

Abstract
Synaptic core complex formation is an essential step in exocytosis, and assembly into a superhelical structure may drive synaptic vesicle fusion. To ascertain how Ca2+ could regulate this process, we examined calmodulin binding to recombinant core complex components. Surface plasmon resonance and pull-down assays revealed Ca2+-dependent calmodulin binding (Kd = 500 nM) to glutathione S-transferase fusion proteins containing synaptobrevin (VAMP 2) domains but not to syntaxin 1 or synaptosomal-associated protein of 25 kDa (SNAP-25). Deletion mutations, tetanus toxin cleavage, and peptide synthesis localized the calmodulin-binding domain to VAMP77–94, immediately C-terminal to the tetanus toxin cleavage site (Q76–F77). In isolated synaptic vesicles, Ca2+/calmodulin protected native membrane-inserted VAMP from proteolysis by tetanus toxin. Assembly of a 35S-SNAP-25, syntaxin 1 GST-VAMP1–96 complex was inhibited by Ca2+/calmodulin, but assembly did not mask subsequent accessibility of the calmodulin-binding domain. The same domain contains a predicted phospholipid interaction site. SPR revealed calcium-independent interactions between VAMP77–94 and liposomes containing phosphatidylserine, which blocked calmodulin binding. Circular dichroism spectroscopy demonstrated that the calmodulin/phospholipid-binding peptide displayed a significant increase in αhelical content in a hydrophobic environment. These data provide insight into the mechanisms by which Ca2+ may regulate synaptic core complex assembly and protein interactions with membrane bilayers during exocytosis.
Transmitter release at the nerve terminal occurs via calcium-dependent exocytosis of the contents of synaptic vesicles at the presynaptic plasma membrane. Synaptic vesicle fusion involves the assembly of a heterotrimeric synaptic core [soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor (SNARE)] complex composed of the vesicle-associated membrane protein (VAMP 2 or synaptobrevin) and two predominantly plasma membrane proteins, syntaxin 1 and synaptosomal-associated protein of 25 kDa (SNAP-25) (1, 2). The fundamental importance of these components is underlined by the fact that the metalloprotease activities of the botulinum (BoNT) and tetanus (TeTx) neurotoxins cleave the synaptic SNARE proteins at well-defined sites and potently inhibit transmitter release (3, 4). Structural studies have demonstrated that the synaptic SNARE complex forms a four-helical parallel bundle with a superhelical twist (5, 6). It has been proposed that the assembly of this structure in a trans configuration, i.e., at the interface between a docked synaptic vesicle and the plasma membrane, pulls the opposing membranes together and may ultimately drive bilayer fusion (7). In support of this hypothesis, reconstitution of purified v-SNARE and t-SNARE proteins into distinct vesicle populations led to an increase in lipid mixing between the two vesicle pools, indicative of fusion (8). Although assembly is thought to be initiated in a trans configuration at the interface between two membranes, complete fusion would result in a cis complex in the plane of a single membrane. Dissociation of the cis core complex to permit a second round of assembly then requires ATP hydrolysis by a specific chaperone: the N-ethylmaleimide sensitive fusion protein (NSF), which binds to the SNARE complex via soluble NSF attachment proteins (SNAPs) (1, 2)
Thus synaptic core complex assembly is a crucial step leading to vesicle fusion, and this process is presumably triggered by Ca2+ influx via voltage-gated channels. Recent studies in “cracked” pheochromocytoma PC12 cells have suggested that Ca2+ initiates trans core complex formation (9). However, the molecular mechanisms by which Ca2+ ions promote assembly and by which subsequent assembly results in the induction of a membrane fusion pore remain obscure. The SNARE complex does not appear to have intrinsic Ca2+-receptor activity, and attention has therefore focused on interacting protein partners with the appropriate properties. These include C2-domain proteins such as the synaptotagmins, which can associate with the core complex and display Ca2+-dependent phospholipid binding (10). Experiments in yeast point to an alternative cascade of events, indicating that homotypic vesicle fusion, induced by Ca2+ efflux from vesicular stores, is mediated by the ubiquitous Ca2+-sensor calmodulin (11). There is little direct evidence to support a role for calmodulin in excitation/secretion coupling in nerve terminals. However, mammalian neuroendocrine cells have often been used to study Ca2+-dependent exocytosis, as they possess the same basic exocytotic machinery as neurones. Furthermore, cell permeabilization permits the introduction of exogenous macromolecules into the cytoplasm without compromising secretion in response to micromolar Ca2+ concentrations. Evidence from functional dissection of the stages leading up to exocytosis in permeabilized bovine chromaffin cells and in the rat pheochromocytoma PC12 cell line is consistent with a requirement for calmodulin at a late triggering step in neurosecretion (12, 13). For these reasons, we have examined whether calmodulin could act as a Ca2+-dependent regulator of exocytosis by binding directly to components of the SNARE complex.
Materials and Methods
Materials.
Biotinylated calmodulin and calmodulin were obtained from Calbiochem and calmodulin–agarose beads from Sigma. The plasmid encoding TeTx light chain–His6 was kindly provided by the late H. Niemann (University of Hanover, Hanover, Germany). Antibodies against the peptide 1–20 of rat VAMP 2 and glutathione-S transferase (GST) fusion proteins were prepared as previously reported (14, 15).
Interactions Between SNARE Proteins and Calmodulin–Agarose.
VAMP 21–96, VAMP 21–90, VAMP 277–90, syntaxin 1A1–261, and SNAP-251- 206 fused to GST were produced. GST-VAMP 21–76 was generated by incubating GST-VAMP 21–96 (2 μM) with recombinant TeTx-LC (400 nM) in 4 mM Hepes–NaOH, pH 7.4/100 mM NaCl/3.5 mM CaCl2/3.5 mM MgCl2 for 1 h at 37°C, and complete cleavage was verified by SDS/ PAGE. GST fusion proteins (2 μM) were incubated for 4 h at 4°C with calmodulin–agarose beads (5 μM Cam) in Tris-buffered saline [(TBS), 25 mM Tris/150 mM NaCl adjusted to pH 7.4 with HCl] containing 0.1% Triton and 1 mM CaCl2 or 5 mM EDTA. Proteins bound to calmodulin–agarose after extensive washing were denatured in 3% SDS in the presence of 10 mM DTT and analyzed by SDS/PAGE and Coomassie blue staining. The amount of GST-VAMP bound to calmodulin–agarose beads at saturation was determined by Coomassie blue staining, densitometry, and interpolation in a standard GST-VAMP curve, within the linear response range (0.005–0.08 nmol). The stoichiometry of the interaction was then calculated by using the calmodulin coupling density specified by the supplier, assuming 100% biological activity of the coupled calmodulin.
Surface Plasmon Resonance.
Binding experiments and kinetic analysis were performed by using a Biacore X apparatus (Uppsala, Sweden) at 25°C with a constant flow rate of 20 μl/min. Rate constants were calculated by global fitting with a single-site binding model by using the bia 3.0 evaluation program (Pharmacia Biosensor, Uppsala, Sweden). A precoated streptavidin biosensor chip (SA) was used to immobilize biotinylated calmodulin (27 fmol/mm2) and then saturated with biotin. Nonspecific binding was evaluated in the same experiment by measuring binding to a surface saturated with biotin or an irrelevant biotinylated peptide and subtracted automatically. GST-VAMP1–96 and the synthetic peptides corresponding to VAMP 2 residues 77–94 were diluted in running buffer (25 mM Hepes, pH 7.4/150 mM NaCl/0.05% Tween 20/either 1 mM CaCl2 or 5 mM EDTA) and injected at a final concentration of 500 nM and 1 μM, respectively. Proteins or peptides were completely dissociated by injecting buffer containing 5 mM EDTA. To study the interaction between calmodulin, phospholipids, and VAMP 2, GST-VAMP1–96 (200 nM) was injected in the presence or absence of dipalmitoyl-l-α-phosphatidyl-choline/dipalmitoyl-l-α-phosphatidyl-l-serine (PC/PS) liposomes (67 μM). Regeneration was performed with buffer containing 5 mM EDTA and 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate.
Immunoisolated Synaptic Vesicles.
Immunoisolated synaptic vesicles were prepared as described (16) by using an anti-synaptotagmin I monoclonal antibody. Vesicles immobilized on Protein A-Sepharose FF beads were resuspended in cleavage buffer and separated into two aliquots. Tetanus toxin (TeTx) light chain (300 pM) was added to both aliquots, and one was supplemented with calmodulin (15 μM). Incubation was carried out at 37°C. Samples were removed at indicated times, denatured in 3% SDS in the presence of 10 mM DTT, and analyzed by Western blotting with a polyclonal antibody directed against the N terminus of VAMP 2.
SNARE Complex Assembly.
35S-SNAP-25 was produced by in vitro translation in the presence of [35S]methionine (TNT T7 Quick Coupled Transcription/Translation System, Promega). GST was removed from GST-syntaxin 1A1–261 by thrombin cleavage and monitored by SDS/PAGE and protein staining. GST or GST-VAMP 21–96 (0.5 μM) immobilized on gluthatione-agarose beads was incubated in TBS/0.1% BSA/0.1% Triton/1 mM CaCl2 with 35S-SNAP-25 in the presence or absence of syntaxin 1A1–261 (0.6 μM). After 3 h at 4°C, the samples were washed by centrifugation, and the radioactivity retained on beads was measured by β counting. To evaluate thermal stability, samples were adjusted to 3% SDS and incubated for 5 min at 37 or 100°C, then analyzed by SDS/PAGE on 5–15% gradient gels. 35S-SNAP-25 and protein complexes containing 35S-SNAP-25 were detected by autoradiography. To study the effects of calmodulin on SNARE complex formation, incubations were performed in the presence or absence of 10 μM calmodulin. Aliquots were removed at the indicated times and counted.
SNARE Complex Binding to Calmodulin–Agarose.
GST-VAMP 21–96 or 1–76 (1 μM), syntaxin 1A1–261 (1 μM) and 35S-SNAP-25 were incubated for 3 h at 4°C in TBS/0.1% BSA/0.1% Triton. Calmodulin–agarose (6 μM) was added, and the incubation was prolonged for 1 h in the same buffer containing 1 mM CaCl2 or 5 mM EDTA, and radioactivity retained on beads after washing was counted.
Phospholipid-Binding Assays.
Unilamellar vesicles of PC or PC/PS, 7.5/2.5, wt/wt, were prepared at a phospholipid concentration of 1 mg/ml. Dry lipid films were hydrated in 150 mM NaCl/1 mM EDTA/25 mM Tris⋅HCl/ 25 mM sodium acetate, adjusted to pH 7.2, and allowed to swell at a temperature above the phase-transition temperature of the phospholipids (typically >45°C). The dispersed lipid (1 ml) was then sonicated for 2 min by using an Ultrasons apparatus (Annemasse, France) with a 2-mm microtip. Liposomes were formed by 19 passes through 100-nm polycarbonate filters by using a Lipofast basic apparatus (Avestin, Ottawa) and stored at 4°C under nitrogen. Phospholipid concentration was assessed according to Wagner et al. (17). Protein–phospholipid interactions were monitored by using the Biacore X apparatus. Anti-GST antibodies were covalently coupled to a CM5 sensor chip by standard amine-coupling procedure. GST alone (reference surface) or GST fusion proteins were injected over the antibody-coated sensorchip surface to obtain the same concentration (i.e., 17 fmol immobilized protein). Liposomes were diluted in running buffer (20 μM) and injected in the presence or absence of VAMP77–94 (50 μM), and regeneration was performed with 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. Anti-GST antibodies were regenerated with 10 μl of 0.1 M glycine, pH 2.2.
Circular Dichroism (CD).
CD spectra were recorded on a Jasco J-600 spectropolarimeter in a range of trifluoroethanol/H2O ratios buffered to pH 7.0 with 10 mM sodium phosphate at 25°C, by using a quartz cell of 1 mm path length.
Results
Calcium-Dependent Calmodulin Binding to VAMP 2.
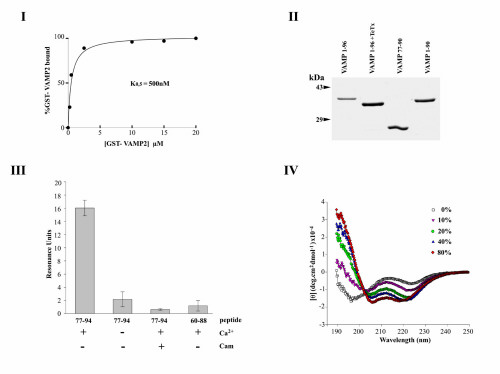
The ability of individual components of the synaptic core complex to interact with calmodulin was evaluated by using bacterially expressed fusion proteins (Fig. 1A) containing the complete sequence of SNAP-25 or the cytoplasmic domains of syntaxin 1A () and VAMP 2 (1–96) fused to GST. These proteins were incubated with calmodulin immobilized on agarose beads in the presence of 1 mM Ca2+ or 5 mM EDTA. The beads were washed by centrifugation and the bound proteins analyzed by SDS/PAGE and Coomassie blue staining (Fig. 1A). The results indicated that GST-VAMP1–96, but not syntaxin 1, SNAP-25, nor GST displayed Ca2+-dependent binding to calmodulin–agarose. Experiments with increasing concentrations of GST-VAMP1–96 in the presence of Ca2+ demonstrated saturable binding, with 50% occupation at 500 nM GST-VAMP1–96, and a VAMP/calmodulin binding stoichiometry close to 1/1 (see supplementary Fig. 5, www.pnas.org). An SPR (Biacore) apparatus was also used to examine the binding of SNARE proteins to biotinylated calmodulin immobilized on a streptavidin-coated sensor chip. No interaction was revealed with GST-SNAP-25, GST-syntaxin 11–261, nor GST (not shown). GST-VAMP1–96 displayed reversible binding to immobilized calmodulin in the presence of Ca2+, and dissociation was accelerated when EDTA was injected in the running buffer (Fig. 1B). Binding rate constants were measured by SPR with a range of GST-VAMP1–96 concentrations and used to calculate the equilibrium dissociation constant (not shown), yielding kon = 7.3 × 103 M−1s−1, koff = 3.8 × 10−3 s−1 and Kd (koff /kon) = 520 nM.
Figure 1.
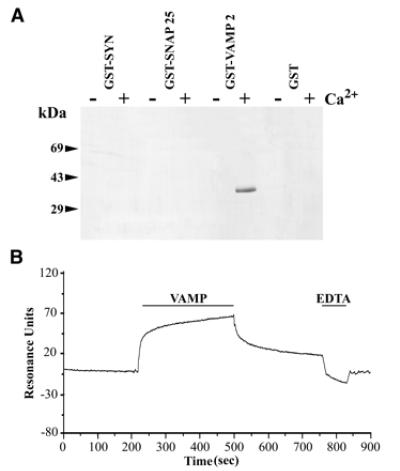
Calcium-dependent calmodulin binding to GST-VAMP 21–96. (A) Bacterially expressed GST-fusion proteins containing SNAP-25, the cytoplasmic domains of syntaxin 1A, VAMP 2, and GST (2 μM) were incubated with calmodulin–agarose (5 μM) in TBS in the presence of 1 mM CaCl2 or 5 mM EDTA. After extensive washing, bound proteins were eluted with sample buffer and analyzed by SDS/PAGE and Coomassie blue staining. (B) GST-VAMP 21–96 was diluted into running buffer containing 1 mM CaCl2 and injected on a streptavidin-coated sensorchip loaded with biotinylated calmodulin. An increase in resonance occurred during VAMP 2 injection (association), followed by decrease at the end of the injection period (dissociation). Injection of EDTA induced an abrupt return to baseline (regeneration). Experiments with a range of VAMP 2 concentrations allowed calculation of the rate constants and Kd reported in the text.
Identification of the Calmodulin-Binding Domain of VAMP 2.
An examination of the amino acid sequence of VAMP 2 revealed a “1–5-8–14 motif” with a net charge of 3+ at residues 77–90 (Fig. 2A), proposed as a consensus for Ca2+-dependent calmodulin binding (18). This sequence is located immediately adjacent, on the C-terminal side, to the peptide bond (Q76–F77) specifically cleaved by TeTx or BoNT/B light chain. To confirm localization of the calmodulin interaction site, GST fusion proteins containing VAMP residues 77–90 or 1–90 were constructed. In addition, GST-VAMP1–96 was incubated with recombinant TeTx light chain to yield GST-VAMP1–76. Binding assays with calmodulin–agarose beads first confirmed that removal of the C-terminal residues 77–96 with TeTx eliminated Ca2+/calmodulin binding (GST-VAMP1–76, Fig. 2B). In contrast constructs containing VAMP residues 77–90 or 1–90 both displayed similar binding, but at a significantly lower level than an equimolar input of VAMP1–96 (Fig. 2B). These observations suggested that additional basic and/or hydrophobic residues in the 91–96 segment contribute to optimal calmodulin-binding activity. Consequently the ability of a synthetic peptide corresponding to VAMP77–94 to compete with GST-VAMP1–96 for binding to calmodulin was measured by SPR assay. The data indicated a Ki = 350 nM for VAMP77–94, thus attaining an apparent Kd similar to that of GST-VAMP1–96 (not shown).. SPR data also showed that binding of VAMP77–94 to immobilized calmodulin was Ca2+ dependent and was displaced by an excess of soluble calmodulin. No interaction was detected with the peptide VAMP60–88, indicating an absolute requirement for 89WW90 (see supplementary Fig. 5, www.pnas.org). These results thus define the optimum calmodulin-binding domain as VAMP77–94. Ca2+-dependent associations between native VAMP 2 and calmodulin were observed by applying solubilized rat brain synaptosomes to calmodulin–agarose beads and detecting adsorbed VAMP 2 by Western blotting (not shown). However, this method does not allow conclusions as to whether binding was direct. The calmodulin-binding site described above (VAMP77–94) overlaps the TeTx cleavage site (Q76–F77), suggesting that calmodulin may interfere with TeTx action. To test this prediction, synaptic vesicles were immunoisolated on an antisynaptotagmin I monoclonal antibody coupled to Protein A-Sepharose beads. Beads were incubated in the presence or absence of TeTx light chain (300 pM), and cleavage was evaluated by using an antibody against the N terminus of VAMP 2. Western blotting of proteins retained on the beads demonstrated that TeTx induced a strong reduction in immunoreactivity, but hydrolysis was prevented when calmodulin (15 μM) was added (Fig. 2C). Inhibition of the action of TeTx is therefore consistent with Ca2+-dependent calmodulin binding to VAMP 2 anchored in the synaptic vesicle membrane.
Figure 2.
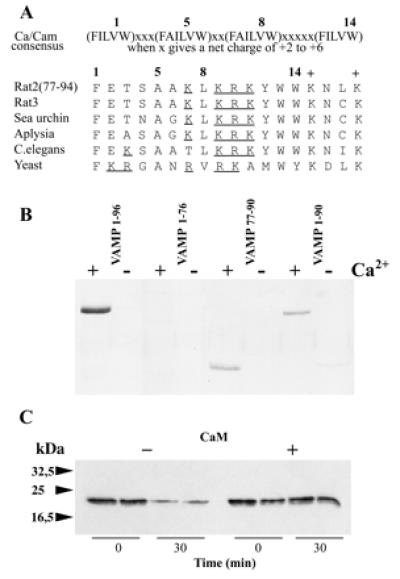
Localization of the calmodulin-binding site to the C-terminal domain of VAMP 2. (A) Residues 77–90 of rat VAMP 2 contain a 1–5-8–14 consensus motif for calcium-dependent calmodulin binding proposed by Rhoads and Friedberg (18). A comparison of VAMP homologues from rat (VAMP 3/cellubrevin), sea urchin (Strongylocentrotus purpuratus) cortical vesicles, aplysia (Aplysia californica), nematode (Caenorhabditis elegans), and yeast (Saccharomyces cerevisiae Snc1) illustrates evolutionary conservation of this motif. Basic residues within the motif are underlined. Two conserved lysines (+) C-terminal to the motif also contribute to calmodulin interactions (see Results). (B) The binding of GST-fusion proteins containing the sequences VAMP1–96, VAMP1–96 cleaved with TeTx light chain (i.e., VAMP1–76), VAMP77–90, and VAMP1–90 to calmodulin–agarose beads was assayed as in Fig. 1A. (C) Immunoisolated synaptic vesicles from rat brain were incubated with TeTx light chain (300 pM) in buffer containing 1 mM CaCl2, in the presence or absence of calmodulin (15 μM). Duplicate aliquots were removed immediately (0 min) or after a 30-min incubation. Samples were denatured and processed for Western blotting with an antibody directed against the N-terminal region of VAMP 2.
Inhibition of SNARE Complex Assembly by Calcium/Calmodulin.
Synaptic vesicle fusion involves assembly of a ternary syntaxin 1/SNAP-25/VAMP complex, and we therefore asked whether Ca2+/calmodulin binding to VAMP affects this process. Ternary complex assembly initially requires formation of a binary syntaxin 1/SNAP-25 complex, which constitutes a high-affinity VAMP-binding site (19). To monitor SNARE complex assembly, 35S-SNAP-25 was produced by translation in vitro in the presence of [35S]methionine. GST was removed from GST-syntaxin 1A1–261 by thrombin cleavage, and the resulting syntaxin fragment was purified and shown to lack intrinsic glutathione binding. Thus when 35S-SNAP-25 and syntaxin 1 are incubated with immobilized GST-VAMP1–96, ternary complex assembly can be quantified as radioactivity bound to glutathione beads via GST-VAMP1–96. When 35S-SNAP-25 alone was incubated with GST immobilized on glutathione beads, only background levels of radioactivity were adsorbed (Fig. 3A). In the presence of GST-VAMP, a slight increase was detected, presumably because of a weak VAMP/SNAP-25 binary interaction; however, addition of syntaxin 1 resulted in an additional 8-fold increase in bound 35S-SNAP-25 (Fig. 3A). Ternary core complexes form parallel helical bundles of high thermostability that can be assayed by their resistance to denaturation by SDS. Analysis of the complexes retained on glutathione beads by SDS/PAGE and autoradiography demonstrated that 35S-SNAP-25 assembled with syntaxin 1 and GST-VAMP to form a heterotrimer migrating at 120 kDa after SDS treatment at 37°C (upper arrow, Fig. 3B), which dissociated at 100°C. Therefore, this assay can be used to monitor SNARE complex formation. In the presence of Ca2+/calmodulin, an approximately 50% reduction in the amount of 35S-SNAP-25 trapped by glutathione beads was apparent (Fig. 3C), indicating that Ca2+-dependent calmodulin binding to VAMP inhibits core complex assembly. If Ca2+/calmodulin inhibits assembly by physically masking syntaxin 1- and/or SNAP-25-binding sites on VAMP, then formation of the stable SNARE complex in vitro should irreversibly occlude calmodulin binding. To test this prediction, 35S-SNAP-25/syntaxin 1/GST-VAMP1–96 complexes were formed, and the ability of calmodulin–agarose beads to trap preassembled complexes was tested. Fig. 3D demonstrates that 30% of the complexes were adsorbed by calmodulin–agarose in a Ca2+-dependent manner, taking as 100% the complexes bound to glutathione beads in parallel experiments. Cleavage of VAMP1–96 by TeTx before assembly eliminated the capacity of calmodulin–agarose to retain SNARE complexes (Fig. 3D). Thus Ca2+/calmodulin binding to VAMP77–94 reduces SNARE complex formation, but this sequence is still accessible for Ca2+/calmodulin binding after assembly, consistent with a noncompetitive inhibitory mechanism. The fact that ternary SNARE complexes were trapped by calmodulin–agarose demonstrates that Ca2+/calmodulin binding did not dissociate SNARE complexes preformed in vitro.
Figure 3.
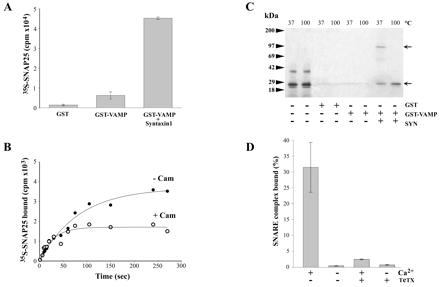
Inhibition of SNARE complex assembly by calcium/calmodulin. (A) In vitro translated 35S-SNAP-25 was incubated in the presence or absence of GST, GST-VAMP1–96, and untagged syntaxin 1A1–261 for 3 h at 4°C. Protein complexes were recovered on glutathione beads and, after washing, bound 35S-SNAP-25 was measured by β counting. Results are means ± SD, n = 3. (B) Samples prepared as in A were analyzed by SDS/PAGE and autoradiography after denaturation in SDS at 37°C or 100°C. Arrows indicate the migration of the trimeric core complex (upper arrow) and 35S-SNAP-25 (lower arrow). The radioactive band at about 40 kDa in the first two lanes is an unidentified translation product that did not interact with VAMP 2 or syntaxin 1A. (C) 35S-SNAP-25, GST-VAMP1–96, and syntaxin 1A1–261 were incubated at 4°C in a buffer containing 1 mM CaCl2, in the presence or absence of calmodulin (10 μM). At the indicated times, samples were removed, and bound 35S-SNAP-25 was evaluated by β counting. The curve is representative of three independent experiments. (D) SNARE assembly was performed as in A, except that complexes were recovered on calmodulin–agarose beads in the presence or absence of 1 mM CaCl2 and after pretreatment of GST-VAMP1–96 with TeTx light chain. 35S-SNAP-25 retained by calmodulin–agarose is shown as a percentage, taking the radioactivity recovered on glutathione beads as 100%, means + SD, n = 3.
Interactions of Phospholipids with the Calmodulin-Binding Site.
The Ca2+/calmodulin-binding site, located close to the transmembrane anchor of VAMP 2, contains a high density of positive charges and a triplet of aromatic residues (88YWW90). It has been suggested that this domain could interact with the phospholipid bilayer during membrane fusion (5), and interactions of acidic phospholipids with calmodulin-binding peptides in other proteins have been reported (20). We therefore examined the phospholipid-binding properties of VAMP 2 by SPR measurements. GST-VAMP constructs were immobilized on the sensorchip via anti-GST antibodies, and mixed PS/PC or pure PC liposomes of approximately 80 nm diameter were injected in the presence or absence of Ca2+. Mixed PS/PC liposomes were found to bind to GST-VAMP1–96, but the association was not Ca2+ dependent (Fig. 4A). Positive control experiments in similar conditions confirmed the Ca2+ dependency of the binding of PS/PC liposomes to the cytoplasmic domain of synaptotagmin I fused to GST (results not shown). When liposomes were injected in the presence of 50 μM VAMP77–94 peptide, the interaction was blocked. No binding to pure PC liposomes was detected (Fig. 4A). GST-VAMP77–90 that contains the calmodulin-binding motif displayed a similar PS/PC binding capacity to VAMP1–96. Cleavage of GST-VAMP1–96 with TeTx to remove the C-terminal residues 77–96 eliminated phospholipid binding. We next examined whether calmodulin and acidic phospholipid binding to VAMP were mutually exclusive. SPR experiments were performed by using immobilized calmodulin. GST-VAMP1–96 displayed binding to Ca2+/calmodulin; however, the interaction was strongly reduced when GST-VAMP1–96 was injected in the presence of PS/PC liposomes (Fig. 4B). Control injections of liposomes alone did not reveal direct interactions between calmodulin and PS/PC. Thus phospholipids block Ca2+-dependent association of VAMP with calmodulin by binding to the consensus motif VAMP77–90. Certain calmodulin- and phospholipid-binding peptides undergo transitions from random coil to α-helix in hydrophobic environment (20). Circular dichroism spectroscopy of the VAMP77–94 peptide by using trifluoroethanol to mime hydrophobicity demonstrated induction of two negative peaks at 208 and 222 nm and a large positive peak at shorter wavelengths, characteristic of increasing α-helical content (supplementary Fig. 5, www.pnas.org). This underlines the tendency of the C-terminal VAMP domain, adjacent to the synaptic vesicle membrane anchor, to form an α-helix in a hydrophobic environment. Thus phospholipid binding could promote a continuous α-helical structure from the presumably α-helical transmembrane region, through the calmodulin- and phospholipid-binding domain and into the superhelical twist.
Figure 4.
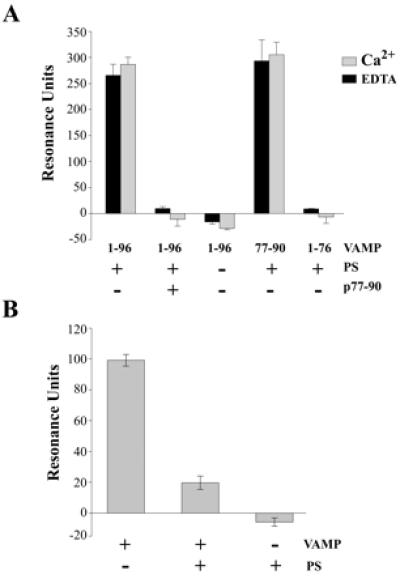
Interactions of phospholipids with the calmodulin-binding domain of VAMP 2. (A) GST-fusion proteins containing the indicated VAMP sequences were immobilized on an CM5 (Biacore) sensor chip coated with anti-GST antibodies. GST-VAMP1–76 was prepared from VAMP1–96 by TeTx cleavage. Liposomes containing 7.5 dipalmitoyl-PC: 2.5 dipalmitoyl-PS; weight: weight (PS+), or pure dipalmitoyl-PC (PS−), were diluted in running buffer and injected in the presence or absence of a synthetic peptide corresponding to VAMP 2 residues 77–94 (p77–94). Experiments were performed in the presence of 1 mM CaCl2 or 5 mM EDTA. Results are shown as means ± SD, n = 3. (B) Biotinylated calmodulin was immobilized on a streptavidin-coated sensorchip, and GST-VAMP1–96 was injected in the absence or presence of PC/PS liposomes. Control injections of liposomes were carried out in the absence of VAMP.
Discussion
Syntaxin 1 and SNAP-25, which are predominantly expressed at the presynaptic plasma membrane associate with VAMP 2, a synaptic vesicle protein to form a highly stable coiled-coil structure (5, 6). It has been suggested that the “zipping-up” of helical bundles, proceeding from the N terminus toward the C-terminal membrane anchors, drives the vesicular membrane toward the plasmalemma, promoting fusion. However, after membrane fusion, fully assembled heterotrimers are incorporated into a single lipid bilayer to form cis SNARE complexes. Cis complexes, possibly undergoing recycling, have been detected in the membrane of isolated synaptic vesicles and shown to be dissociated by N-ethylmaleimide sensitive fusion protein (16, 21).
Although synaptic exocytosis is triggered by Ca2+ influx, little is known about the molecular mechanisms by which Ca2+ regulates this cycle of events. We have now identified an evolutionarily conserved calmodulin- and phospholipid-binding domain between residues 77–94 of rat VAMP 2, which is also found in Nyv1p, the yeast v-SNARE involved in homotypic vacuolar fusion (22), a process mediated by Ca2+/calmodulin (11).
The Kd for the interaction of calmodulin with rat VAMP 2 was 500 nM. A wide range of calmodulin-binding affinities to target proteins has been reported. Whereas calcium-independent binding of calmodulin to neuromodulin (GAP 43) displays a Kd close to 1 μM, calcium-dependent interactions for many proteins are within the nanomolar range (18). Calcium-dependent interactions with caldesmon and α-fodrin that contain a 1–8-14 type B motif have Kds of 1 and 3 μM, respectively (18). Peptides from skeletal muscle light-chain kinase and nitric oxide synthase that constitute a 1–8-14 type A motif (i.e., 1–5-8–14) like that of VAMP displayed significantly higher affinity with Kds = 1 and 4 nM respectively (23, 24). Thus whereas the Kd for calmodulin binding to VAMP (500 nM) is within the general range of published interactions and consistent with cellular calmodulin concentrations, the affinity is relatively low compared with other 1–8-14 type A motifs. This may be because of differences in the positions of charged residues which also contribute to binding.
The calmodulin interaction site is located in a segment located immediately C-terminal to the TeTx or BoNT/B cleavage site (Q76-F77) and close to the transmembrane region. Calmodulin binding protected VAMP in isolated rat brain synaptic vesicles from TeTx action, consistent with the conclusion that this interaction occurs when native VAMP is membrane anchored. These findings may have implications for the mechanism of action of clostridial toxins. Proteolysis by TeTx and BoNT/B does not block the assembly of heterotrimeric SNARE complexes, and these toxins are thought to inhibit vesicle fusion simply by severing the link between the coiled-coil SNARE bundle and the VAMP membrane anchor (5, 25). If so, there is no obvious advantage to clipping any particular peptide bond. Thus it may be significant that both TeTx and BoNT/B cleave the peptide bond (Q76-F77) adjacent to the first residue (F77) of the calmodulin consensus motif and precisely separate the calmodulin/phospholipid interaction domain from the superhelical structure. However, the reasons why cleavage of this particular peptide bond may lead to a more efficient block of exocytosis are not apparent.
A pronounced basic charge distribution at the membrane-anchored end of the synaptic core complex suggests that during assembly, cumulative electrostatic potential may promote membrane fusion (5). Additional factors may intervene, including association with the membrane interface of the aromatic triplet (88YWW90) located in the C-terminal region of VAMP 2 (5). This triplet, flanked by basic residues, is part of the calmodulin/phospholipid-binding site, and our data suggest that calmodulin could mask a potential site for protein/membrane interaction during SNARE complex assembly or dissociation. Domains that interact with calmodulin and phospholipids in a mutually exclusive fashion have been described in other proteins, including GAP-43 (26), eNOS (20), the MARCKS protein (27), and the plasma membrane Ca2+-ATPase (28). In the Ca2+-ATPase, a phospholipid interaction domain constitutes a secondary calmodulin-binding site that is not directly implicated in pump activation by calmodulin. This secondary site binds calmodulin with lower affinity (Kd = 800 nM), comparable to that of VAMP77–94 (Kd = 500 nM) (28).
Our data support the view that calmodulin may mediate Ca2+-dependent regulation of interactions between vesicular VAMP and its protein and lipid partners. It is unlikely that calmodulin alone could constitute a Ca2+ sensor for exocytosis. Functional assays have defined Ca2+ receptors that trigger the fusion of synaptic vesicles at 10s to 100s of micromolar concentrations (29, 30), i.e., one to two orders of magnitude higher than the ranges in which Ca2+ binding to calmodulin occurs. Nevertheless, Ca2+/calmodulin binding to VAMP may constitute a step that is subsequently permissive for the downstream action of lower affinity calcium sensors such as synaptotagmin I, which dissociates from the SNARE complex in the 100 μM Ca2+ concentration range (16).
What are the implications of our observations in the context of the hypothesis that SNARE complex assembly drives membrane fusion (5, 7)? At first sight, our data demonstrating that Ca2+/calmodulin antagonizes core complex assembly are contradictory to reports that Ca2+ induces trans complex formation (9) and that calmodulin promotes a late step in vesicular fusion (11–13). However, our experiments monitor assembly of SNARE proteins lacking membrane anchors, probably providing a closer approximation to cis complex assembly in a single membrane plane, rather than the trans conformation in which additional mechanical constraints are imposed. We can speculate that calmodulin-mediated inhibition of cis complex formation in the plane of the synaptic vesicle membrane may promote trans SNARE interactions at the vesicle/plasma membrane interface. It is also possible that calmodulin displaces low-affinity cis interactions between phospholipids of the external vesicle leaflet and the C-terminal region of VAMP, subsequently allowing higher affinity trans interactions with the inner leaflet of the plasma membrane, which is enriched in acidic phospholipids. These issues must be addressed in the future by using purified SNARE proteins inserted into liposomes of defined phospholipid composition, approximating that of different subcellular compartments.
It may also be useful to consider an alternative hypothesis, namely that calmodulin could mediate Ca2+-dependent dissociation of trans SNARE complexes before vesicle fusion. Recent studies in both yeast and sea urchin eggs suggest that disruption of SNARE complexes does not subsequently preclude homotypic vesicular fusion, thus challenging the concept that complex assembly drives fusion in these systems (31, 32). Intriguingly, Ca2+ dissociates heterotrimeric SNARE complexes in cortical vesicles of sea urchin eggs (33). Although the precise molecular mechanism is not known, it may be significant that earlier reports have underlined a requirement for calmodulin in Ca2+-dependent exocytosis of cortical granules (34). Further investigation will be required both to examine whether calmodulin acts as an inhibitor of cis complex formation promoting trans complex assembly, or initiates trans SNARE complex disruption, and to determine the functional consequences on vesicle fusion.
Supplementary Material
Acknowledgments
We thank J. C. Mani and M. Pugnière for technical advice concerning Biacore experiments and Cecile Iborra for preparing the figures. Funding was provided by the Association Française contre les Myopathies and by a research grant from the Human Frontiers Science Program.
Abbreviations
- TBS
Tris-buffered saline
- BoNT
botulinum neurotoxin
- GST
glutathione S-transferase
- PS
dipalmitoyl-l-α-phosphatidyl-l-serine
- PC
dipalmitoyl-l-α-phosphatidyl-choline
- SNAP-25
synaptosomal associated protein of 25 kDa
- SNARE
soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor
- SPR
surface plasmon resonance
- TeTx
tetanus toxin
- VAMP
vesicle-associated membrane protein
References
- 1.Sollner T, Bennett M K, Whiteheart S W, Scheller R H, Rothman J E. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 2.Sollner T, Whiteheart S W, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman J E. Nature (London) 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 3.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta B R, Montecucco C. Nature (London) 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 4.Niemann H, Blasi J, Jahn R. Trends Cell Biol. 1994;4:179–185. doi: 10.1016/0962-8924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 5.Sutton R B, Fasshauer D, Jahn R, Brunger A T. Nature (London) 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 6.Poirier M A, Xiao W, Macosko J C, Chan C, Shin Y K, Bennett M K. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 7.Hanson P I, Heuser J E, Jahn R. Curr Opin Neurobiol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 8.Weber T, Zemelman B V, McNew J A, Westermann B, Gmachl M, Parlati F, Sollner T H, Rothman J E. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y A, Scales S J, Patel S M, Doung Y C, Scheller R H. Cell. 1999;97:165–174. doi: 10.1016/s0092-8674(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 10.Sudhof T C, Rizo J. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 11.Peters C, Mayer A. Nature. 1998;396:575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain L H, Roth D, Morgan A, Burgoyne R D. J Cell Biol. 1995;130:1063–1070. doi: 10.1083/jcb.130.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y A, Duvvuri V, Schulman H, Scheller R H. J Biol Chem. 1999;274:26469–26476. doi: 10.1074/jbc.274.37.26469. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Moutot N, Charvin N, Leveque C, Sato K, Nishiki T, Kozaki S, Takahashi M, Seagar M. J Biol Chem. 1996;271:6567–6570. doi: 10.1074/jbc.271.12.6567. [DOI] [PubMed] [Google Scholar]
- 15.Charvin N, LeVeque C, Walker D, Berton F, Raymond C, Kataoka M, Shoji-Kasai Y, Takahashi M, De Waard M, Seagar M J. EMBO J. 1997;16:4591–4596. doi: 10.1093/emboj/16.15.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leveque C, Boudier J A, Takahashi M, Seagar M. J Neurochem. 2000;74:367–374. doi: 10.1046/j.1471-4159.2000.0740367.x. [DOI] [PubMed] [Google Scholar]
- 17.Wagner H, Lissou A, Holzi J, Horammer L. J Lipid Res. 1962;3:117–180. [Google Scholar]
- 18.Rhoads A R, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 19.Pevsner J, Hsu S C, Braun J E, Calakos N, Ting A E, Bennett M K, Scheller R H. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 20.Matsubara M, Titani K, Taniguchi H. Biochemistry. 1996;35:14651–14658. doi: 10.1021/bi9613988. [DOI] [PubMed] [Google Scholar]
- 21.Otto H, Hanson P I, Jahn R. Proc Natl Acad Sci USA. 1997;94:6197–6201. doi: 10.1073/pnas.94.12.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nichols B J, Ungermann C, Pelham H R, Wickner W T, Haas A. Nature (London) 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- 23.Edelman A M, Takio K, Blumenthal D K, Hansen R S, Walsh K A, Titani K, Krebs E G. J Biol Chem. 1985;260:11275–11285. [PubMed] [Google Scholar]
- 24.Venema R C, Sayegh H S, Kent J D, Harrison D G. J Biol Chem. 1996;271:6435–6440. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof T C, Niemann H. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi N, Matsubara M, Titani K, Taniguchi H. J Biol Chem. 1997;272:7639–7645. doi: 10.1074/jbc.272.12.7639. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Shishido T, Jiang X, Aderem A, McLaughlin S. J Biol Chem. 1994;269:28214–28219. [PubMed] [Google Scholar]
- 28.Filoteo A G, Enyedi A, Penniston J T. J Biol Chem. 1992;267:11800–11805. [PubMed] [Google Scholar]
- 29.Heidelberger R, Heinemann C, Neher E, Matthews G. Nature (London) 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 30.Tandon A, Bannykh S, Kowalchyk J A, Banerjee A, Martin T F, Balch W E. Neuron. 1998;21:147–154. doi: 10.1016/s0896-6273(00)80522-x. [DOI] [PubMed] [Google Scholar]
- 31.Coorssen J R, Blank P S, Tahara M, Zimmerberg J. J Cell Biol. 1998;143:1845–1857. doi: 10.1083/jcb.143.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ungermann C, Sato K, Wickner W. Nature (London) 1998;396:543–548. doi: 10.1038/25069. [DOI] [PubMed] [Google Scholar]
- 33.Tahara M, Coorssen J R, Timmers K, Blank P S, Whalley T, Scheller R, Zimmerberg J. J Biol Chem. 1998;273:33667–33673. doi: 10.1074/jbc.273.50.33667. [DOI] [PubMed] [Google Scholar]
- 34.Steinhardt R A, Alderton J M. Nature (London) 1982;295:154–155. doi: 10.1038/295154a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}