

Abstract
Background
Amyotrophic lateral sclerosis (ALS) is an age-dependent neurodegenerative disease that causes motor neuron degeneration, paralysis and death. Mutations in Cu, Zn superoxide dismutase (SOD1) are one cause for the familial form of this disease. Transgenic mice expressing mutant SOD1 develop age-dependent motor neuron degeneration, skeletal muscle weakness, paralysis and death similar to humans. The mechanism whereby mutant SOD1 induces motor neuron degeneration is not understood but widespread mitochondrial vacuolation has been observed during early phases of motor neuron degeneration. How this vacuolation develops is not clear, but could involve autophagic vacuolation, mitochondrial permeability transition (MPT) or uncharacterized mechanisms. To determine which of these possibilities are true, we examined the vacuolar patterns in detail in transgenic mice expressing mutant SOD1G93A.
Results
Vacuolar patterns revealed by electron microscopy (EM) suggest that vacuoles originate from the expansion of the mitochondrial intermembrane space and extension of the outer mitochondrial membrane. Immunofluorescence microscopy and immuno-gold electron microscopy reveal that vacuoles are bounded by SOD1 and mitochondrial outer membrane markers, but the inner mitochondrial membrane marker is located in focal areas inside the vacuoles. Small vacuoles contain cytochrome c while large vacuoles are porous and lack cytochrome c. Vacuoles lack lysosomal signal but contain abundant peroxisomes and SOD1 aggregates.
Conclusion
These findings demonstrate that mutant SOD1, possibly by toxicity associated with its aggregation, causes mitochondrial degeneration by inducing extension and leakage of the outer mitochondrial membrane, and expansion of the intermembrane space. This could release the pro-cell death molecules normally residing in the intermembrane space and initiate motor neuron degeneration. This Mitochondrial Vacuolation by Intermembrane Space Expansion (MVISE) fits neither MPT nor autophagic vacuolation mechanisms, and thus, is a previously uncharacterized mechanism of mitochondrial degeneration in mammalian CNS.
Background
Amyotrophic lateral sclerosis (ALS) is an age-dependent neurodegenerative disease that causes progressive motoneuron degeneration, skeletal muscle atrophy, paralysis and death [1-3]. Although clinically indistinguishable, the majority of cases are sporadic (SALS) and approximately 10 % are due to inherited causes (familial ALS or FALS). Of the FALS cases about 20% are due to mutations in SOD1 [4]. Rodents transgenic for mutant human SOD1 develop progressive skeletal muscle atrophy, paralysis and death similar to human cases [5-8]. The disease causing property of mutant SOD1 is independent of the normal SOD1 activity, and thus, mutant SOD1 kills motor neurons by gaining a toxic property (reviewed in [9,10]).
Pathological examination of transgenic mice revealed early changes that are not prominent in human spinal cord autopsies at the terminal disease stage. These changes include astrogliosis, fragmentation of Golgi apparatus, SOD1 aggregation and vacuolar degeneration [7,11-14]. Vacuolar degeneration is most prominent at the onset of the disease and precedes motoneuron death by two to three months [15]. Although the high expresser line of SOD1G93A showed vacuoles derived from both endoplasmic reticulum and mitochondria [13], the lower expresser line of SOD1G93A, which more closely mimics the human SOD1 levels [16], showed vacuoles that are nearly all developed from degenerating mitochondria [15,17]. Mitochondrial vacuolation was also reported in another transgenic line SOD1G37R [7].
The early onset of mitochondrial degeneration suggests that mutant SOD1 damages mitochondria and this damage plays a causative role in motoneuron degeneration. Increasing evidence supports this proposal. Mutant SOD1 causes dysfunction and structural damage of mitochondria in cultured neuronal cells [18-20] and at early disease stages in mutant SOD1 transgenic mice [21,22]. Human ALS also shows mitochondrial damage, dysfunction and loss [23,24]. The mechanism whereby mutant SOD1 causes mitochondrial damage has not been determined, but recent evidence shows that mutant SOD1 is imported into mitochondria [21,25,26], and this mitochondrial localization may cause direct damage to mitochondria and induce cell death [27]. Consistent with this possibility, previous work suggested that mitochondrial vacuolation was developed from expansion of the intermembrane space [17]. However, the possibility that the vacuoles represent autophagic vacuolation has not been ruled out, because the source of the vacuolar membrane was not known.
In the current work we sought to determine whether the vacuoles are developed from autophagy, MPT or another mechanism. We examined vacuoles using transmission electron microscopy (TEM), immunogold electron microscopy and immunoflouresence microscopy. We present new evidence that, together with the published literature [17], unequivocally demonstrates that vacuoles are developed by expansion of mitochondrial intermembrane space and extension of the outer membrane. This is neither autophagy nor MPT, but a new mitochondrial degeneration mechanism in the CNS.
Results
Patterns of mitochondrial abnormalities revealed by transmission electron microscopy (TEM) suggest a model of vacuole formation by extension of mitochondrial outer membrane and expansion of the intermembrane space (Fig. 1). Vacuoles might be initiated at focal weakening of inner and outer mitochondrial membrane contact, resulting in small protrusions from the outer membrane (Fig. 1A, arrow). This protrusion might then expand (Fig. 1B,1C, arrows), leading to further detachment of the outer membrane from the inner membrane (Fig. 1D,1E, unfilled arrowheads). Eventually the mature vacuoles form (Fig. 1F,1G,1H,1I), which contain inner membrane remnants (Fig. 1F,1G,1I, large arrows) and vesicular structures (Fig. 1H,1I, unfilled arrows). Often noticeable is the discontinuity in the vacuolar membrane in large vacuoles (Fig. 1D,1I, small arrows), suggesting that as the vacuoles expand the vacuolar membrane becomes porous.
Figure 1.
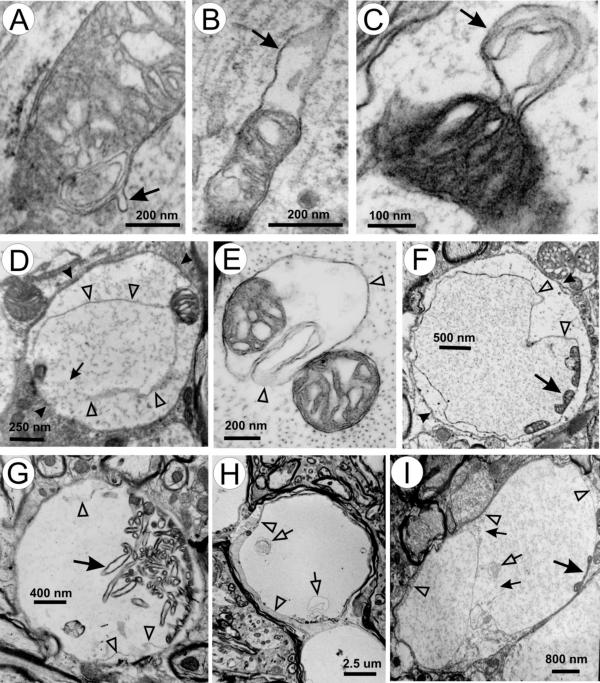
Patterns of mitochondrial vacuoles. (A) a small outer membrane bud (arrow); (B-D) enlargement of the buds (arrows) and expansion of the intermembrane space; (E) further separation of outer and inner membrane and intermembrane space expansion; (F-I) patterns of mature vacuoles. Unfilled arrowheads in D-I mark the vacuolar membrane. Large arrows in F-I indicate remnants of inner membrane. Small arrows in D and I indicate broken areas of vacuolar membrane. Small unfilled arrows in H and I point to vesicular membranous elements within the vacuoles. Arrowheads in D and F indicate dendritic membranes.
To test this model, we immunologically located the inner and outer membrane components in the vacuoles. We took advantage of a previous observation that SOD1 marks the boundary of vacuoles [17,28] and doubly stained spinal cord sections with antibodies against SOD1 and cytochrome c oxidase subunit 1 (CO1), an inner mitochondrial membrane marker. CO1 and the inner membrane structure are located focally within the boundary of vacuoles (Fig. 2A,2B,2C,2D). Intense SOD1 signal is located both at the boundary of the vacuoles and inside (Fig. 2A,2B,2C,2D), most of which appear to be associated with membranes (Fig. 2C,2D).
Figure 2.
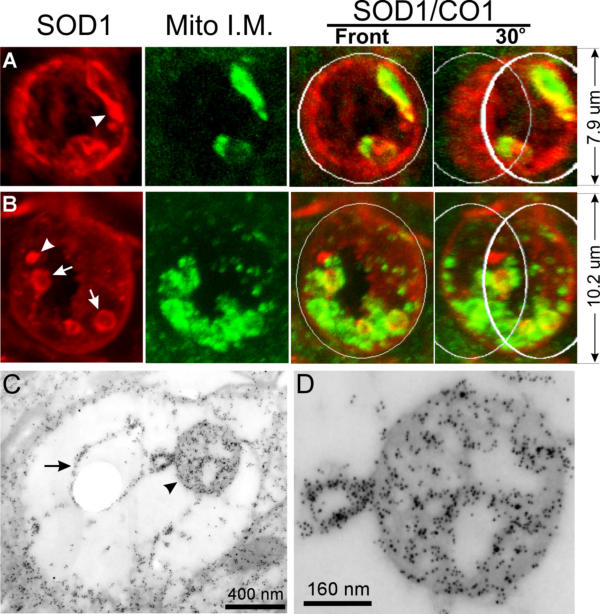
SOD1 is concentrated at the boundary of vacuoles and in focal areas inside vacuoles, while cytochrome c oxidase subunit 1 (CO1) is located only in focal areas inside vacuoles. (A, B) Spinal cord sections from SODG93A mice were doubly stained for SOD1 (red) and CO1 (green). Vacuoles are photographed using a confocal microscope and viewed by 3-d reconstruction. The right panels show the superimposition of the red and green signals. The two right-most panels are viewed from a 30° angel. White circles are added to the top and bottom to aid visualization of 3-d structure. Arrows point to small vesicular structure inside vacuoles. Arrowheads point to SOD1 aggregates.(C, D) Spinal cord sections from SODG93A mice were doubly stained for SOD1 (small particles) and CO1 (large particles). D is a high magnification view of the mitochondrial matrix remnants pointed by the arrowhead in C. The arrow points to a vesicular structure inside the vacuole. The arrowhead points to the inner membrane remnants within this vacuole.
Next, we doubly stained spinal cord sections with antibodies against SOD1 and two protein subunits of TOM (translocator of outer membrane), TOM20 and TOM40. Both are outer mitochondrial membrane proteins [29]. Similar to Figure 2, SOD1 signal marked the boundary of a barrel-shaped vacuole (Fig. 3A,3D). In contrast to the location of the CO1 signal (Fig. 2), TOM20/40 signals are at the boundary of the vacuole (Fig. 3B,3E), partially overlapping with the SOD1 signal (Fig. 3C,3F, small arrows). TOM20/40 signals are patchy, concentrated in certain areas of the vacuolar boundary (large arrows).
Figure 3.
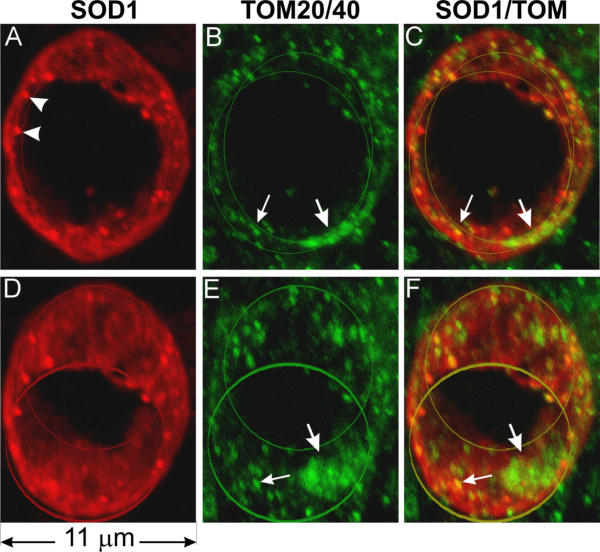
Mitochondrial outer membrane markers TOM20 and TOM40 are localized in the vacuolar wall. Spinal cord sections were doubly stained with anti-SOD1 (red) and a mixture of anti-TOM40 and anti-TOM20 antibodies (green), photographed using a confocal microscope, and visualized by 3-D reconstruction. This vacuole is barrel-shaped. The front and the back openings of the vacuole are marked by two line circles. D-F are the same vacuole as A-C but rotated downwards 30 degrees. Staining using either TOM40 or TOM20 alone yielded the same staining pattern except that the signals were somewhat weaker than using both antibodies together. Arrowheads in A point to SOD1 aggregates, large arrows show patches of TOM and small arrows point to where TOM overlaps with SOD1.
Because TOM20/40 staining also labeled many normal mitochondria outside of the vacuoles, the signal near the vacuole obscured the signal associated with the vacuole. This left open the possibility that the TOM20/40 signal at the boundary of the vacuole represents surrounding mitochondria, which are compressed by the expanding vacuole. Although this possibility is inconsistent with the absence of CO1 signal at the vacuolar boundary (Fig. 2), we further examined this possibility by cutting a middle section of the vacuole out and visualized it in three dimensions (Fig. 4). The patches of TOM20/40 signals (Fig. 4B,4E, arrows) did form a ring around the vacuole, and this ring overlaps with the inner boundary of the ring formed by the SOD1 signal (Fig. 4C,4F). This indicates that mitochondrial outer membrane forms the boundary of vacuoles and mutant SOD1 may be attached to the outside of the vacuolar membrane.
Figure 4.
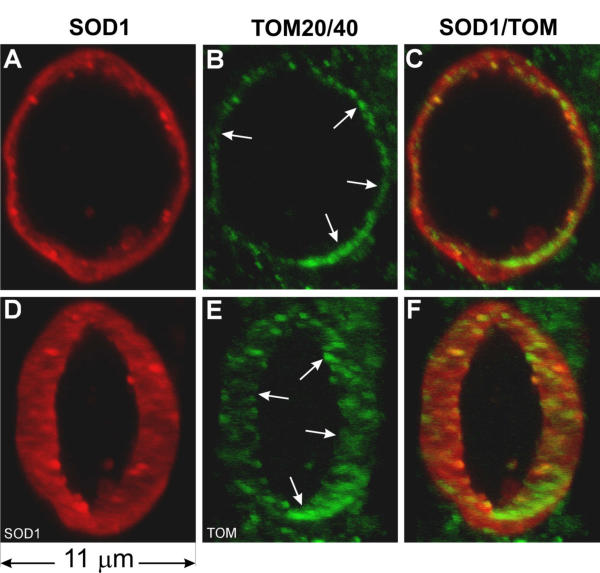
Mitochondrial outer membrane markers TOM40 and TOM20 are localized in the vacuolar wall, inside the SOD1 signal at the vacuolar boundary. The panels are the mid section of the vacuole shown in Figure 3. D-F are the same as A-C but rotated to the right by 45°. Arrows point to patches of TOM signal.
Is cytochrome c, an intermembrane space protein, present in the expanded intermembrane space? To answer this question, we doubly stained spinal cord sections with antibodies against SOD1 and cytochrome c. Inside large vacuoles cytochrome c signal is absent (Fig. 5A,5B,5C,5D). On the other hand, small vacuoles contain abundant cytochrome c signal, which nearly completely overlaps with the SOD1 signal (Fig. 5E,5F,5G,5H, arrows). As a control, we also examined small vacuoles doubly stained with SOD1 and CO1 antibodies. CO1 is still localized focally in these small vacuoles (Fig. 5I,5J,5K,5L, arrows) and only partially overlaps with SOD1 signal. Together, these observations suggest that cytochrome c is present in the intermembrane space of the early, small vacuoles and either being diluted to below the detection levels in large, intact vacuoles, or leaked out in the late, large vacuoles because of porous nature of the large vacuoles (Figs. 1, 2 and 5).
Figure 5.
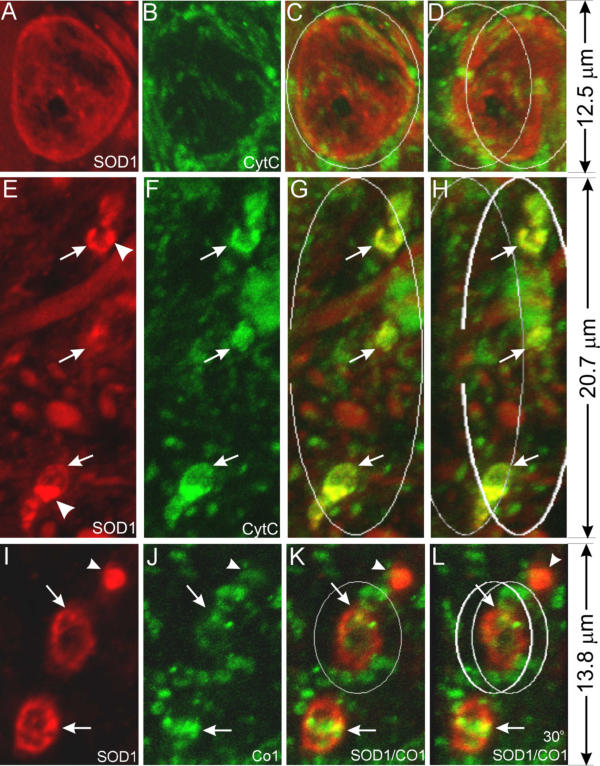
Early vacuoles contain cytochrome c but the late ones do not. Spinal cord sections from SOD1G93A mice at the pre-muscle-weakness (PMW) stage were stained with anti-SOD1 (red) and anti-cyt c (green) antibodies (A-H), or anti-SOD1 (red) and anti-CO1 (green) antibodies (I-L), and shown as 3-D reconstructed confocal images. C, G and K are superimpositions of the red and green signals. D, H and L are the same as C, G and K, respectively, but rotated to the right for 30°. Arrows point to small vacuoles and arrowheads point to SOD1 aggregates. Early vacuoles contain cytochrome c (E-H) but the late ones do not (A-D). Notice that the inner member marker CO1 does not colocalize with SOD1 (I-L).
In normal mitochondria the outer membrane wraps closely around the inner membrane and is not folded like the inner membrane. Therefore, for the outer membrane to extend, additional membrane would be needed. Because, in addition to mitochondria, peroxisomes are also involved in phospholipid synthesis and metabolism (see Discussion), we examined whether peroxisomes might participate in vacuole formation. Vacuoles contain many catalase-positive granules, the vast majority of which are positioned closely against the inner vacuolar wall (Fig. 6B,6C,6D,6F,6G,6H, filled arrows). A small minority of these granules are attached to the outside of the vacuolar wall (Fig. 6B,6C,6D,6F,6G,6H, unfilled arrows). Similarly, numerous PMP70-positive granules are observed close to the inner wall of the vacuole (Fig. 6J,6K,6L, filled arrows) and a small number of these granules are attached to the outside (Fig. 6J,6K,6L, unfilled arrows). Furthermore, catalase signal is present together with CO1 signal in the same vacuoles (Fig. 7). Finally, catalase signal is also detected in both small and large, SOD1-positive vacuoles at early disease stages (Fig. 8). These results suggest that peroxisomes actively participate in the formation of mitochondrial vacuoles at the early stages.
Figure 6.
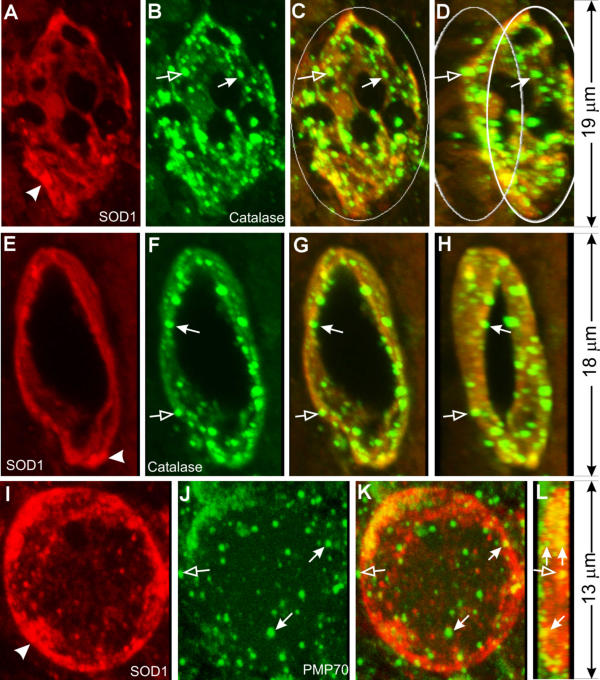
Peroxisomes participate in mitochondrial vacuolation. Spinal cord sections from SOD1G93A mice were stained with anti-SOD1 (red) and anti-catalase (green) antibodies (A-H), or anti-SOD1 (red) and anti-PMP70 (green) antibodies (I-L), and shown as 3-D reconstructed confocal images. C, G and K are superimpositions of the red and green signals. D and H are the same as C and G, respectively, but rotated to the right for 30°. L is the same as K but rotated for 90°. Arrows in B-D and F-H point to small, catalase-positive granules that line against the inner wall of vacuoles. Arrows in J-L point to small, PMP70-positive granules that line against the inner wall of vacuoles. Notice one PMP-70 granule in J and K represents two granules when viewed at the 90° in L. Unfilled arrows point to granules that lie against the outer wall of the vacuoles.
Figure 7.
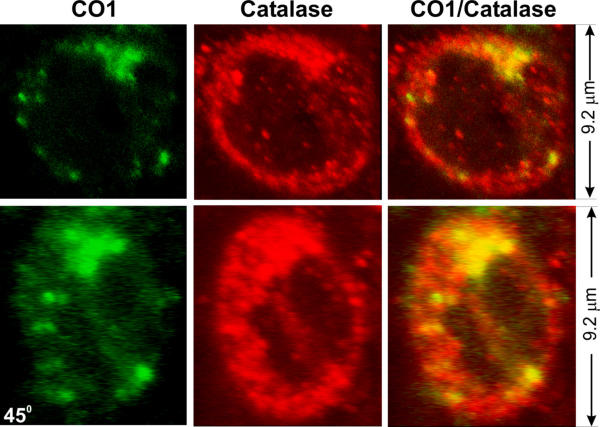
Colocalization of catalase and CO1 in vacuoles.Spinal cord sections from SOD1G93A mice were stained with anti-catalase (red) and anti-CO1 (green) antibodies. Lower panels show the same vacuoles as in the upper panels except that the vacuole is turned 45 degrees.
Figure 8.
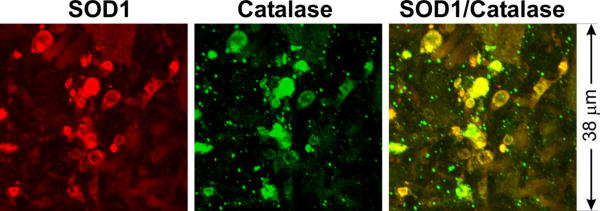
Peroxisoms are involved in early vacuoles. Spinal cord sections from a SOD1G93A mouse at early disease stage (pre-muscle weakness stage; see [15]) were stained with anti-SOD1 (red) and anti-catalase (green) antibodies.
Because lysosomes participate in autophagy [30], we examined whether lysosomes participate in vacuole formation. We doubly stained spinal cord sections with antibodies against SOD1 and cathepsin D, a lysosomal protease. Vacuoles do not contain high levels of cathepsin D signal (Fig. 9). For example, the vacuole in figure 9B contains no more cathepsin D signal than a normal motor neuron cell body in figure 9F. Noticeably, no cathepsin D signal is associated with small vacuoles (Fig. 9E,9F,9G,9H, arrows). These results indicate that lysosomes do not actively participate in vacuole formation.
Figure 9.
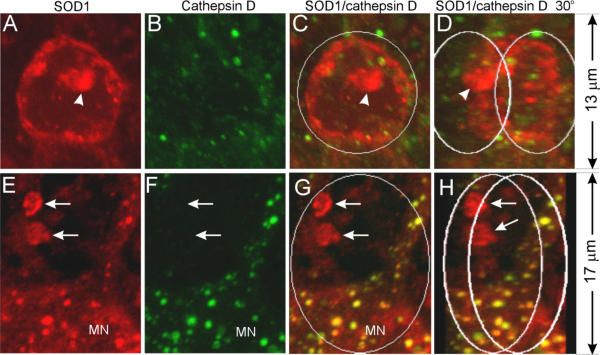
Lysosomes do not actively participate in vacuole formation. Spinal cord sections from SOD1G93A mice were stained with anti-SOD1 (red) and anti-cathepsin D (green) antibodies, and visualized by 3-D reconstruction of confocal images. C and G are superimpositions of the red and green signals. D and H are the same as C and G, respectively, but rotated 30° to the right. Arrows point to small vacuoles that contain SOD1 but not cathepsin D signals. MN marks a motor neuron cell body. Arrows point to small vacuoles. The arrowhead points to SOD1 aggregates.
The absence of matrix expansion and the involvement of peroxisomes but not lysosomes together indicate that mutant SOD1-induced mitochondrial vacuolation is neither MPT nor autophagy. Because mutant SOD1 is prone to unfold [31-34] and form aggregates [11,14,35,36], it has been proposed that abnormal SOD1 aggregation generates toxicity [10]. To determine whether SOD1 aggregates are involved in mitochondrial vacuolation, we examined whether vacuoles contain SOD1 aggregates by immunofluorescence and immuno EM. SOD1 aggregates are in the majority of vacuoles (Fig. 10; also see figures 2, 3, 5, 6 and 9, arrowheads). In all cases, the aggregates were found to be on or near the boundary of the vacuoles (Fig. 10, arrowheads). Some SOD1 aggregates overlap with CO1 signal while others do not (see Fig. 2A and 2B). In small vacuoles, SOD1 aggregates overlap with cytochrome c signal (see Fig. 5E,5F,5G). These results suggest a role of mutant SOD1 aggregation in Mitochondrial Vacuolation by Intermembrane Space Expansion (MVISE).
Figure 10.
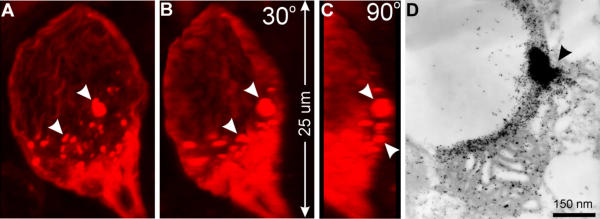
Vacuoles contain focal SOD1 aggregates. Panels A-C are 3-D reconstructed images of a vacuole from SOD1G93A mouse spinal cord sections stained with anti-SOD1 antibodies (red). The vacuole is bounded by SOD1 and contains many SOD1 aggregates (arrowheads). Panel D is an immunogold electron microscopic image from SOD1G93A mouse spinal cord stained with an anti-SOD1 antibody (5 nm gold) and an anti-cytochrome oxidase subunit 4 antibody (10 nm gold). The arrowhead indicates a region of SOD1 aggregation.
Discussion
Our EM observations (Fig. 1) suggest that mutant SOD1G93A causes MVISE in vivo. These observations agree with an earlier suggestion that vacuoles in the SOD1G93A mouse model of ALS are derived from the expansion of mitochondrial intermembrane space [17]. Our subsequent experiments confirm this model. First, the inner mitochondrial membrane does not constitute the vacuole boundary and is contained within the vacuoles (Fig. 2). Second, the outer mitochondrial membrane composes a large part of the vacuolar membrane (Figs. 3, 4). Third, cytochrome c colocalizes with SOD1 in small vacuoles. Both molecules are known intermembrane space proteins. Although cytochrome c signal is lost in the subsequent vacuolar expansion (Fig. 5), this absence could be due to a dilution of cytochrome c by the increased vacuolar volume [17] and leakage of the outer mitochondrial membrane. Fourth, catalase- and PMP70-positive granules are concentrated in the vacuoles (Fig. 6,7,8) but lysosomes are not (Fig. 9). This suggests that peroxisomes actively participate in the vacuole formation but lysosomes do not play a significant role. These observations indicate that neither MPT (expansion of matrix) nor autophagy (in which lysosomes participate) is responsible for mutant SOD1-induced mitochondrial vacuolation. Thus, MVISE represents a previously uncharacterized mechanism of mitochondrial degeneration in the CNS.
How mutant SOD1 causes MVISE is not known but could be associated with SOD1 localization. Fridovich and colleagues first noticed the presence of SOD1 in the intermembrane space in liver mitochondria [37]. Although this was disputed later [38,39], recent experiments in yeast and mammalian cells reaffirmed the presence of both wild type and mutant SOD1 in the intermembrane space of mitochondria [21,25,26,40]. This finding suggests that mutant SOD1 could directly damage mitochondria by its toxicity. The increased cell death induced by direct targeting of mutant SOD1 to mitochondria supports this possibility [27]. Interestingly, low levels (compared with the levels caused by mutant SOD1) of mitochondrial vacuolation can also be caused by expression of high levels of wild type SOD1 [7,13,15,41], suggesting that the mitochondria-damaging property also exists in the wild type protein and this property strengthens by the mutations.
The toxicity produced by mutant SOD1 that leads to mitochondrial damage remains to be identified. One possibility is that mutant SOD1 catalyzes abnormal redox reactions, thereby producing toxic reactive oxygen species (ROS) and damaging cellular structures in its vicinity. Two such activities have been proposed. The first proposes that the mutated SOD1 interacts with NO to produce peroxynitrite (ONOO-), which damages proteins by modifying tyrosine to nitrotyrosine residues [42-44]. The second postulates that the mutant SOD1 possesses an enhanced peroxidase activity, which causes oxidative modification of nucleic acids, proteins and lipids [45-47]. Both of these activities require copper at the active center of SOD1. However, dramatic reduction of copper in SOD1 by knocking out the copper chaperon for SOD1 (CCS), or by mutating the copper chelating residues, did not affect ALS progression in vivo [36,48]. Therefore, the role of copper-mediated oxidative damage remains unclear [49,50].
An alternative proposal is that the toxicity stems from the propensity of the mutated SOD1 to aggregate. SOD1 aggregates have been widely observed in mutant SOD1 transgenic mice and in human ALS caused by SOD1 mutations [11,14,35,36]. In cultured cells, mutant SOD1 forms high molecular weight aggregates when the cells are stressed by high mutant protein expression [51], proteosome inhibition [52] and serum withdraw [27]. The role of mutant SOD1 aggregation, however, remains unclear. One study reports that SOD1 aggregation and cell death stimulating activity are dissociable in mutant SOD1-transfected PC12 cells [53].
All studies conducted on SOD1 aggregation thus far have been on the large aggregates that might be composed of long fibrils. The role of small, oligomeric SOD1 aggregation has not been investigated. Recent studies on neurodegenerative diseases, including Parkinson's, Alzheimer's and prion diseases, suggest that it is not the long fibrils that produce toxicity and kill neurons, rather, it is the oligomeric aggregation intermediates that produce toxicity [54,55]. Interestingly, interactions between biomembranes and toxic proteins produce a dual effect of both promoting oligomeric aggregation of the proteins and membrane damage [56-59], suggesting that one possible toxicity of oligomeric aggregates is mediated through damaging membranes. In this context, our observation that SOD1 aggregates are associated with vacuolar membrane (Figs. 2, 10) suggests that mutant SOD1 interacts with mitochondrial membrane and this interaction promotes aggregation. The form of these aggregates is not yet known but could include both the large aggregates (e.g. Fig. 10, arrowheads) that bind to thioflavin S [36], and oligomers, which may be represented by the intense SOD1 immuno-reactive signals associated with vacuolar membrane (Fig. 10).
The presence of peroxisomes in mutant SOD1-induced MVISE is a surprise. How this occurs is unclear. Nevertheless, the higher concentration of peroxisomes in and around vacuoles suggests that peroxisomes actively migrate to or proliferate in the vicinity of abnormal mitochondria and participate in the vacuolation process. What might be the role of peroxisome? The answer is not known but the normal peroxisome function might provide some clues. Peroxisomes are indispensable in lipid metabolism and maintenance of lipid homeostasis. Peroxisomes shorten chains of very long fatty acids, unsaturated fatty acids and branched fatty acids by β-oxidation [60,61]. They play critical roles in synthesis of ether phospholipids and cholesterol [62,63]. Peroxisomal abundance is associated with high membrane demand such as myelination in the central nervous system [64,65] and peroxisome diseases result in defective myelination [66]. Because the outer mitochondrial membrane normally tightly wraps around mitochondria, the extension of the outer membrane during vacuolation requires new membranes. Peroxisomes could provide the new membranes by either synthesis of lipids or fusing themselves with the vacuolar membrane. Alternatively, peroxisomes might be involved in membrane breakdown in the mitochondrial degeneration process. Interestingly, accumulation of ceramides and cholesterol esters has been observed in ALS patients and mice [67], which could be associated with peroxisomal proliferation.
The mitochondrial vacuolation could in part be the result of persistent mitochondrial dysfunction and further facilitate the downstream motor neuron degeneration events. Our previous study showed that functional decline of mitochondria appears in the SOD1G93A mice as early as 60 to 100 days of age [22]. This precedes the peak of mitochondrial vacuolation and the onset of the disease by two to three months [15]. Mitochondrial dysfunction can lead to energy deficiency and ionic imbalance [68], elevated ROS and oxidative damage [69], and increased sensitivity of neurons to excitotoxicity [19,70-72]. These effects could precipitate MVISE, which trigger cell death programs by releasing pro-apoptotic proteins resided in the mitochondrial intermembrane space, such as cytochrome c, AIF, SMAC/DIABLO, endo G and Htra/Omi [73]. The absence of cytochrome c in the porous large vacuoles and the presence of cytochrome c in the small vacuoles (Fig. 5) are consistent with this possibility. Because the majority of vacuoles develop in distal small dendrites [28], the release of these proapoptotic molecules may not cause typical apoptotic changes in cell bodies including chromatin condensation and cytoplasmic blebbing. Indeed, the typical changes are not observed by EM (Higgins, Kong and Xu, unpublished observation; also see [74,75]), but widespread caspase activation is detected in spinal cords [75-77], suggesting the occurrence of a neuritic death program [78].
Our results as well as others' show that mitochondrial vacuolation provides an important window for examination of the mechanism whereby mutant SOD1 causes mitochondrial damage and motor neuron degeneration. However, some mutant SOD1 causes motor neuron degeneration without mitochondrial vacuolation [8,11,36]. One difference between the mutants that induce vacuoles and the ones that do not, is that the former accumulates at higher levels in the mouse spinal cord than that latter. It is possible that mitochondrial vacuolation represents an accelerated form of mitochondrial damage caused by high levels of mutant SOD1 accumulation. This does present a challenge to examine and define mitochondrial damage in low mutant SOD1 expressing transgenic lines.
Conclusions
Taken together, this study reveals a mitochondrial degeneration mechanism in the CNS, mitochondrial vacuolation by intermembrane space expansion or MVISE. The basic features of MVISE are expansion of mitochondrial intermembrane space, extension of mitochondrial outer membrane, collapse and/or disintegration of the matrix and inner membrane, and involvement of peroxisomes. These features bare resemblances to mitochondrial degeneration in other neurodegenerative conditions, including Alzheimer's disease [79], prion disease [80], Rett syndrome [81], excitotoxic neuronal death [70] and Mn++ superoxide dismutase (SOD2) deletion [82]. Although in none of these conditions are sufficiently detailed observations available for a comprehensive comparison, MVISE could play a role in other neurodegenerative disorders. Mitochondrial vacuolation may contribute to several non-apopototic cell death pathways, but its role in a wide spectrum of neurodegenerative diseases remains to be defined [83,84].
Methods
Transgenic mice
The low expression line of transgenic mice expressing human mutant SOD1G93A (C57BL/6J-TgN(SOD1-G93A)1Gurdl) were purchased from Jackson Lab (Bar Harbor, ME) and bred onto FVB background at University of Massachusetts Medical School animal facility. Non-transgenic littermates of SODG93A transgenic mice were used as wild type controls (WT mice). All transgenic mice were identified using PCR according to Gurney et al. [85]. Mice were maintained at the University of Massachusetts Medical School animal facility according to the guidelines set forth by the Institutional Animal Care and Use Committee (IACUC).
Immunofluorescence Microscopy
Mice were perfused under anesthesia with fixative (4% paraformaldehyde, 0.1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.5). Sections of lumbar spinal cords from fixed mice were dissected out and 30 μm sections were cut using a vibratome. Sections were processed for antibody staining using the following antibodies: rabbit [86] and sheep (BioDesign) anti-SOD1 (both recognize mouse and human SOD1), mouse anti-cytochrome c oxidase subunit CO1 and CO4 (Molecular Probes, OR), rabbit anti-TOM20 and TOM40 (Santa Cruz Biotechnology, CA), mouse anti-cytochrome c (BD PharMingen, San Diego, CA), sheep anti-catalase (The Binding Site, Birmingham, England), rabbit anti-catalase (Calbiochem, SanDiego, CA), rabbit anti-PMP70 (ABR, Golden, CO or Axxora, SanDiego, CA), and rabbit anti-cathepsin D (DAKO, Carpinteria, CA) according to the protocol described previously [26,28]. The stained sections were examined and digitized using a confocal microscope (Leica TCS-SP). Imaging analysis and three-dimensional reconstruction were conducted using MetaMorph (Universal Imaging Corp.).
Transmission electron microscopy (TEM)
Animal fixation, tissue dissection, and microscopic analysis were performed as described previously [15]. In brief, mice were anesthetized and perfused with a solution of 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.6. Tissues were kept in the same solution for further fixation. The L4 and L5 spinal nerve roots and lumbar spinal cords (a 2 mm segment centered at the L5 root entry level) were dissected out and post-fixed with 2% osmium tetroxide in 100 mM cacodylate buffer, pH 7.6. After dehydration in graded alcohol, the tissue blocks were embedded in Epon. Thin sections of ventral horn were cut from the Epon tissue blocks, stained with uranyl acetate and lead citrate, and visualized using a Philips CM10 transmission electron microscope.
Immuno-electron microscopy
Mice were perfused under anesthesia with fixative (4% paraformaldehyde, 0.1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.5). Sections of lumbar spinal cords from fixed mice were dissected out, dehydrated through an ethanol series to 100% ethanol and embedded in hard grade LR White resin (EMS, Fort Washington, PA) by polymerization overnight at 60 deg. C. Sixty five nm sections were cut from the embedded tissue using a Reichert-Jung Ultracut E microtome and collected onto gold grids (200 mesh) (SPI Inc., Westchester, PA). Sections on grids were processed for immunogold electron microscopy according to recommendations by the manufacturer of the gold-conjugated antibodies (Amersham Pharmacia Biotech, Piscataway, NJ) with some modification. Sections were etched in 0.1 N HCl for 5 min., rinsed three times for five min each in TBS (25 mM Tris, 140 mM NaCl, 2.7 mM KCl, pH 8.0), placed in blocking buffer for 30 min (0.1% gelatin, 1% normal goat serum, 0.3% Triton-X-100 in TBS), placed in primary antibody (1:10 diluted in blocking buffer for all antibodies used) for 2 hours at room temperature, rinsed 3 times for 5 min each in TBS, placed in gold-conjugated secondary antibodies (10 nm gold anti-mouse and 5 nm gold anti-rabbit) for 1 hour, rinsed 3 times in TBS, fixed in 2% glutaraldehyde, rinsed 3 times in TBS, rinsed in water, stained with Reynold=s lead citrate followed by aqueous 2% uranyl acetate, dried on filter paper and viewed using a Philips CM10 transmission electron microscope. The following antibodies and sera were used, normal rabbit serum (Vector Laboratories Inc., Burlingame, CA), rabbit polyclonal anti-SOD1 [86], a second rabbit polyclonal anti-SOD1 (Biodesign, Saco, ME) and a mouse monoclonal antibody against CO1. All antibodies and serum were used at a 1:10 dilution.
Authors' contributions
CMJH carried out immunofluorescence and EM, analyzed data, made figures and drafted the manuscript. CWJ assisted in immunofluorescence and EM. ZX carried out immunofluorescence and EM, analyzed data, made figures and wrote the manuscript.
Acknowledgments
Acknowledgements
We thank Ms. Ellen Trang for maintaining transgenic mice and Dr. Paul Mathews for sharing cathepsin D antibodies. The EM work was carried out with the support of the Core Electron Microscopy Facility of the University of Massachusetts Medical School. We thank Dr. Gregory Hendricks and Mr. John Nunnari for their expert advice and technical assistance with electron microscopy. We also thank members of Xu lab for support and helpful discussion. This work is supported by grants from the ALS Association, NINDS (RO1 NS41739, NS35750) and the Robert Pachard Center for ALS Research at Johns Hopkins to ZSX. CMJH is supported by NIH Training Grant 5 T32 NS07366-05. The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the NINDS.
Contributor Information
Cynthia MJ Higgins, Email: cynthia.higgins@umassmed.edu.
Cheolwha Jung, Email: cheolwha_jung@uml.edu.
Zuoshang Xu, Email: zuoshang.xu@umassmed.edu.
References
- Brown R. H., Jr., Robberecht W. Amyotrophic lateral sclerosis: pathogenesis. Semin Neurol. 2001;21:131–139. doi: 10.1055/s-2001-15260. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Siddique T, Lalani I. Genetic aspects of amyotrophic lateral sclerosis. Adv Neurol. 2002;88:21–32. [PubMed] [Google Scholar]
- Hand CK, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:135–159. doi: 10.1002/mus.10001. [DOI] [PubMed] [Google Scholar]
- Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Zuoshang. Mechanism and treatment of motoneuron degeneration in ALS: what have SOD1 mutants told us? Amyotrophic Lateral Sclerosis and other Motor Neuron Disorders. 2000;1:225–234. doi: 10.1080/14660820050515052. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS) Brain Research. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Gonatas NK. Aggregation of ubiquitin and a mutant ALS-linked SOD1 protein correlate with disease progression and fragmentation of the Golgi apparatus. J Neurol Sci. 2000;173:53–62. doi: 10.1016/s0022-510x(99)00300-7. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. The Journal of Neuroscience. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathologica. 1997;93:537–550. doi: 10.1007/s004010050650. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berl) 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Carri MT, Ferri A, Battistoni A, Famhy L, Cabbianelli R, Poccia F, Rotilio G. Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Letters. 1997;414:365–368. doi: 10.1016/s0014-5793(97)01051-x. [DOI] [PubMed] [Google Scholar]
- Kruman ,II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Experimental Neurology. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, Sheehan JP, Maguire RS, Pattee G, Juel VC, Philips LH, Tuttle JB, J. P. Bennett Jr., Davis RE, Parker Jr. WD. Mitochondria in sporadic amyotrophic lateral sclerosis. Experimental Neurology. 1998;153:135–142. doi: 10.1006/exnr.1998.6866. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80:616–625. doi: 10.1046/j.0022-3042.2001.00731.x. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:RC215.. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Kobayashi Y, Ishigaki S, Doyu M, Sobue G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:50966–50972. doi: 10.1074/jbc.M209356200. [DOI] [PubMed] [Google Scholar]
- Levine John B., Kong Jiming, Nadler Mark, Xu Zuoshang. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia. 1999;28:215–224. [PubMed] [Google Scholar]
- Neupert W, Brunner M. The protein import motor of mitochondria. Nat Rev Mol Cell Biol. 2002;3:555–565. doi: 10.1038/nrm878. [DOI] [PubMed] [Google Scholar]
- Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem. 2000;69:303–342. doi: 10.1146/annurev.biochem.69.1.303. [DOI] [PubMed] [Google Scholar]
- Cardoso RM, Thayer MM, DiDonato M, Lo TP, Bruns CK, Getzoff ED, Tainer JA. Insights into Lou Gehrig's disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J Mol Biol. 2002;324:247–256. doi: 10.1016/s0022-2836(02)01090-2. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Hayward LJ. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem. 2003;278:5984–5992. doi: 10.1074/jbc.M210419200. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, Brown R. H., Jr., Hayward LJ. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- Lindberg MJ, Tibell L, Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci U S A. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. Journal of Biological Chemistry. 1973;248:4793–4796. [PubMed] [Google Scholar]
- Geller BL, Winge DR. Rat liver Cu,Zn-superoxide dismutase. Subcellular location in lysosomes. Journal of Biological Chemistry. 1982;257:8945–8952. [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxinitrate. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y-X, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. Journal of Neurochemistry. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang YX, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- Yim MB, Kang J-H, Yim H-S, Kwak H-S, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proceedings of the National Academy of Sciences USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim HS, Kang JH, Chock PB, Stadtman ER, Yim MB. A familial amyotrophic lateral sclerosis-associated A4V Cu, Zn-superoxide dismutase mutant has a lower Km for hydrogen peroxide. Correlation between clinical severity and the Km value. Journal of Biological Chemistry. 1997;272:8861–8863. doi: 10.1074/jbc.272.14.8861. [DOI] [PubMed] [Google Scholar]
- Subramaniam JR, Lyons WE, Liu J, Bartnikas TB, Rothstein J, Price DL, Cleveland DW, Gitlin JD, Wong PC. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- Wong PC. Is ALS caused by an altered oxidative activity of mutant superoxide dismutase--reply. Nat Neurosci. 2002;5:919–920. doi: 10.1038/nn1002-919a. [DOI] [PubMed] [Google Scholar]
- Bush AI. Is ALS caused by an altered oxidative activity of mutant superoxide dismutase? Nat Neurosci. 2002;5:919; author reply 919–20. doi: 10.1038/nn1002-919a. [DOI] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. Journal of Neuropathology & Experimental Neurology. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JP, Gerin C, Bindokas VP, Miller R, Ghadge G, Roos RP. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;82:1229–1238. doi: 10.1046/j.1471-4159.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Lansbury P. T., Jr., Brice A. Genetics of Parkinson's disease and biochemical studies of implicated gene products. Curr Opin Genet Dev. 2002;12:299–306. doi: 10.1016/s0959-437x(02)00302-7. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci U S A. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury P. T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Gong JS, Fan QW, Sawamura N, Yanagisawa K. A novel action of alzheimer's amyloid beta-protein (Abeta): oligomeric Abeta promotes lipid release. J Neurosci. 2001;21:7226–7235. doi: 10.1523/JNEUROSCI.21-18-07226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veldhoven PP, Casteels M, Mannaerts GP, Baes M. Further insights into peroxisomal lipid breakdown via alpha- and beta-oxidation. Biochem Soc Trans. 2001;29:292–298. doi: 10.1042/0300-5127:0290292. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Filppula SA, Koivuranta KT, Siivari K, Qin YM, Hayrinen HM. Peroxisomal beta-oxidation and polyunsaturated fatty acids. Ann N Y Acad Sci. 1996;804:116–128. doi: 10.1111/j.1749-6632.1996.tb18612.x. [DOI] [PubMed] [Google Scholar]
- Hajra AK, Das AK. Lipid biosynthesis in peroxisomes. Ann N Y Acad Sci. 1996;804:129–141. doi: 10.1111/j.1749-6632.1996.tb18613.x. [DOI] [PubMed] [Google Scholar]
- Kovacs WJ, Olivier LM, Krisans SK. Central role of peroxisomes in isoprenoid biosynthesis. Prog Lipid Res. 2002;41:369–391. doi: 10.1016/s0163-7827(02)00002-4. [DOI] [PubMed] [Google Scholar]
- Adamo AM, Aloise PA, Soto EF, Pasquini JM. Neonatal hyperthyroidism in the rat produces an increase in the activity of microperoxisomal marker enzymes coincident with biochemical signs of accelerated myelination. J Neurosci Res. 1990;25:353–359. doi: 10.1002/jnr.490250312. [DOI] [PubMed] [Google Scholar]
- Lazo O, Singh AK, Singh I. Postnatal development and isolation of peroxisomes from brain. J Neurochem. 1991;56:1343–1353. doi: 10.1111/j.1471-4159.1991.tb11431.x. [DOI] [PubMed] [Google Scholar]
- Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;8:101–120. doi: 10.1111/j.1750-3639.1998.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Klivenyi P, Klein AM, Shinobu LA, Epstein CJ, Beal MF. Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann Neurol. 2000;47:447–455. [PubMed] [Google Scholar]
- Ikonomidou C, Qin Qin Y, Labruyere J, Olney JW. Motor neuron degeneration induced by excitotoxin agonists has features in common with those seen in the SOD-1 transgenic mouse model of amyotrophic lateral sclerosis. Journal of Neuropathology & Experimental Neurology. 1996;55:211–224. doi: 10.1097/00005072-199602000-00010. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Ikonomidou C. Glutamate in neurologic diseases. Journal of Child Neurology. 1997;12:471–485. doi: 10.1177/088307389701200802. [DOI] [PubMed] [Google Scholar]
- Kaal EC, Vlug AS, Versleijen MW, Kuilman M, Joosten EA, Bar PR. Chronic mitochondrial inhibition induces selective motoneuron death in vitro: a new model for amyotrophic lateral sclerosis. Journal of Neurochemistry. 2000;74:1158–1165. doi: 10.1046/j.1471-4159.2000.741158.x. [DOI] [PubMed] [Google Scholar]
- Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191:25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Guegan C, Przedborski S. Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest. 2003;111:153–161. doi: 10.1172/JCI17610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown R. H., Jr., Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci. 2001;21:6569–6576. doi: 10.1523/JNEUROSCI.21-17-06569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Duan W. "Apoptotic" biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, Scott JR, Fraser H. Scrapie inoculation of mice: light and electron microscopy of the superior colliculi. Acta Neuropathol (Berl) 1991;81:562–571. doi: 10.1007/BF00310139. [DOI] [PubMed] [Google Scholar]
- Ruch A, Kurczynski TW, Velasco ME. Mitochondrial alterations in Rett syndrome. Pediatr Neurol. 1989;5:320–323. doi: 10.1016/0887-8994(89)90027-1. [DOI] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Golstein P. More than one way to go. Proc Natl Acad Sci U S A. 2001;98:11–13. doi: 10.1073/pnas.98.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci U S A. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]