

Abstract
Interactions with magnesium (Mg2+) ions are essential for RNA folding and function. The locations and function of bound Mg2+ ions are difficult to characterize both experimentally and computationally. In particular, the P456 domain of the Tetrahymena thermophila group I intron, and a 58 nt 23s rRNA from Escherichia coli have been important systems for studying the role of Mg2+ binding in RNA, but characteristics of all the binding sites remain unclear. We therefore investigated the Mg2+ binding capabilities of these RNA systems using a computational approach to identify and further characterize their Mg2+ binding sites. The approach is based on the FEATURE algorithm, reported previously for microenvironment analysis of protein functional sites. We have determined novel physicochemical descriptions of site-bound and diffusely bound Mg2+ ions in RNA that are useful for prediction. Electrostatic calculations using the Non-Linear Poisson Boltzmann (NLPB) equation provided further evidence for the locations of site-bound ions. We confirmed the locations of experimentally determined sites and further differentiated between classes of ion binding. We also identified potentially important, high scoring sites in the group I intron that are not currently annotated as Mg2+ binding sites. We note their potential function and believe they deserve experimental follow-up.
INTRODUCTION
The importance of RNA and Mg2+
RNA molecules form complex three-dimensional folds that ultimately determine their biological role. Metal ions, particularly Mg2+, are crucial to stabilizing RNA structure and hence, its function. They counter the negatively charged phosphate backbone and participate in the catalytic reactions of some ribozymes (1–3). Reliably locating these binding sites is important for understanding RNA folding, catalysis and ultimately in drug development (4–10).
Two main modes of Mg2+ binding have been proposed (11). In diffuse binding, fully hydrated Mg2+ ions interact with RNA through long-range electrostatic interactions, while site-bound Mg2+ ions are bound directly via anionic ligands from the RNA. Site binding is energetically costly, as it requires 10– 30 kcal/mol to remove a single water molecule from the outer and inner hydration layers of an ion, respectively, and therefore requires a large electrostatic contribution to binding (12).
Locating Mg2+ binding sites
Mg2+ binding sites in RNA are typically identified through X-ray crystallography or NMR methods. However, electron densities of metal ions can be difficult to distinguish and NMR techniques have only just recently given direct evidence of ion binding in RNA (13). Many sites can be easily mislabeled as waters or may be missing entirely from fully refined crystal structures or solution structures of RNA, and must be located through further experimentation. Ion binding sites of high affinity can be probed using cleavage experiments, where hydrolytic or oxidative metals cleave surrounding RNA (14). Additionally, mutation experiments can create the loss of a functional Mg2+ binding site (15,16). Another approach involves rescuing phosphorothioate effects in the presence of thiophilic metal ions such as Cd2+ or Mn2+ (17,18). This method can detect site-bound ions, as they require direct coordination between metal ions and phosphoryl oxygens in the RNA. If a structure is known, computational methods may be useful for predicting and characterizing Mg2+ binding sites in RNA structures and can aid crystallographers in distinguishing ion densities.
A straightforward and computationally fast algorithm is valence screening for metal ions (19,20). This method essentially calculates the valence potential of oxygens within a defined radius along a fine three-dimensional grid laid over a molecular structure. A more rigorous approach would be to use electrostatic calculations, where solutions to the Poisson–Boltzmann equation, or in the case of nucleic acids, the non-linear Poisson–Boltzmann (NLPB) equation, can be used to identify negative potential on the surface of an RNA molecule where ions most likely aggregate (4,21). In addition to calculating electrostatic potentials, Brownian-Dynamics (BD) simulations can be used to further elucidate ion binding sites in RNA folds (22–24). However, none of these computational approaches are suitable for analyzing large complexes without significant calculation (25).
A novel approach in the context of structural genomics
The Protein Data Bank (PDB; http://www.rcsb.org/pdb/) now includes over 1900 structures of nucleic acids, ranging from simple helices to complex, entangled, assemblies of RNA and protein (26). With advances in structure determination techniques, and the many structural genomic efforts currently underway, the number of solved RNA structures and complexes is rapidly increasing (see http://www.rcsb.org/pdb/strucgen.htmlWorldwide for projects worldwide) (27,28). A major goal of structural genomics is to classify all the possible molecular folds that exist in nature. Although, the number of known RNA structural motifs is less than those for proteins, structures of RNA–protein complexes, such as the ribosome, are providing numerous examples of these motifs (29–34), and causing increased interest in computational methods for analysis of RNA structural motifs (35,36). Computational approaches that can scale to analyze databases of RNA structures, including large assemblages, will be able to mine the structural data and provide examples of these motifs.
FEATURE was originally developed for the analysis of protein microenvironments (37). The FEATURE method uses a supervised learning algorithm to recognize sites from a training set of sites and non-sites. Details of the algorithm and its performance characteristics have been published elsewhere (37–43). Site models have been created for different types of functional sites in proteins, such as various metal binding sites, serine protease active sites and ATP binding sites (40,41). These sites were detected with high sensitivity and specificity. Until now, FEATURE has not been applied to analyzing nucleic acid structures.
Although Mg2+ binding is driven mostly by electrostatics, it has been shown that non-coulombic properties can influence the electrostatic field (44). We believe that Mg2+ binding sites can be differentiated not only by charge alone, but also by the biochemical and structural properties surrounding the binding site. We used FEATURE to study the differences between site-bound and diffusely bound Mg2+ ions in complex RNA folds. To confirm our hypothesis that sites could be differentiated by these microenvironments, we used our statistical models to identify sites in well-characterized RNA systems where both types of binding occur. These RNA structures were not included in the training sets and served as an independent test set. FEATURE analysis was compared with the results of valence and electrostatic calculations on these various systems.
MATERIALS AND METHODS
RNA modified FEATURE
FEATURE was originally developed and implemented for analyzing protein structures (37,41). We extended the property set to include RNA biochemical and structural properties. The resulting properties are listed in Figure 1.
Figure 1.

The microenvironments of site-bound and diffusely bound Mg2+ ions in RNA. Physicochemical properties of significance (at P = 0.05) are listed down the left-most column. Increasing volumes move away from the center of the site and are 2 Å in width. Colored squares represent property-volume pairs of statistical significance (see legend). Properties that were statistically insignificant between sites and non-sites for both models are not shown. These include: atom-based properties: Sulfur, Other, Positive Charge; residue-based properties: T, Sugar Pucker C1′ endo, Sugar Pucker C4′ endo; secondary structure based properties: Adjacent Base Pair, A-Form Helix, X-Form Helix, Strand is Other; and tertiary structure based properties: Loop–Strand, Helix–Strand, Bulge–Loop, Bulge–Helix, Internal Loop–Bulge, Internal Loop–Loop, Other and Pseudoknot.
To create the diffuse binding model, we defined sites as Mg2+ ions not within 3 Å of any RNA atom. The atomic density surrounding each site was calculated and averaged over the total number of sites. We generated non-sites by randomly choosing grid points laid over an RNA structure. Only grid points not within 10 Å of a Mg2+ ion, and whose radial density was within one standard deviation of the average atomic density of the sites were included as non-sites. These selection criteria prevent bias for sites or non-sites based on atomic density alone. Mg2+ ions within 3 Å of any three RNA ligands (site bound) were also included as non-sites. A total of 126 sites and 334 non-sites were used to generate the diffuse binding model. For the site-bound model, Mg2+ ions within 3 Å of any three RNA ligands were considered as sites, while Mg2+ ions not within 3 Å of an RNA ligand were used as non-sites. The randomly generated non-sites from the diffuse model were also used as non-sites for the site-bound model. A total of 30 sites and 330 non-sites were used to generate the site-bound model. The structures used for training are listed in Table 1. FEATURE source code and documentation is available at http://feature.stanford.edu/. A web interface for FEATURE (http://feature.stanford.edu/webfeature/) (45) is available for real-time scanning and visualization of functional sites, including Mg2+ binding sites, in both RNA and protein structures.
Table 1. A list of the structures used for training site-bound and diffusely bound models.

For scans of the group I intron, sites from 1gid and 1hr2 were removed from the training set.
FEATURE results consist of two separate outputs. The result of training is the site model, visualized as a two-dimensional plot of properties against volumes in Figure 1. Statistical significance is determined using the Wilcoxon rank-sum test. Statistically significant property-volume pairs (at P = .05) are colored blue, while statistically deficient pairs are colored green. Insignificant pairs are left blank. The second output is the result of scanning a query structure for sites. Detected hits are scored using a log-odds scoring function (41) as shown below (Equation 1); where higher scoring hits are more likely to be a site of interest.
![]()
Score cut-offs for the site-bound and diffusely bound models were determined using a ROC (Receiver Operating Curve) plot of 1-specificity against sensitivity; where sensitivity and specificity were calculated by FEATURE’s ability to detect crystallographically bound Mg2+ ions in a set of RNA structures. Sensitivity provided a measure of the method’s ability to identify all crystallographical Mg2+ ions, while specificity provided a measure of the false positive rate. As crystallographical Mg2+ ions are not comprehensively determined, we opted for score cut-offs that gave a high specificity at the loss of sensitivity so as to assure the reliability of predicted sites. The ROC plot is shown in Figure 2. Details of the statistical model, learning method, and inference method are available in previous publications (37,38,40–43).
Figure 2.

ROC curve for FEATURE detection of site-bound and diffusely bound Mg2+ ions in RNA were used to determine score cut-offs. Higher sensitivity and specificity is achieved for site-bound ions as their locations are fewer, more localized, and thus, more easily detected in RNA. The score cut-offs used in our analysis are labeled on the curve in relation to the resulting sensitivity and specificity of detection. Cut-offs were chosen to maximize specificity over sensitivity, so as to assure reliable detection of sites.
MC-Annotate
We used a modified version of MC-Annotate (45), to calculate secondary and tertiary RNA structural properties. MC-Annotate takes RNA pdb files as input and generates a list of RNA secondary and tertiary structure properties. These properties were translated into a FEATURE readable format and then assigned to each base within an RNA molecule. For analysis of proteins, FEATURE previously used DSSP (46) files to calculate properties such as solvent accessibility, mobility and secondary structural properties. FEATURE uses a proprietary format of the MC-Annotate output to count the secondary and tertiary structural properties. The current implementation of FEATURE can read both DSSP and our formatted version of MC-Annotate files in order to support complexes of both RNA and proteins. MC-Annotate is accessible at http://www-lbit.iro.umontreal.ca/mcannotate/.
ViewFEATURE
The results of FEATURE scans were analyzed in ViewFEATURE, a visualization tool developed previously (39). ViewFEATURE allows the user to view the FEATURE hits as spheres within the RNA structure of interest. Radial shells, sites and non-sites, as well as the properties of interest, can be visualized with an interactive graphical user interface. ViewFEATURE is available as an extension to the molecular modeling tool, Chimera (47). The ViewFEATURE source code can be downloaded from http://feature.stanford.edu/. Chimera is available at http://www.cgl.ucsf.edu/chimera/.
Electrostatics
The non-linear solution for the Poisson–Boltzmann equation was used to calculate the electrostatic potential of each RNA system. The specific program we used, Qnifft, was downloaded from http://pylelab.org/research/computation/electrostatics/. Calculations were performed as published previously (21).
Valence calculations
For valence calculations (19), parameters were varied to try and predict both site-bound and diffusely bound ions. For site-bound ions, grid spacing was varied from 0.1 to 0.15 Å, ligation sphere from 3 to 4 Å, and number of ligands from one to three. For diffusely bound ions, the ligation sphere was increased to 6–7 Å to simulate a hydration layer. Valence source code was downloaded from http://biochem.wustl.edu/~enrico/vale.htm/.
RESULTS
In order to support analysis of RNA structures, in addition to proteins, we selected a set of physicochemical and structural properties that pertain to either or both types of molecules, which resulted in a total of 112 of properties. These properties provided descriptions for the statistical models of the microenvironments surrounding site-bound and diffusely bound Mg2+ ions, and are summarized in Figure 1.
Our statistical models reveal, in addition to charge, the major differences between the microenvironments of site-bound and diffusely bound Mg2+ ions. We found that diffusely bound Mg2+ ions are coordinated by a larger repertoire of potential ligands than are site-bound ions. However, site-bound Mg2+ ions are more often located near ‘irregular’ or non-helical RNA tertiary interactions. To verify these structural motifs, we used FEATURE to identify binding sites in a number of RNA systems not included in the training set. We compared FEATURE with two other computational methods for ion binding site detection and found that often FEATURE could better pinpoint crystallographically observed Mg2+ binding sites. Because of their unique environment, site-bound Mg2+ ions were easier to positively predict than diffusely bound Mg2+ ions in complex RNA systems.
Microenvironment analysis of Mg2+ binding
To differentiate between site-bound and diffusely bound Mg2+ ions, we computed distances between crystallographically observed Mg2+ ions and the surrounding RNA. For diffuse binding, we trained FEATURE on Mg2+ ions that were either fully hydrated or not within 3 Å of any RNA-derived atom. Ions that were within 3 Å of at least three possible coordinating ligands, such as phosphate oxygens or amine nitrogens, were considered site-bound. In high-resolution structures (∼2 Å), inner-sphere contacts with the ion can be observed, as in the 50s ribosomal subunit (33). Site-bound Mg2+ ions as well as randomly chosen non-sites served as controls (see Materials and Methods for description of how these non-sites were generated). For the site-bound model, diffusely bound Mg2+ ions and randomly generated non-sites served as controls. The resulting statistical models revealed that biochemical and structural properties, in addition to charge, contribute to differentiating between site-bound and diffusely bound Mg2+ ions. The site-bound and diffusely bound models are plotted in Figure 1 as property-volume pairs of statistical significance.
Mg2+ binding site detection
To detect Mg2+ binding sites based on their microenvironments, we used FEATURE to identify and differentiate between sites in simple and complex RNA folds. The P5b stem–loop (1ajf) (48) and P456 domain (1gid and 1hr2) (49,50) from the Tetrahymena thermophila group I intron, and a 58 nt sequence from Escherichia coli and Bacillus stearothermophilus 23s rRNA (1qa6 and 1hc8) (51,52) provided examples of known, important metal binding sites in RNA. We performed electrostatic and valence calculations on these structures and compared results with those of FEATURE. Previous studies differentiated between the modes of Mg2+ binding in these structures (4,21). In our analyses, we used these examples of site-bound and diffusely bound Mg2+ ions as test cases and found that we were able to identify both types of sites occurring in simple (P5b stem–loop) and complex systems (23s rRNA and group I intron). Table 2 summarizes the results of the various computational methods for Mg2+ binding site prediction on the test structures. In most cases, FEATURE identified the crystallographically bound ion more precisely than the other methods. Figures 3–5 display the results of these predictions superimposed upon the RNA test structures.
Table 2. A list of the cyrstallographical Mg2+ ions in the group I intron and 23s rRNA for which FEATURE was able to distinguish between site-bound and diffusely bound sites.

The first three columns, from left, list the structures and their PDB identifiers. The fourth column lists crystallographical Mg2+ ions as labeled in their respective structures. Site-bound ions are labeled with a single asterisk. Columns 5 through to 8 describe the FEATURE hits near the crystallographically bound ion. Column 5 provides the FEATURE hit’s rank, by score, with respect to the total number of hits above the defined cut-offs of 50 and 35 for the site-bound and diffusely bound models, respectively. Column 6 has the percentile rank of the hit’s score compared with the hits above cut-off. Column 7 lists the score of the hit while column 8 gives the hit’s distance to the crystallographical Mg2+ ion. Columns 9 and 10 provide the rank and percentile, with respect to all detected locations above cut-off, for valence calculations. The values in columns 9 and 10 provide a measure of comparison for FEATURE’s values in columns 5 and 6. Valence calculations were not able to detect many of the crystallographical sites, while those that were detected were below the cut-off of 1.8. Column 10 shows the valence score, above cut-off, of a detected location, while column 11 provides its distance to the crystallographical ion. The highest scoring valence positions, as labeled in the legend, did not agree with electrostatics and FEATURE. As electrostatics calculations do not provide single locations for potential sites, column 12 refers to the figures for a visual comparison of the results of all three methods. B/C, below cutoff; ***, distance to nearest site-bound ion; **, as labelled in PDB; *, site bound ion; ∧, highest valence points, not near any crystallographically bound magnesium.
Figure 3.

The results of FEATURE detection for site-bound Mg2+ ions (a), valence calculations (b) and electrostatic calculations (c) on the P456 domain of the Tetrahymena group I intron. Crystallographically bound Mg2+ ions are shown as yellow spheres. FEATURE successfully detects [red spheres in (a)] the well-known ion binding core of the intron. Valence hits [blue spheres in (b)] do not correlate well with the ion binding core while electrostatics (c), in agreement with FEATURE, detects this site as the most negative potential in the molecule.
Figure 5.

The results of FEATURE detection for diffusely bound Mg2+ ions (a), valence calculations (b) and electrostatic calculations (c) on the P5b stem–loop of the group I intron. The crystallographically determined cobalt hexamine, located at the center of the stem–loop is displayed in yellow in (a) and (b). Cobalt hexamine is representative of diffuse ion binding in RNA. Feature’s hits [red spheres in (a)] above cut-off nearly superimpose the cobalt hexamine. Highest valence values [blue spheres in (b)] range from 1.0–1.2 and miss the diffusely bound ion. Electrostatics calculations (c) show a negatively charged pocket (red = approximately –20 kT/e) where the diffusely bound ion locates.
Predicted sites in the Tetrahymena group I intron
We predicted two binding sites in the P456 domain of the group I intron. The hits at the sites and their potential coordinating ligands are highlighted in Figure 6 and labeled as ‘Site 1’ and ‘Site 2’. Table 3 contains the coordinates and scores of the FEATURE hits. Site 1 was observed using both the site-bound and diffusely bound models for Mg2+ binding. Site 2 was seen only using the diffusely bound model. Although the scores of the hits were just above the defined cut-off, they nevertheless ranked higher than other sites for crystallographically observed Mg2+ ions. Interestingly, the hits at Site 1 overlap with a crystallographically observed water molecule in 1hr2.
Figure 6.
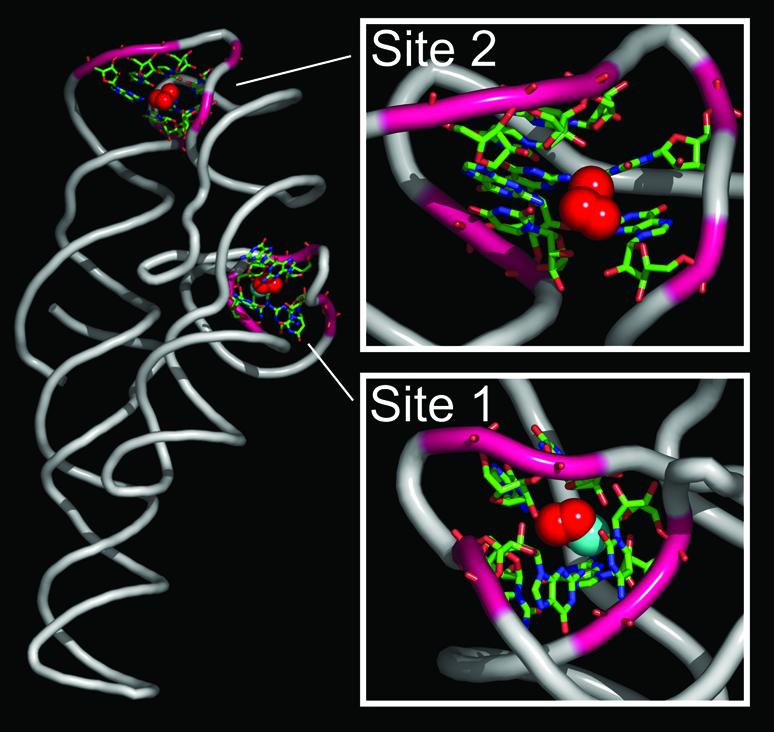
Two predicted Mg2+ binding sites in the group I intron. FEATURE hits (red spheres) at Site 1 overlap the crystallographically bound water (cyan colored sphere, WAT 123 in PDB ID 1hr2). Site 1 is potentially site-bound (micro view of Site 1 is rotated 180 degrees to provide a better view of the site). Residues that could potentially participate in coordination of an ion are displayed as sticks. Site 2 shows a potential diffusely bound Mg2+ site. Atoms are colored by type: C = green, N = blue, O = red.
Table 3. Coordinates and scores of predicted Mg2+ binding sites.

DISCUSSION
Statistical models for Mg2+ binding
A major advantage FEATURE has over both electrostatic and valence calculations is that FEATURE reveals detailed statistical differences of the physicochemical properties between the two site models for Mg2+ binding in RNA. At a significance of P = 0.05, the following description of these statistical differences can also be visualized in Figure 1. A major difference between the site models for both types of Mg2+ binding was the presence of most of the atom-derived properties in the first two shells (0–4 Å) in the site-bound model versus a lack thereof in the diffuse model. The hydration layer surrounding diffusely bound Mg2+ may explain this result. Diffusely bound Mg2+ ions tend to sit in large pockets or grooves on the surface of the RNA. Whereas site-bound Mg2+ ions locate in tighter, more electronegative pockets where potential coordinating ligands can directly bind the ion, and penetrate the surrounding layer of water molecules (53). Our results confirm the notion that site binding occurs in highly electronegative pockets. In contrast to the diffuse binding model, many of the significant properties in the site bound model occur within the innermost 4 Å of a site. Specifically, phosphate groups, which are most often the coordinating ligand to Mg2+ ions, are significant from 0 to 8 Å. Also, a significance of oxygen atoms continuously surround the Mg2+ ion from 2 to 10 Å. Hydroxyl groups are significant at 4 Å and extend to 10 Å. Our site-bound model shows other potential ligands, such as amines and carbonyls, occurring only in the outermost shell from 8 to 10 Å, suggesting that phosphates and sometimes hydroxyl groups are the predominant ligand donors for site-bound Mg2+ ions. Negative charge is significant from 2 to 4 Å and then from 6 to 10 Å within the site model. This is most likely due to the fact that negatively charged atoms that contribute to the site-bound pocket are covalently bound to non-polar atoms, such as carbons. This creates a binding pocket in the 2–4 Å layer surrounded immediately by a non-polar layer from 4 to 6 Å. This contrasts the diffuse model, where negative charge only first appears significant outside 4 Å from a Mg2+ ion.
Of all the bases, adenosines were the most significant from 2 to 10 Å in the site-bound model. This suggests that adenosines are highly prevalent near site-bound Mg2+ ions. This may be explained by adenosine, of all the bases, having the most number of negatively charged atoms that could behave as ligands. Another explanation is the regular participation of adenosines in irregular RNA conformations that cause the tight twisting of the phosphate backbone, such as in the A-minor motif, as seen in the Tetrahymena group I intron. In this motif, flipped adenosines are stabilized through the interaction with the minor groove of an adjacent helix and inner sphere contacts of phosphate oxygens with a Mg2+ ion. This contrasts the diffusely bound model where adenosines had no significant role in the site and was deficient in the first two shells (0–4 Å). Instead, uridines and guanosines were significant in the outer two to three shells of the diffusely bound model. As non-Watson–Crick base pairs were also significant in the outer two shells, it may be that this significance of G’s and U’s are actually base paired in a non-canonical fashion. Interestingly, it has been described that the 5′-UG/GU sequence is the most common and stable among non-Watson–Crick pairs, and that GU tandem pairs provide potential metal binding sites with a unique structural environment (54). This GU tandem metal binding motif is seen in the group I intron as well as other smaller RNA structures (48,49). Our analysis provides further evidence of GU tandems providing locations for ion binding.
Finally, many of the properties that might be associated with irregular, non-helical, RNA structure were significant from 2 to 10 Å in the site-bound model. These properties included base conformation in syn, non-WC base pairing, base triples, non-adjacent base stacking, single-stranded RNA, and many tertiary RNA interactions. These results suggests that irregular RNA conformations most likely generate regions where site-bound Mg2+ ions would be needed to counteract highly negative pockets formed by the twisting RNA backbone and bases. This is seen in both the Tetrahymena group I intron and Escherichia coli 23s rRNA where the phosphate backbone forms a tight kink and becomes buried beneath the surface of the RNA, thus requiring a site-bound Mg2+ to stabilize the fold (see Figs 3 and 4).
Figure 4.

FEATURE detects both site-bound (a) and diffusely bound (b) ions in the E.coli 58 nucleotide 23s rRNA. Crystallographically bound Mg2+ ions are shown as yellow spheres. FEATURE also detects the K+ ion (orange sphere), suggested to help stabilize the buried RNA backbone (52). Valence hits [values 1.6–1.7 as blue spheres in (c)] are observed in the site-bound region, but are below cut-off. Electrostatic calculations (d) reveal the highly negative potential (red surface color) contributing to the binding of a site-bound ion near the buried RNA backbone.
FEATURE site detection
The P456 domain from the group I intron, and the 58 nt 23s rRNA provided well-characterized examples of known site-bound Mg2+s (see Table 2). In all cases, using the site-bound model, FEATURE was able to locate the site-bound Mg2+ ions. The top scoring hits, above the cut-off, were always located within several Å of the crystallographically bound Mg2+ and in most cases overlapped the documented site-bound ion. In all these examples, the highest scoring hits were well above the cut-off, which gave us confidence in predicting those locations as site-bound. Lower scoring hits also located near the true binding site. In theory, site-bound locations should be easier to predict as their microenvironments are somewhat unique compared with diffuse binding sites, as seen in the statistical models.
Diffuse sites have been described to be more pervasive throughout RNA structure and bind in grooves and pockets on the surface of the RNA. Although this makes their binding sites more difficult to pinpoint to a single location, diffuse binding sites have been observed in crystal structures, such as those in the aforementioned structures, as well as in less complex systems such as in the P5b stem–loop, hairpin ribozyme, lead-dependent ribozyme and 7s RNA of human SRP. These sites might be described as more ‘specific’ than most diffusely bound ions involved in counter ion effects, yet not quite site-bound (by definition) since they do not make contact with non-water ligands. Using the diffusely bound model, FEATURE was able to locate diffuse sites in the aforementioned test structures. Because FEATURE was trained on diffuse sites in crystal structures, it should be successful in predicting diffuse sites to single locations in query structures. In the case of structures containing site-bound Mg2+ ions, the diffuse model also identified those locations as high-scoring sites.
Comparison of methods for Mg2+ site prediction
P456 domain of Tetrahymena group I intron. Figure 3 shows the results of a FEATURE analysis, as well as valence and electrostatics calculations for the P456 domain of the Tetrahymena group I intron. Near the center of the molecule is the well-known ion binding core, implicated in folding, stability and function of this ribozyme (2). The electrostatic potential is highly negative over this region of the RNA, where the tight kink in the phosphate backbone results in the placement of many electronegative groups near one another. The Mg2+ ions observed in the crystal structure have an important role in countering the negative potential and stabilizing the fold. FEATURE’s top hits, above the score cut-off, also lie in this region, and nearly superimpose the experimentally observed Mg2+ ions. In contrast, we found that valence calculations did not perform as well in picking out site-bound Mg2+ ions. The highest valence hits (valence values of 2.0–2.1) above the defined cut-off of 1.8 (19) were not located at the ion binding core of the group I intron, in disagreement with electrostatic and FEATURE calculations. Some valence hits above the cut-off did locate near the ion binding core, but other hits of the same value also scattered over the surface of the molecule without much correlation with the crystallographically bound ions, electrostatic and FEATURE predictions. Although valence calculations should theoretically work on RNA structures, we show that inclusion of other properties in addition to the coordinating ligands can improve ion binding site detection.
The two predicted binding sites we observed in the group I intron had scores just above the defined cut-off for both the site-bound and diffusely bound model. Our results suggest that a partially site-bound ion may exist at Site 1, while more diffuse ion binding interactions might take place at Site 2 (see Fig. 6). Valence calculations detected hits at Site 1 (valence values 1.4–1.6), but were below the required cut-off. Valence hits were not detected at Site 2. Electrostatics calculations show negative potential at both locations, but at cut-offs too low to locate a specific binding site. Using the site-bound model, we observed that Site 1 overlaps a crystallographically observed water. Visual inspection of Site 1 shows that the potential site is surrounded by three 2′ hydroxyl groups from the ribose groups of three nearby guanosines that could act as potential ligands to a site-bound ion. Site 2, on the other hand, was only observed when using the diffusely bound model. As in Site 1, 2′ hydroxyl groups from nearby ribose groups may also be the major coordinating ligands in diffuse interactions with a bound ion at Site 2.
23s rRNA
Figure 4 shows the results of the same calculations on the 23s rRNA. All the known site-bound Mg2+ ions are successfully detected by both electrostatics and FEATURE. Diffusely bound Mg2+ ions were also detected by FEATURE. However, FEATURE missed two of the diffuse binding sites because the cut-off was set high enough to maintain a reasonable false positive rate. It was more difficult to pinpoint diffuse binding sites using electrostatics calculations since the RNA surface is so negatively charged. As the kT/e cut-off was moved towards zero, the entire surface of the RNA molecule quickly became covered by negative potential and describes more of a distribution in which binding could occur. In the case of the 23s rRNA, electrostatics had a harder time detecting the specific locations of more diffusely bound Mg2+ ions on the whole RNA structure.
P5b stem–loop
Figure 5 shows our binding site analysis of the P5b stem–loop, a simple RNA fold. The crystal structure reveals a single cobalt hexamine, representative of a diffusely bound Mg2+ ion. Both electrostatic calculations and valence calculations only give general indications of where diffusely bound ions can be observed. In the case of valence calculations, sites of low valence (values of 1.1–1.2) were detected along the phosphate backbone, while the crystallographically bound site was missed completely. Theoretically, in solution, diffusely bound Mg2+ ions can migrate through deep grooves and along the phosphate backbone in RNA (4). At a cut-off around 10–20 kT/e, characteristic of diffuse ion binding, electrostatic calculations reveal that most of the major groove and phosphate backbone are regions where diffusely bound Mg2+ could migrate. However, FEATURE analysis of this RNA molecule predicts a highly localized binding site, precisely at the location of the crystallographically observed ion. FEATURE’s ability to pinpoint binding sites is most likely due to the fact that our training set consists of sites from crystal structures. It is therefore more likely to pick out sites that could potentially be observed in actual crystal structures versus those in solution. Thus we find that the computational methods may be complementary to each other as they provide different pieces of evidence towards predicting binding sites.
FEATURE is complementary to other computational methods for predicting Mg2+ binding sites in RNA. FEATURE provides statistical evidence for ion binding in crystal structures, whereas electrostatics and valence calculations provide physical quantification of the electrostatic potential of regions within the molecule. Together these methods can provide complementary evidence for site binding of Mg2+. We have analyzed RNA systems whose site-bound and diffusely bound Mg2+ binding sites are well characterized. FEATURE was successful at detecting both site-bound and diffusely bound Mg2+ ions. We find that site-bound ions are easier to detect because of their unique microenvironment. Such is the case in both the E.coli 23s rRNA and the P456 domain from the Tetrahymena group I intron. In simple RNA folds, such as the P5b stem–loop, FEATURE is successful at detecting diffuse Mg2+ binding sites and locates them to specific areas, whereas the other techniques give more general descriptions of ion binding. We also report the observation of two predicted Mg2+ binding sites in the P456 domain of the Tetrahymena group I intron; one site-bound and one diffusely bound. Presently, we do not report overall sensitivity and specificity for FEATURE’s performance, as it is unclear as to the number of missing or mislabeled Mg2+ ions in known RNA crystal structures. In other words, there lacks a standard for statistical comparison. As the number of known Mg2+ ions in RNA crystal structures increases, and as structure determination methods improve, we will be able to report better performance statistics for FEATURE. Since FEATURE runs in linear time, we propose that FEATURE be used as a screening tool for Mg2+ binding site detection. High scoring hits can be further explored with rigorous NLPB calculations. As structure determination methods improve, the types of RNA structures and complexes being solved become increasingly sophisticated. FEATURE lends itself well to the analysis of large complexes, such as the ribosome, where there are thousands of interactions. Once site-models are built, databases can be quickly screened for functional sites (38). Computational methods, like FEATURE, provide a high level annotation of function to molecular structures, and are needed as structural genomics projects provide a wealth of structural data. Finally, as the importance of RNA as a tool and target for drug discovery increases, our approach provides a foundation for the analysis of other RNA functional sites, such as RNA–RNA, RNA–protein and RNA–small molecule interactions.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Mike Liang, Dr Sean Mooney and Dr Andrew Bogan for helpful discussions on this work and manuscript. We also thank Dr Francois Major and Dr Patrick Gendron for providing a modified version of MC-Annotate. This work is supported by NIH Grants LM-05652 and LM-06422 (R.B. Altman P.I.). D.R.B. is supported by NIH 2 R25 GM056847-04 and is a PhD candidate in the Graduate Program in Biological and Medical Informatics at the University of California, San Francisco.
REFERENCES
- 1.Hanna R. and Doudna,J.A. (2000) Metal ions in ribozyme folding and catalysis. Curr. Opin. Chem. Biol., 4, 166–170. [DOI] [PubMed] [Google Scholar]
- 2.Cate J.H., Hanna,R.L. and Doudna,J.A. (1997) A magnesium ion core at the heart of a ribozyme domain. Nature Struct. Biol., 4, 553–558. [DOI] [PubMed] [Google Scholar]
- 3.Brannvall M. and Kirsebom,L.A. (2001) Metal ion cooperativity in ribozyme cleavage of RNA. Proc. Natl Acad. Sci. USA, 98, 12943–12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misra V.K. and Draper,D.E. (2002) The linkage between magnesium binding and RNA folding. J. Mol. Biol., 317, 507–521. [DOI] [PubMed] [Google Scholar]
- 5.Shan S., Yoshida,A., Sun,S., Piccirilli,J.A. and Herschlag,D. (1999) Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc. Natl Acad. Sci. USA, 96, 12299–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tinoco I. Jr, and Bustamante,C. (1999) How RNA folds. J. Mol. Biol., 293, 271–281. [DOI] [PubMed] [Google Scholar]
- 7.Hermann T., Auffinger,P., Scott,W.G. and Westhof,E. (1997) Evidence for a hydroxide ion bridging two magnesium ions at the active site of the hammerhead ribozyme. Nucleic Acids Res., 25, 3421–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikkelsen N.E., Johansson,K., Virtanen,A. and Kirsebom,L.A. (2001) Aminoglycoside binding displaces a divalent metal ion in a tRNA-neomycin B complex. Nature Struct. Biol., 8, 510–514. [DOI] [PubMed] [Google Scholar]
- 9.Hermann T. and Westhof,E. (1998) Aminoglycoside binding to the hammerhead ribozyme: a general model for the interaction of cationic antibiotics with RNA. J. Mol. Biol., 276, 903–912. [DOI] [PubMed] [Google Scholar]
- 10.Lind K.E., Du,Z., Fujinaga,K., Peterlin,B.M. and James,T.L. (2002) Structure-based computational database screening, in vitro assay and NMR assessment of compounds that target TAR RNA. Chem. Biol., 9, 185–193. [DOI] [PubMed] [Google Scholar]
- 11.Misra V.K. and Draper,D.E. (1998) On the role of magnesium ions in RNA stability. Biopolymers, 48, 113–135. [DOI] [PubMed] [Google Scholar]
- 12.Peschke M., Blades,A.T. and Kebarle,P. (1998) Hydration energies and entropies for Mg2+, Ca2+, Sr2+, Ba2+. J. Phys. Chem. A, 102, 9978–9985. [Google Scholar]
- 13.Tanaka Y., Kojima,C., Morita,E.H., Kasai,Y., Yamasaki,K., Ono,A., Kainosho,M. and Taira,K. (2002) Identification of the metal ion binding site on an RNA motif from hammerhead ribozymes using (15)N NMR spectroscopy. J. Am. Chem. Soc., 124, 4595–4601. [DOI] [PubMed] [Google Scholar]
- 14.Sigel R.K., Vaidya,A. and Pyle,A.M. (2000) Metal ion binding sites in a group II intron core. Nature Struct. Biol., 7, 1111–1116. [DOI] [PubMed] [Google Scholar]
- 15.Rasmussen T.A. and Nolan,J.M. (2002) G350 of Escherichia coli RNase P RNA contributes to Mg2+ binding near the active site of the enzyme. Gene, 294, 177–185. [DOI] [PubMed] [Google Scholar]
- 16.Tzokov S.B., Murray,I.A. and Grasby,J.A. (2002) The role of magnesium ions and 2′-hydroxyl groups in the VS ribozyme-substrate interaction. J. Mol. Biol., 324, 215–226. [DOI] [PubMed] [Google Scholar]
- 17.Crary S.M., Kurz,J.C. and Fierke,C.A. (2002) Specific phosphorothioate substitutions probe the active site of Bacillus subtilis ribonuclease P. RNA, 8, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christian E.L. and Yarus,M. (1993) Metal coordination sites that contribute to structure and catalysis in the group I intron from Tetrahymena. Biochemistry, 32, 4475–4480. [DOI] [PubMed] [Google Scholar]
- 19.Nayal M. and Di Cera,E. (1994) Predicting Ca(2+)-binding sites in proteins. Proc. Natl Acad. Sci. USA, 91, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayal M. and Di Cera,E. (1996) Valence screening of water in protein crystals reveals potential Na+ binding sites. J. Mol. Biol., 256, 228–234. [DOI] [PubMed] [Google Scholar]
- 21.Chin K., Sharp,K.A., Honig,B. and Pyle,A.M. (1999) Calculating the electrostatic properties of RNA provides new insights into molecular interactions and function. Nature Struct. Biol., 6, 1055–1061. [DOI] [PubMed] [Google Scholar]
- 22.Madura J.D., Davis,M.E., Gilson,M.K., Wade,R.C., Luty,B.A. and McCammon,J.A. (1994) Biological applications of electrostatic calculations and Brownian Dynamics simulations. Rev. Comput. Chem., 5, 229–267. [Google Scholar]
- 23.Hermann T. and Westhof,E. (1998) Exploration of metal ion binding sites in RNA folds by Brownian-dynamics simulations. Structure, 6, 1303–1314. [DOI] [PubMed] [Google Scholar]
- 24.van EBuuren B.N., Hermann,T., Wijmenga,S.S. and Westhof,E. (2002) Brownian-dynamics simulations of metal-ion binding to four-way junctions. Nucleic Acids Res., 30, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker N.A., Sept,D., Joseph,S., Holst,M.J. and McCammon,J.A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Hashimi H.M., Gorin,A., Majumdar,A., Gosser,Y. and Patel,D.J. (2002) Towards structural genomics of RNA: rapid NMR resonance assignment and simultaneous RNA tertiary structure determination using residual dipolar couplings. J. Mol. Biol., 318, 637–649. [DOI] [PubMed] [Google Scholar]
- 28.Burley S.K. and Bonanno,J.B. (2002) Structuring the universe of proteins. Annu. Rev. Genomics Hum. Genet., 3, 243–262. [DOI] [PubMed] [Google Scholar]
- 29.Hansen J.L., Ippolito,J.A., Ban,N., Nissen,P., Moore,P.B. and Steitz,T.A. (2002) The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell, 10, 117–128. [DOI] [PubMed] [Google Scholar]
- 30.Yusupov M.M., Yusupova,G.Z., Baucom,A., Lieberman,K., Earnest,T.N., Cate,J.H. and Noller,H.F. (2001) Crystal structure of the ribosome at 5.5 Å resolution. Science, 292, 883–896. [DOI] [PubMed] [Google Scholar]
- 31.Wimberly B.T., Brodersen,D.E., Clemons,W.M. Jr, Morgan-Warren,R.J., Carter,A.P., Vonrhein,C., Hartsch,T. and Ramakrishnan,V. (2000) Structure of the 30S ribosomal subunit. Nature, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 32.Ban N., Nissen,P., Hansen,J., Moore,P.B. and Steitz,T.A. (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science, 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 33.Klein D.J., Schmeing,T.M., Moore,P.B. and Steitz,T.A. (2001) The kink-turn: a new RNA secondary structure motif. EMBO J., 20, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nissen P., Ippolito,J.A., Ban,N., Moore,P.B. and Steitz,T.A. (2001) RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA, 98, 4899–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doudna J.A. (2000) Structural genomics of RNA. Nature Struct. Biol., 7 (Suppl), 954–956. [DOI] [PubMed] [Google Scholar]
- 36.Klosterman P.S., Tamura,M., Holbrook,S.R. and Brenner,S.E. (2002) SCOR: a structural classification of RNA database. Nucleic Acids Res., 30, 392–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagley S.C. and Altman,R.B. (1995) Characterizing the microenvironment surrounding protein sites. Protein Sci., 4, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waugh A., Williams,G.A., Wei,L. and Altman,R.B. (2001) Using meta computing tools to facilitate large-scale analyses of biological databases. Pac. Symp. Biocomput., 6, 360–371. [DOI] [PubMed] [Google Scholar]
- 39.Banatao D.R., Huang,C.C., Babbitt,P.C., Altman,R.B. and Klein,T.E. (2001) ViewFeature: integrated feature analysis and visualization. Pac. Symp. Biocomput., 6, 240–250. [DOI] [PubMed] [Google Scholar]
- 40.Bagley S.C. and Altman,R.B. (1996) Conserved features in the active site of nonhomologous serine proteases. Fold Des., 1, 371–379. [DOI] [PubMed] [Google Scholar]
- 41.Wei L. and Altman,R.B. (1998) Recognizing protein binding sites using statistical descriptions of their 3D environments. Pac. Symp. Biocomput., 3, 497–508. [PubMed] [Google Scholar]
- 42.Wei L., Altman,R.B. and Chang,J.T. (1997) Using the radial distributions of physical features to compare amino acid environments and align amino acid sequences. Pac. Symp. Biocomput., 2, 465–476. [PubMed] [Google Scholar]
- 43.Wei L., Huang,E.S. and Altman,R.B. (1999) Are predicted structures good enough to preserve functional sites? Structure Fold Des., 7, 643–650. [DOI] [PubMed] [Google Scholar]
- 44.van Buuren B.N., Schleucher,J. and Wijmenga,S.S. (2000) NMR structural studies on a DNA four-way junction: stacking preferences and localization of the metal-ion binding site. J. Biomol. Struct. Dyn., 237–244. [DOI] [PubMed] [Google Scholar]
- 45.Liang M.P., Banatao,D.R., Klein,T.E., Brutlag,D.L. and Altman,R.B. (2003) WebFEATURE: an interactive web tool for identifying and visualizing functional sites on macromolecular structures. Nucleic Acids Res., 31, 3324–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gendron P., Lemieux,S. and Major,F. (2001) Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol., 308, 919–936. [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W. and Sander,C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 48.Huang C.C., Couch,G.S., Pettersen,E.F. and Ferrin,T.E. (1996) Chimera: an extensible molecular modeling application constructed using standard components. Pac. Symp. Biocomput., 1, 724. [Google Scholar]
- 49.Kieft J.S. and Tinoco,I.,Jr (1997) Solution structure of a metal-binding site in the major groove of RNA complexed with cobalt (III) hexammine. Structure, 5, 713–721. [DOI] [PubMed] [Google Scholar]
- 50.Cate J.H., Gooding,A.R., Podell,E., Zhou,K., Golden,B.L., Kundrot,C.E., Cech,T.R. and Doudna,J.A. (1996) Crystal structure of a group I ribozyme domain: principles of RNA packing. Science, 273, 1678–1685. [DOI] [PubMed] [Google Scholar]
- 51.Juneau K., Podell,E., Harrington,D.J. and Cech,T.R. (2001) Structural basis of the enhanced stability of a mutant ribozyme domain and a detailed view of RNA–solvent interactions. Structure, 9, 221–231. [DOI] [PubMed] [Google Scholar]
- 52.Conn G.L., Draper,D.E., Lattman,E.E. and Gittis,A.G. (1999) Crystal structure of a conserved ribosomal protein–RNA complex. Science, 284, 1171–1174. [DOI] [PubMed] [Google Scholar]
- 53.Conn G.L., Gittis,A.G., Lattman,E.E., Misra,V.K. and Draper,D.E. (2002) A compact RNA tertiary structure contains a buried backbone-K+ complex. J. Mol. Biol., 318, 963–973. [DOI] [PubMed] [Google Scholar]
- 54.Misra V.K. and Draper,D.E. (2001) A thermodynamic framework for Mg2+ binding to RNA. Proc. Natl Acad. Sci. USA, 98, 12456–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serra M.J., Baird,J.D., Dale,T., Fey,B.L., Retatagos,K. and Westhof,E. (2002) Effects of magnesium ions on the stabilization of RNA oligomers of defined structures. RNA, 8, 307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]