Tpl2 in the activation of NF-κB in CD40-stimulated B cells has been excluded on the basis of several observations. Thus, neither NF-κB binding activity nor the induction of NF-κB-dependent cell surface markers such as CD54 or CD95 were affected in Tpl2–/– cells. Moreover, the suppression of CD40-mediated NF-κB activation has been reported to result in marked defects in B-cell proliferation and in the humoral response to both T cell-dependent and -independent antigens (Horwitz et al., 1999; Ren et al., 2002) but B-cell proliferation is not affected by the absence of Tpl2 (Figure 6). Extensive genetic and biochemical evidence suggests a key role for NF-κB in lymphocyte growth and development, as demonstrated by the severe phenotype and short lifespan of mice deficient in key components of the NF-κB pathway, such as p65, IKKγ, IKKα/β and T2K/TBK1/NAK. Unlike these animals, Tpl2–/– mice are viable and have none of the phenotypic defects that denote impairment in NF-κB transcriptional activity (Dumitru et al., 2000).
Activation signals induced by CD40L, TNF-α and LPS promote the recruitment of TRAF proteins to the cytoplasmic tails of their cognate receptors and the formation of a TRAF-dependent multiprotein signaling complex. A number of MAPK kinase kinases, such as NIK, MEKK1, MEKK3 and Ask1, are recruited to this complex promoting signal activation by mechanisms that are not fully understood. CD40-induced ERK MAPK activation has been shown to depend on TRAF6, inasmuch as a dominant-negative TRAF6 mutant suppresses CD40-mediated ERK signals. Furthermore, ectopic expression of wild-type TRAF6, but not other TRAFs, promotes ERK activation in the absence of CD40 engagement (Reinhard et al., 1997; Kashiwada et al., 1998). However, an alternative, TRAF6-independent pathway that is engaged by CD40 ligation and results in ERK activation may also exist, as a CD40 mutant lacking the TRAF6 but containing the TRAF2 binding site retains some ability to activate ERK (Kashiwada et al., 1998). This pathway may involve the ligand-dependent membrane raft-restricted recruitment of TRAF2 or TRAF3 to the cytoplasmic tail of CD40 and/or of src family kinase members within this intracellular compartment to initiate ERK signaling (Vidalain et al., 2000; Basaki et al., 2002). Biochemical evidence also suggests that TRAF2 directly interacts with TRAF6 (Cao et al., 1996), and the possibility that TRAF6 contributes to CD40 and TNFR1 signaling downstream of TRAF2 cannot be excluded. TRAF6 is also a critical component of LPS-induced signaling (Lomaga et al., 1999). In the present report, we have provided evidence that Tpl2 is recruited to a TRAF6-containing signaling complex and that it plays a critical role in TRAF6-mediated ERK but not NF-κB activation. Thus, endogenous Tpl2 and TRAF6 are detected in anti-CD40 immunoprecipitates from CD40L-stimulated BJAB lymphoma cells and the two proteins coimmunoprecipitate when transiently coexpressed in HEK 293 cells. Moreover, whereas transiently transfected TRAF6 activated ERK in Tpl2+/+ MEFs and keratinocytes, it failed to do so in Tpl2–/– cells. Although these results do not demonstrate conclusively that Tpl2 interacts directly with TRAF6, they do show that Tpl2 is recruited to the TRAF6 signalosome and through this interaction may control CD40-mediated ERK signaling. However, the TRAF6/Tpl2 complex is either not involved in NF-κB activation or is only one of many complexes that activate NF-κB in response to CD40 stimulation. Consistent with the latter possibility, we have shown previously that overexpression of a catalytically inactive Tpl2 inhibits CD40-induced NF-κB activation in HEK 293 cells (Eliopoulos et al., 2002b).
CD40 and its ligand play a critical role in the regulation of both the innate and adaptive immunity. Humans and mice carrying mutations in the gene encoding CD40L develop a characteristic immunodeficiency syndrome (Allen et al., 1993; Aruffo et al., 1993; DiSanto et al., 1993; Korthauer et al., 1993; Kawabe et al., 1994; Xu et al., 1994). This syndrome, which in humans is called X-linked hyper-IgM syndrome, is characterized by an increased susceptibility to pathogens that is caused by impaired macrophage activation by type 1 helper T cells (Grewal et al., 1995). Humans and mice with mutations in CD40L also exhibit a severe defect in humoral immunity. Th2 cells from these individuals are defective in their ability to provide help for the response of B cells to thymus-dependent protein antigens. CD40-dependent T-cell help controls both the level of the antibodies synthesized by B cells and Ig isotype switching. As a result, patients with hyper-IgM syndrome produce only low levels of IgM and IgD antibodies in response to foreign antigens. However, the overall IgM levels in these individuals are high, perhaps because they respond to thymus-independent antigens that are produced by pathogens that chronically infect them. CD40 stimulation of B cells has also been linked to the induction of several B-cell activation markers and to B-cell proliferation. Proliferation of B cells following immunization with thymus-dependent antigens gives rise to germinal centers that arise in the B-cell areas of the draining lymph nodes. As lack of functional CD40L compromises the proliferation and activation of B cells, lymphoid tissues of patients with hyper-IgM syndrome and of CD40L knockout mice are devoid of germinal centers (Allen et al., 1993). The profound inability of individuals with a defect in CD40L to produce IgE antibodies requires special mention, because it appears to be controlled in part by a parallel autostimulatory loop. IgE produced by activated B cells binds FcεRII receptors on the surface of mast cells, basophils and eosinophils and induces the expression of CD40L and IL-4. In turn, mast cells, basophils and eosinophils expressing these molecules further stimulate IgE secretion by B cells (Gauchat et al., 1993).
Understanding the biological importance of individual signaling pathways activated by CD40 stimulation will depend on the analysis of mutations that selectively inhibit some but not all of these pathways. The inability of Tpl2–/– B cells to activate ERK in response to CD40 stimulation provides the opportunity to assess the contribution of ERK to key CD40 functions. We have found that the deficiency in ERK signaling observed in CD40-stimulated B cells from Tpl2–/– mice affects IgE production. IgE synthesis requires synergistic signals induced by IL-4, a Th2 cytokine, and CD40 (Bacharier and Geha, 2000). IL-4-induced STAT6 activation is essential but not sufficient for germline Cε Ig gene transcription, a prerequisite for antibody switching to IgE (Shimoda et al., 1996; Takeda et al., 1996). Additional signals are provided by CD40 and may include NF-κB, which has been proposed to cooperate with STAT6 in regulating IgE gene transcription (Iciek et al., 1997; Linehan et al., 1998). However, NF-κB activation by CD40 is intact in Tpl2–/– B cells, and IL-4 does not influence this response (Figures 2 and 7). The defects in ERK activation and IgE synthesis observed in IL-4 and anti-CD40-stimulated Tpl2–/– B cells, and the effects of the MEK inhibitor UO126 on IgE synthesis in wild-type cells, suggest that IgE production is also regulated by Tp12-dependent ERK activation signals. These data are consistent with the results of a recent study addressing the role of CD40-activated TRAF6 in Ig isotype switching and IgE production. It showed that, whereas a transgene encoding a mutant CD40 that lacks binding sites for TRAF2 and TRAF3 partially corrects the isotype switch defect of CD40–/– mice, a transgene encoding a mutant CD40 that lacks binding sites for TRAF2, TRAF3 and TRAF6 is totally inactive (Jabara et al., 2002). The precise mechanism by which ERK regulates Ig production will be a subject of future studies.
Recent work from this laboratory revealed that ovalbumin-immunized Tpl2–/– mice express higher than normal levels of IgE antibodies in vivo, an effect reproduced in culture by exposure of total splenocytes from immunized mice to ovalbumin (C.C.Wang, A.G.Eliopoulos, C.D.Dumitru, C.Daskalakis and P.N.Tsichlis, submitted). The above findings do not contradict the data of the present report that address the response of CD40-stimulated purified Tpl2+/+ and Tpl2–/– splenic B cells to the same concentration of recombinant IL-4. We have found that the exaggerated production of IgE antibodies in ovalbumin-immunized Tpl2–/– mice reflects the elevated levels of IL-4 produced by the T cells of these animals, which is caused by the Th2 polarization of the immune response to ovalbumin. Given that the amount of IgE secreted by anti-CD40 and IL-4-stimulated B cells critically depends on the concentration of IL-4, the Th2 polarization of the immune response caused by the ablation of Tpl2 provides a strong enough signal to compensate for the B-cell defect in Tpl2–/– B cells. These findings demonstrate how the crosstalk between different cell lineages in the intact animal may alter the effects of a genetic mutation in a single lineage. Although the genetic ablation of Tpl2 in B cells downregulates IgE production in vitro, the inactivation of Tpl2 throughout the immune system leads to the overexpression of cytokines and costimulatory molecules that promote the IgE switch. As a result, even though IgE synthesis is downregulated in Tpl2–/– B cells, the overproduction by other cells of molecules that favor the production of IgE compensates for the B-cell defect. We should add here that ovalbumin immunization did not induce a permanent B-cell change that favors IgE synthesis, because purified B cells from both naïve and ovalbumin-immunized Tpl2–/– mice were equally defective in their ability to synthesize IgE following stimulation with anti-CD40 and IL-4.
In summary, the data presented here demonstrate a critical role for Tpl2 in CD40 and TNFR-transduced signals that result in ERK activation and Ig class switching. CD40 recruits both Tpl2 and TRAF6 in a complex that plays a critical role in the transduction of signals that activate ERK. The ability of Tpl2 to control ERK activation downstream of TRAF6 raises the possibility that Tpl2 may have a broader role in MAPK regulation by affecting signaling from a plethora of receptors and viral proteins that signal through TRAF6.
Materials and methods
Animals and animal cell isolation
Tpl2–/– mice and bone marrow-derived macrophage isolation and culture have been described previously (Dumitru et al., 2000). B cells were isolated from mouse spleens by negative magnetic separation. Total splenocytes were resuspended in ACK hypotonic buffer to remove red blood cells and washed in RPMI supplemented with 10% FBS. Cells were then incubated with avidin-coated magnetic beads (Immunotech, Paris, France) carrying biotin-labeled anti-CD4 and anti-CD8 antibodies (Pharmingen, San Diego, CA). and T lymphocytes were magnetically removed. Macrophages were then left to adhere to Petri dishes, and the resulting non-adherent cells were collected and analyzed for expression of the B-cell marker B220 by flow cytometry or cultured for subsequent experiments. Cell suspensions routinely consisted of >85% B220+ cells. Keratinocytes were cultured from the tongues of sacrificed mice. Tongues were cut sideways and placed in dispase (Roche) O/N. The following day, the epithelial cells were scraped off, washed three times with PBS supplemented with 1% penicillin/streptomycin and incubated in trypsin for 1 h. Cells were then collected, resuspended in keratinocyte media (Invitrogen) without EGF and plated out on plates with irradiated NIH 3T3 fibroblasts as feeders. Confluent keratinocyte cultures were infected with a lentivirus expressing a temperature-sensitive SV40 T antigen to obtain immortalized cells.
Supplementary data
Supplementary Materials and methods and other Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
This work was supported by the NIH grant RO1 CA38047 to P.N.T. and by the Cancer Research UK project grant SP2584 to A.G.E.; A.G.E. is supported by a Medical Research Council (UK) Career Development Award, C.-C.W. was supported by an NIH training grant (5-T32-CA09662) and C.D.D. is a special fellow of the Leukemia and Lymphoma Society, USA. Part of this work was carried out at the Kimmel Cancer Center, Thomas Jefferson University, prior to the relocation of the P.N.T. laboratory to Tufts-NEMC.
References
- Afford S.C., Randhawa,S., Eliopoulos,A.G., Hubscher,S.G., Young,L.S. and Adams,D.H. (1999) CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface Fas ligand expression and amplifies Fas-mediated hepatocyte death during allograft rejection. J. Exp. Med., 189, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahonen C., Manning,E., Erickson,L.D., O’Connor,B., Lind,E.F., Pullen,S.S., Kehry,M.R. and Noelle,R.J. (2002) The CD40–TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol., 3, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R.C. et al. (1993) CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science, 259, 990–993. [DOI] [PubMed] [Google Scholar]
- Armitage R.J., Macduff,B.M., Spriggs,M.K. and Fanslow,W.C. (1993) Human B cell proliferation and Ig secretion induced by recombinant CD40 ligand are modulated by soluble cytokines. J. Immunol., 150, 3671–3680. [PubMed] [Google Scholar]
- Aruffo A. et al. (1993) The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell, 72, 291–300. [DOI] [PubMed] [Google Scholar]
- Bacharier L.B. and Geha,R.S. (2000) Molecular mechanisms of IgE regulation. J. Allergy Clin. Immunol., 105, S547–S558. [DOI] [PubMed] [Google Scholar]
- Basaki Y., Ikizawa,K., Kajiwara,K. and Yanagihara,Y. (2002) CD40-mediated tumor necrosis factor receptor-associated factor 3 signaling upregulates IL-4-induced germline Cepsilon transcription in a human B cell line. Arch. Biochem. Biophys., 405, 199–204. [DOI] [PubMed] [Google Scholar]
- Cao Z., Xiong,J., Takeuchi,M., Kurama,T. and Goeddel,D.V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature, 383, 443–446. [DOI] [PubMed] [Google Scholar]
- Ceci J.D., Patriotis,C.P., Tsatsanis,C., Makris,A.M., Kovatch,R., Swing,D.A., Jenkins,N.A., Tsichlis,P.N. and Copeland,N.G. (1997) Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev., 11, 688–700. [DOI] [PubMed] [Google Scholar]
- Challa A. et al. (2002) Population depletion activates autonomous CD154-dependent survival in biopsylike Burkitt lymphoma cells. Blood, 99, 3411–3418. [DOI] [PubMed] [Google Scholar]
- Chung J.Y., Park,Y.C., Ye,H. and Wu,H. (2002) All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci., 115, 679–688. [DOI] [PubMed] [Google Scholar]
- DiSanto J.P., Bonnefoy,J.Y., Gauchat,J.F., Fischer,A. and de Saint Basile,G. (1993) CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature, 361, 541–543. [DOI] [PubMed] [Google Scholar]
- Dumitru C.D. et al. (2000) TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell, 103, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Eliopoulos A.G. et al. (1996) CD40-induced growth inhibition in epithelial cells is mimicked by Epstein–Barr virus-encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene, 13, 2243–2254. [PubMed] [Google Scholar]
- Eliopoulos A.G., Davies,C., Knox,P.G., Gallagher,N.J., Afford,S.C., Adams,D.H. and Young,L.S. (2000) CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol. Cell. Biol., 20, 5503–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos A.G., Dumitru,C.D., Wang,C.-C., Cho,J. and Tsichlis,P.N. (2002a) Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J., 21, 4831–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos A.G., Davies,C., Blake,S.S., Murray,P., Najafipour,S., Tsichlis,P.N. and Young,L.S. (2002b) The oncogenic protein kinase Tpl-2/Cot contributes to Epstein–Barr virus-encoded latent infection membrane protein 1-induced NF-κB signaling downstream of TRAF2. J. Virol., 76, 4567–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny K.M., Peli,J., Lambert,J.F., Muller,V. and Diggelmann,H. (1996) Involvement of the Tpl-2/cot oncogene in MMTV tumorigenesis. Oncogene, 13, 2015–2020. [PubMed] [Google Scholar]
- Funakoshi S. et al. (1994) Inhibition of human B-cell lymphoma growth by CD40 stimulation. Blood, 83, 2787–2794. [PubMed] [Google Scholar]
- Gauchat J.F. et al. (1993) Induction of human IgE synthesis in B cells by mast cells and basophils. Nature, 365, 340–343. [DOI] [PubMed] [Google Scholar]
- Grell M. et al. (1999) Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J., 18, 3034–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal I.S., Xu,J. and Flavell,R.A. (1995) Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature, 378, 617–620. [DOI] [PubMed] [Google Scholar]
- Horwitz B.H., Zelazowski,P., Shen,Y., Wolcott,K.M., Scott,M.L., Baltimore,D. and Snapper,C.M. (1999) The p65 subunit of NF-κB is redundant with p50 during B cell proliferative responses and is required for germline CH transcription and class switching to IgG3. J. Immunol., 162, 1941–1946. [PubMed] [Google Scholar]
- Iciek L.A., Delphin,S.A. and Stavnezer,J. (1997) CD40 cross-linking induces Ig epsilon germline transcripts in B cells via activation of NF-κB: synergy with IL-4 induction. J. Immunol., 158, 4769–4779. [PubMed] [Google Scholar]
- Inoue J., Ishida,T., Tsukamoto,N., Kobayashi,N., Naito,A., Azuma,S. and Yamamoto,T. (2000) Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp. Cell Res., 254, 14–24. [DOI] [PubMed] [Google Scholar]
- Ishida T. et al. (1996) Identification of TRAF6, a novel tumour necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J. Biol. Chem., 271, 28745–28748. [DOI] [PubMed] [Google Scholar]
- Jabara H.H., Fu,S.M., Geha,R.S. and Vercelli,D. (1990) CD40 and IgE: synergism between anti-CD40 monoclonal antibody and interleukin 4 in the induction of IgE synthesis by highly purified human B cells. J. Exp. Med., 172, 1861–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara H. et al. (2002) The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity, 17, 265–276. [DOI] [PubMed] [Google Scholar]
- Jupp O.J., McFarlane,S.M., Anderson,H.M., Littlejohn,A.F., Mohamed,A.A., MacKay,R.H., Vandenabeele,P. and MacEwan,D.J. (2001) Type II tumour necrosis factor-α receptor (TNFR2) activates c-Jun N-terminal kinase (JNK) but not mitogen-activated protein kinase (MAPK) or p38 MAPK pathways. Biochem. J., 359, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwada M., Shirakata,Y., Inoue,J.-I., Nakano,H., Okazaki,K., Okumura,K., Yamamoto,T., Nagaoka,H. and Takemori,T. (1998) Tumor necrosis factor receptor-associated factor 6 (TRAF6) stimulates extracellular signal-regulated kinase (ERK) activity in CD40 signalling along a Ras-independent pathway. J. Exp. Med., 187, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T., Naka,T., Yoshida,K., Tanaka,T., Fujiwara,H., Suematsu,S., Yoshida,N., Kishimoto,T. and Kikutani,H. (1994) The immune responses in CD40 deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity, 1, 167–178. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D. et al. (2002) Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J. Exp. Med., 196, 1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthauer U. et al. (1993) Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature, 361, 539–541. [DOI] [PubMed] [Google Scholar]
- Leo E., Zapata,J.M. and Reed,J.C. (1999) CD40-mediated activation of Ig-Cγ1- and Ig-cε germ-line promoters involves multiple TRAF family proteins. Eur. J. Immunol., 29, 3908–3913. [DOI] [PubMed] [Google Scholar]
- Lin X., Cunningham,E.T., Mu,Y., Geleziunas,R. and Greene,W.C. (1999) The proto-oncogene Cot kinase participates in CD3/CD28 induction of NF-κB acting through the NF-κB-inducing kinase and IκB kinases. Immunity, 10, 271–280. [DOI] [PubMed] [Google Scholar]
- Linehan L.A., Warren,W.D., Thompson,P.A., Grusby,M.J. and Berton,M.T. (1998) STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J. Immunol., 161, 302–310. [PubMed] [Google Scholar]
- Lomaga M.A. et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40 and LPS signaling. Genes Dev., 13, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriotis C., Makris,A., Bear,S.E. and Tsichlis,P.N. (1993) Tumor progression locus 2 (Tpl-2) encodes a protein kinase involved in the progression of rodent T-cell lymphomas and in T-cell activation. Proc. Natl Acad. Sci. USA, 90, 2251–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriotis C., Makris,A., Chernoff,J. and Tsichlis,P.N. (1994) Tpl-2 acts in concert with Ras and Raf-1 to activate mitogen-activated protein kinase. Proc. Natl Acad. Sci. USA, 91, 9755–9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkerson J. and Isakson,P. (1992) A two-signal model for regulation of immunoglobulin isotype switching. FASEB J., 6, 3245–3252. [DOI] [PubMed] [Google Scholar]
- Reinhard C., Shamoon,B., Shyamala,V. and Williams,L.T. (1997) Tumour necrosis factor-α-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J., 16, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H., Schmalstieg,A., Yuan,D. and Gaynor,R.B. (2002) I-κB kinase β is critical for B cell proliferation and antibody response. J. Immunol., 168, 577–587. [DOI] [PubMed] [Google Scholar]
- Salmeron A., Ahmad,T.B., Carlile,G.W., Pappin,D., Narsimhan,R.P. and Ley,S.C. (1996) Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J., 15, 817–826. [PMC free article] [PubMed] [Google Scholar]
- Shimoda K. et al. (1996) Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature, 380, 630–633. [DOI] [PubMed] [Google Scholar]
- Takeda K. et al. (1996) Essential role of Stat6 in IL-4 signalling. Nature, 380, 627–630. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C., Patriotis,C., Bear,S.E. and Tsichlis,P.N. (1998a) The Tpl-2 protooncoprotein activates the nuclear factor of activated T cells and induces interleukin 2 expression in T cells. Proc. Natl Acad. Sci. USA, 95, 3827–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsanis C., Patriotis,C. and Tsichlis,P.N. (1998b) Tpl-2 induces IL-2 expression in T cell lines by triggering multiple signaling pathways that activate NFAT and NF-κB. Oncogene, 17, 2609–2618. [DOI] [PubMed] [Google Scholar]
- Tsukamoto N., Kobayashi,N., Azuma,S., Yamamoto,T. and Inoue,J.-I. (1999) Two differently regulated nuclear factor κB activation pathways triggered by the cytoplasmic tail of CD40. Proc. Natl Acad. Sci. USA, 96, 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kooten C. and Banchereau,J. (2000) CD40–CD40 ligand. J. Leukoc. Biol., 67, 2–17. [DOI] [PubMed] [Google Scholar]
- Vidalain P.O., Azocar,O., Servet-Delprat,C., Rabourdin-Combe,C., Gerlier,D. and Manie,S. (2000) CD40 signaling in human dendritic cells is initiated within membrane rafts. EMBO J., 19, 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterfield M.R., Zhang,M., Norman,L.P. and Sun,S.C. (2003) NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell, 11, 685–694. [DOI] [PubMed] [Google Scholar]
- Xu J., Foy,T.M., Laman,J.D., Elliot,E.A., Dun,J.J., Waldscmidt,T.J., Elsemore,J., Noelle,R.J. and Flavell,R.A. (1994) Mice deficient for the CD40 ligand. Immunity, 1, 423–431. [DOI] [PubMed] [Google Scholar]