

Abstract
TREK channels are unique among two-pore-domain K+ channels. They are activated by polyunsaturated fatty acids (PUFAs) including arachidonic acid (AA), phospholipids, mechanical stretch and intracellular acidification. They are inhibited by neurotransmitters and hormones. TREK-1 knockout mice have impaired PUFA-mediated neuroprotection to ischemia, reduced sensitivity to volatile anesthetics and altered perception of pain. Here, we show that the A-kinase-anchoring protein AKAP150 is a constituent of native TREK-1 channels. Its binding to a key regulatory domain of TREK-1 transforms low-activity outwardly rectifying currents into robust leak conductances insensitive to AA, stretch and acidification. Inhibition of the TREK-1/AKAP150 complex by Gs-coupled receptors such as serotonin 5HT4sR and noradrenaline β2AR is as extensive as for TREK-1 alone, but is faster. Inhibition of TREK-1/AKAP150 by Gq-coupled receptors such as serotonin 5HT2bR and glutamate mGluR5 is much reduced when compared to TREK-1 alone. The association of AKAP150 with TREK channels integrates them into a postsynaptic scaffold where both G-protein-coupled membrane receptors (as demonstrated here for β2AR) and TREK-1 dock simultaneously.
Keywords: G-protein-coupled receptors, ion channels, proteomics, scaffolding
Introduction
Potassium channels allow the passive and selective transport of K+ through the cell membranes. They regulate cell volume and potassium uptake and control the flow of salt across epithelia. They are also involved in cell excitability and control neuronal signaling, heart rate, vascular tone and hormone secretion. This large functional diversity is underscored by the number of channelopathies related to mutations in K+ channel genes: diseases of the brain (benign familial neonatal convulsions, episodic ataxia with myokimia), skeletal muscle (Andersen's syndrome), heart (arrhythmia), ear (deafness), kidney (Bartter's syndrome) and pancreas (hyperinsulinemic hypoglycemia of infancy, diabetes).
Growing evidence indicates that trafficking, addressing as well as functional properties of native ion channels depend on their lipidic and proteic environments. In particular, the specificity and the speed of their regulations by membrane receptors require a promiscuous organization of the constitutive channel subunits with membrane receptors and complexes of intracellular molecules (Levitan, 2006). The role of scaffolding proteins in orchestrating these regulatory microdomains is crucial.
TREK-1 and TREK-2 are closely related channels that belong to the family of two-pore-domain K+ (K2P) channels (Lesage and Lazdunski, 2000; Patel and Honoré, 2001; Goldstein et al, 2005; Kim, 2005). They produce outwardly rectifying currents with low basal activity in classical conditions of heterologous expression (Fink et al, 1996; Patel et al, 1998; Bang et al, 2000; Lesage et al, 2000). Mechanical stretch, cell swelling, intracellular acidification, temperature, polyunsaturated fatty acids (PUFAs) including arachidonic acid (AA), lysophospholipids and phosphatidylinositol 4,5-bisphosphate (PIP2) are natural stimulators of TREK channels (Patel et al, 1998; Maingret et al, 1999, 2000a; Honoré et al, 2002; Chemin et al, 2005; Lopes et al, 2005). On the contrary, neurotransmitters and hormones that activate Gq- or Gs-coupled receptors decrease their activity (Lesage et al, 2000; Chemin et al, 2003). In terms of pharmacology, TREK-1 and TREK-2 are opened by clinical concentrations of volatile anesthetics (Patel et al, 1999; Lesage et al, 2000) and by riluzole (Duprat et al, 2000; Lesage et al, 2000), a neuroprotective drug used to protect motoneurons in amyotrophic lateral sclerosis. Mice with an inactivated TREK-1 gene display an increased vulnerability to epileptic seizures and brain ischemia. They have lost neuroprotection afforded by PUFAs and are more resistant to volatile anesthetics (Heurteaux et al, 2004). TREK-1 is also present in sensory neurons, particularly in nociceptors, and is involved in polymodal pain perception (Alloui et al, 2006).
During the past years, a particular effort has been made to understand the gating mechanism of these TREK channels. The cytosolic carboxy-terminal (C-ter) domain of TREK-1, immediately following the fourth transmembrane segment (M4), plays a key structural role in the activation mechanisms. The protonation of a glutamate residue of this domain (E306) by cytosolic acidification controls the pressure dependency and the activation by internal acidification (Maingret et al, 1999; Honoré et al, 2002). A cluster of positively charged residues surrounding E306 interacts with membrane phospholipids, including PIP2, inducing a structural rearrangement that is the key step in mechanoactivation of the channel and in transforming the outwardly rectifying and low-activity TREK-1 channel into a leak K+ channel (Chemin et al, 2005). This regulatory post-M4 region also contains two serine residues, S300 and S333, that are crucial for phosphorylation by protein kinase C (PKC) (Murbartian et al, 2005) and protein kinase A (PKA) (Patel et al, 1998) and for channel inhibition by neurotransmitter receptors.
This report constitutes the first identification and characterization of a partner protein for TREK channels. Here, we show that both TREK-1 and TREK-2 interact with AKAP150. The binding of AKAP150 is associated with radical modifications of the channel behavior and regulation. The mapping of the AKAP150 interacting site to the regulatory post-M4 region of TREK-1 provides a molecular basis for the observed effects.
Results
AKAP150 identification by proteomics
To identify proteins interacting with TREK-1, we designed a proteomic approach based on the immunoprecipitation and mass spectrometry analysis of native channel complexes. Affinity-purified antibodies directed against TREK-1 (Maingret et al, 2000a) were covalently crosslinked to protein A-Sepharose to produce anti-TREK-1 immunobeads. These beads were incubated with brain synaptosomal proteins solubilized in a buffer containing a mild detergent, then briefly washed. Bound proteins were eluted and separated by SDS–PAGE. The precipitated proteins were identified via direct nanoLC-ESI-MS/MS analysis of trypsin-digested gel bands. From the whole brains of C57Bl6J wild-type (WT) mice, more than 100 different proteins were identified. The precipitation of many of these proteins was due to nonspecific binding to immunobeads and to the crossreactivity of antibodies to epitopes unrelated to TREK-1. For this reason, the same experiment was carried out by using solubilized synaptosomal proteins prepared from C57Bl6J mice in which TREK-1 has been genetically inactivated (TREK-1−/− mice) (Heurteaux et al, 2004). The majority of the proteins precipitated from WT mice were identical to the proteins isolated from TREK-1−/− mice with the exception of four of them. One of these proteins was AKAP150. AKAP150 is a scaffolding protein known to organize signaling complexes in neurons. AKAP150 interacts with PKA, PKC, protein phosphatase 2B (PP2B), as well as with membrane receptors, ion channels and postsynaptic proteins PSD95 and SAP97 (for a review, see Colledge and Scott, 1999). In mouse brain, TREK-1 (Fink et al, 1996; Maingret et al, 2000a) and AKAP150 (Supplementary Figure 1) have different but significantly overlapping distributions. In P7 and adult mice, they are both expressed at high levels in striatum, cortex and hippocampus. TREK-1 is also present in thalamus and cerebellum, whereas the level of AKAP150 level is lower in these areas, particularly at the adult stage. These expression patterns, together with the complex regulation of TREK channels by neurotransmitter and hormone membrane receptors coupled to G-proteins (Fink et al, 1996; Patel et al, 1998; Lesage et al, 2000; Chemin et al, 2003; Murbartian et al, 2005), prompted us to further characterize the role of AKAP150 as a TREK-1-interacting protein.
AKAP150 interaction and upregulation of TREK channels
First, we verified that cloned TREK-1 and AKAP150 proteins interact in heterologous expression systems. In Mabin Darby canine kidney (MDCK) cells that constitute a good cell system for immunocytochemistry (Decressac et al, 2004), TREK-1 and AKAP150 colocalized perfectly (Figure 1A). The colabeling was particularly strong at the plasma membrane (Supplementary Figure 2). AKAP150 was co-immunoprecipitated with TREK-1, confirming the physical interaction between the two proteins (Figure 1B). In the absence of TREK-1 expression, anti-TREK-1 antibodies did not precipitate AKAP150 (Figure 1B), a result that is in agreement with the specific precipitation of AKAP150 from WT mice and not from TREK-1−/− mice.
Figure 1.

AKAP150 interacts with TREK-1 and increases its basal current activity. (A) Immunolocalization of TREK-1 (green labeling) and AKAP150 (red labeling) in permeabilized MDCK cells. The cell nuclei are in blue. In the merge panel, overlapping green and red labelings are in yellow. (B) Co-immunoprecipitation of AKAP150 by anti-TREK-1 antibodies from transfected MDCK cells. (C) Effect of AKAP150 expression on TREK-1 currents. Currents were elicited by voltage ramps (from −120 to +50 mV, 1 s in duration) from COS cells expressing TREK-1 in the presence or absence of AKAP150, as displayed. In the inset, the histograms represent the ratio of the mean currents recorded at 0 mV in the presence (I) or absence (Icontrol) of AKAP150. (D) Current–voltage (I–V) relationships deduced from whole-cell recordings in symmetrical K+ conditions and 3 mM external Mg2+. Steady-state currents were elicited by voltage pulses from a holding potential of 0 mV. In the inset, the histograms represent the ratio (absolute values) of the mean currents recorded at −80 and +80 mV, in the presence or absence of external Mg2+.
The functional effect of AKAP150 on TREK-1 activity was studied by electrophysiology from transfected COS cells. As shown in Figure 1C, AKAP150 expression was associated with a strong increase of the whole-cell current amplitude (5.3±0.7-fold; the number of tested cells is indicated in the figures). This increase is not related to any change in the cell volume. The values of the normalized currents are 23.6±2.6 pA/pF (n=56) for TREK-1 and 112.8±13.9 pA/pF (n=62) for TREK-1+AKAP150. In symmetrical K+ conditions, TREK-1 produced more outward currents for depolarizations than inward currents for the corresponding hyperpolarized states (Figure 1D). This outward rectification of the I–V relationship constitutes a hallmark of TREK-1 and has been attributed to a pore-blocking and voltage-dependent effect of external Mg2+ (Maingret et al, 2002). As expected, the outward rectification was strongly reduced when Mg2+ was removed from the external medium. I−80 mV/I+80 mV shifted from 0.4±0.1 to 0.7±0.1 (Figure 1D, inset). AKAP150 coexpression resulted also in a very significant decrease in the voltage dependence of TREK-1 activation in physiological Mg2+ concentration (I−80 mV/I+80 mV=0.9±0.1) (Figure 1D).
Is AKAP150 able to modulate other K2P channels? To answer this question, we coexpressed AKAP150 with the other lipid and mechano-gated channels TREK-2 (Lesage et al, 2000) and TRAAK (Fink et al, 1998), and with the acid-sensitive channel TASK-1 (Duprat et al, 1997). Immunocytochemistry labelings of AKAP150 and TREK-2 showed a good overlay (Figure 2A). However, neither TRAAK (Figure 2C) nor TASK-1 (not shown) immunoreactivities overlapped the AKAP150 staining. AKAP150 induced a strong increase of the TREK-2 current amplitude (5.0±1.2-fold at 0 mV) (Figure 2B), whereas no significant effect was seen on TRAAK (Figure 2D) and TASK-1 currents (Figure 3A) (0.8±0.2-fold for TRAAK and 0.9±0.2-fold for TASK-1). These results confirm that AKAP150 interacts with TREK-2, which is closely related to TREK-1, but not with the more distant channels TRAAK and TASK-1.
Figure 2.
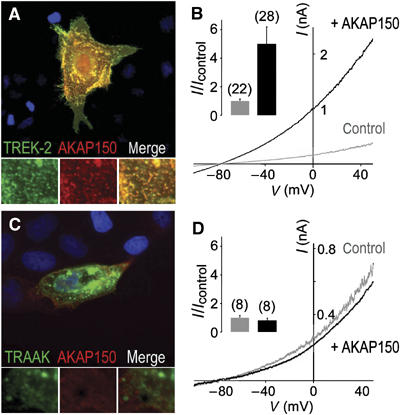
AKAP150 interacts physically and functionally with TREK-2, but not with TRAAK, a more distantly related K2P channel. (A, C) Immunolocalization of AKAP150 and TREK-2 (A) and TRAAK (C) in permeabilized MDCK cells. The channels were labeled in green and AKAP150 in red. Overlapping green and red labelings are in yellow. Close-up images in the green, red and merge channels are displayed below the main merge panels. (B, D) Effects of AKAP150 coexpression on TREK-2 (B) and TRAAK (D). Currents were elicited by voltage ramps in transfected COS cells (from −120 to +50 mV, 1 s in duration). In the inset, histograms represent the ratio of the mean currents recorded at 0 mV in the presence (I) or absence (Icontrol) of AKAP150.
Figure 3.

AKAP150 interacts with the cytoplasmic C-ter region of TREK-1. (A) Effect of AKAP150 coexpression on TASK-1 and (B) on a chimeric channel TASK-1/CtTREK-1 containing the core region of TASK-1 fused to the cytoplasmic C-ter region of TREK-1. Currents were elicited by voltage ramps (from −120 to +50 mV, 1 s in duration) in transfected COS cells. In the inset, histograms represent the ratio of the mean currents recorded at 0 mV in the presence (I) or absence (Icontrol) of AKAP150.
Binding of AKAP150 to the regulatory post-M4 region of TREK-1
The next step was to identify the AKAP150-binding site in TREK-1. The C-ter following the M4 is the major cytoplasmic domain of K2P channels (Lesage et al, 1996a, 1996b). To test the possibility that this region of TREK-1 interacts with AKAP150, we designed a TASK-1/CtTREK-1 chimeric channel composed of TASK-1 and TREK-1 parts. In TASK-1, the cytoplasmic C-ter was replaced by the corresponding C-ter of TREK-1 to form TASK-1/CtTREK-1 (Figure 4A). Unlike TASK-1 (Figure 3A), TASK-1/CtTREK-1 was sensitive to AKAP150 (Figure 3B). Whereas AKAP150 had a stimulatory effect on the TREK-1 channel, it had an inhibitory effect on TASK-1/CtTREK-1 channel (ITASK-1/CtTREK-1=1.2±0.2 nA at +0 mV in the presence of AKAP150 compared with 3.7±0.1 nA at +0 mV in control conditions). This result indicates that an AKAP150-binding site was successfully transferred from TREK-1 to TASK-1. To map this site more precisely, we used a series of TREK-1 mutants deleted of their last 51, 76, 100 or 113 amino-acid residues (Figure 4A) and we tested their ability to colocalize with AKAP150 in transfected MDCK cells. A perfect overlap was observed for TREK-1Δ51 (not shown) and TREK-1Δ76 (Figure 4B) and only a partial overlap for TREK-1Δ100 (Figure 4C). The colocalization was completely lost for TREK-1Δ113 (Figure 4D) indicating that the region extending from residues V298 to R311 is crucial for AKAP150 binding. The post-M4 region of TREK-1 extending from V298 to S333 and its corresponding region in TREK-2 have been shown to be a key domain for regulation by PUFAs, lysophospholipids, temperature, pH and mechanical stretch (Patel et al, 1998; Maingret et al, 1999, 2000b; Kim et al, 2001; Honoré et al, 2002), as well as for outward rectification (Maingret et al, 2002). Recently, it has been shown that a cluster of basic residues located in this region forms the phospholipid-sensing domain of TREK-1 (Chemin et al, 2005). We coexpressed AKAP150 with a mutant of TREK-1 (TREK-1-5A) in which this particular cluster of positively charged residues has been replaced by a cluster of alanine residues (R297A, K301A, K302A, K304A and R311A) (Figure 4A). Figure 4E shows that this mutant did not interact with AKAP150. Taken together, these results demonstrate that the interaction site in TREK-1 is located between V298 and R311 and that AKAP150 association with this site induces a major modification of TREK-1 and TASK-1/CtTREK-1 channel activities. The primary sequence of this site is perfectly conserved in TREK-2 (Lesage et al, 2000). In mouse and human TREK-1, the sequences of this site are identical. Human TREK-1 is able to interact with AKAP79, the human ortholog of mouse AKAP150 (Supplementary Figure 4).
Figure 4.

The post-M4 region of TREK-1 is crucial for binding to AKAP150. (A) Cartoon depicting TREK-1 mutants and TASK-1/CtTREK-1 chimera. (B–E) Immunolocalization of TREK-1 mutants and myc-tagged AKAP150 in permeabilized MDCK cells. The cytoplasmic C-ter region of TREK-1 was progressively deleted of the last 76 amino-acid residues in (B), 100 in (C) and 113 in (D) or mutated in the post-M4 regulatory domain in (E). Overlapping green and red labelings are in yellow. Colocalization with AKAP150 was lost for TREK-1Δ113 and TREK-1-5A. As quantified by Western blot analysis, the expression levels of the different TREK-1 mutants were not significantly different (Supplementary Figure 5).
Conversion of TREK-1 into a novel type of leak current
Does binding of AKAP150 to the regulatory post-M4 region alter TREK-1 regulation by its stimulators? In transfected COS cells and in the inside-out patch configuration, acidification from pH 7.2 to 5.5 of the perfusing internal solution induced a 25±5-fold increase of the current amplitude in the absence of AKAP150 (Figure 5A). When AKAP150 was coexpressed, this stimulatory effect of intracellular protonation was lost (IpH 5.5/IpH 7.2=1±0.5). A similar observation was made with AA. Application of AA was without effect on the TREK-1 current in the presence of AKAP150 (IAA/Icontrol=1±0.1), whereas the same AA concentration induced a 21±7-fold increase of the current amplitude in control cells (Figure 5C). One of the most remarkable properties of TREK channels is to be mechano-gated and activated by negative pressures (Patel et al, 1998; Maingret et al, 1999; Lesage et al, 2000). A mechanical stretch of the membrane had essentially no effect on the TREK-1/AKAP150 complex (1.2±0.1-fold of stimulation compared with 6±2-fold for TREK-1 expressed alone) (Figure 5B). These electrophysiological data indicate that once associated to AKAP150, TREK-1 is fully activated and cannot be further stimulated by AA, acidic pH and mechanical stress. AKAP150 binding to the regulatory post-M4 domain and the resulting activation of TREK-1 provide a molecular basis for explaining the increase of current amplitude (Figure 1C). AKAP79, the human ortholog of mouse AKAP150, has been shown to increase the amplitude of voltage-gated Ca2+ currents produced by Cav1.2 by directing its expression at the cell surface (Altier et al, 2002). Such a chaperone-like effect can be excluded for TREK-1 because expression of AKAP150 had no significant effect on the levels of total TREK-1 protein or TREK-1 at the cell surface (Supplementary Figure 3).
Figure 5.
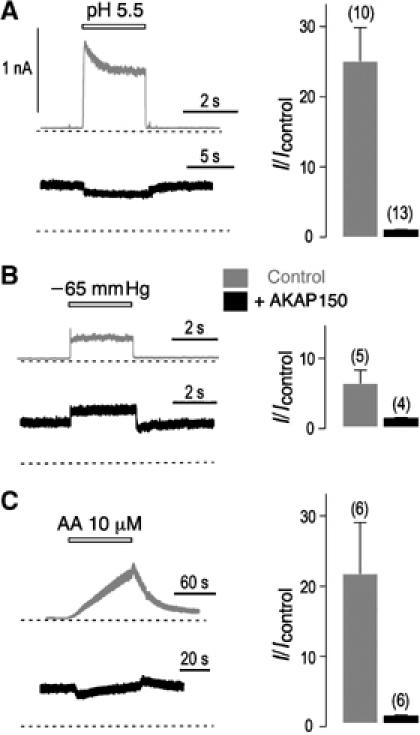
AKAP150 binding changes TREK-1 channel regulation. (A–C) Effects of AKAP150 coexpression on TREK-1 stimulation by intracellular acidification (pH 5.5) (A), negative pressure (–65 mmHg) (B) and AA (10 μM) (C). Inside-out patch currents were recorded at 0 mV from transfected COS cells. The zero current levels are indicated by a dotted line. The histograms represent the ratio of the mean currents recorded before (Icontrol) or after (I) intracellular acidification, membrane stretching or AA application.
TREK-1/AKAP150 currents are resistant to Gq receptor activation
TREK channels are inhibited by cAMP and PKC-activator phorbol-12 myristate-acetate (PMA) (Fink et al, 1998) and by Gs-protein and Gq-protein-coupled membrane receptors (Patel et al, 1998; Lesage et al, 2000; Chemin et al, 2003). As demonstrated for TREK-1, the effect of cAMP and Gs-coupled receptors depends on the phosphorylation of S333 by PKA (Patel et al, 1998; Maingret et al, 2000a), whereas the effect of PMA and thyrotropin-releasing hormone and orexin Gq-coupled receptors involves PKC phosphorylation of S300 (Murbartian et al, 2005). Signaling pathways may differ depending on the conditions and Gq-coupled receptors. Inhibition of AA-activated TREK-1 and TREK-2 by group I glutamate receptors has been related to direct inhibitory effects of diacylglycerol (DAG) and phosphatidic acid (PA) generated by Gq-activated phospholipase C (Chemin et al, 2003). The role of AKAP150 in TREK-1 current inhibition by cAMP and PMA was examined in COS cells. As shown in Figure 6A, AKAP150 did not affect maximal inhibition induced by the membrane-permeant cAMP analog chlorophenylthio-cAMP (CPT-cAMP) (80±2% of inhibition versus 84±3%) but the effect of PMA application was strongly affected. Inhibition reached 72±4% in control conditions, and only 20±4 % when AKAP150 was coexpressed (Figure 6A). We next studied the effect of G-coupled receptor activation in Xenopus oocytes. 5HT4sR and β2AR are Gs-coupled receptors that stimulate adenylyl cyclase, leading to an increase of cAMP concentration and ultimately to PKA activation. As shown in Figure 6B, AKAP150 did not modify maximal TREK-1 current following 5HT4sR or β2AR activation. 5HT2bR and mGluR5 are Gq-coupled receptors. In the presence of AKAP150, the extent of inhibition induced by 5HT2bR decreased from 60±5 to 17±7% (Figure 6B), and inhibition by mGluR5 from 76±8 to 27±9% (Figure 6B). Binding of AKAP150 to the post-M4 region between V298 and R311 may prevent access of PKC to S300 or/and access of inhibitory DAG and PA, thus decreasing inhibition by the PKC activator PMA and the Gq-coupled receptors 5HT2bR and mGluR5. S333, which constitutes the major PKA phosphorylation site in TREK-1, is located far from the AKAP150-interacting site, explaining why cAMP and Gs receptors 5HT4sR and β2AR retain their inhibitory capacity in the presence of AKAP150.
Figure 6.

AKAP150 changes TREK-1 regulation and promotes TREK-1 association with β2AR. (A) Effect of AKAP150 coexpression on TREK-1 current inhibition induced by application of CPT-cAMP (500 μM) or PMA (50 nM) in transfected COS cells. The histograms represent the percentage of inhibition in the absence (in gray) or presence (in black) of AKAP150. (B) Effect of AKAP150 coexpression on TREK-1 current inhibition by 5HT4sR, 5HT2bR, β2AR and mGluR5 in Xenopus oocytes. 5HT4sR and 5HT2bR were activated by application of 30 μM 5-HT, β2AR by 50 μM noradrenaline (NA) and mGluR5 by 30 μM glutamate. (C, D) Representative examples of TREK-1 current inhibition induced by β2AR (C) and 5HT2bR (D) in oocytes. (E) Immunolocalization of β2AR and TREK-1 in the absence (left panel) or presence of AKAP150 (right panel). Overlapping green and red labelings are in yellow. Close-up images in the green, red and merge channels are displayed below the main merge panels. (F) Co-precipitation of AKAP150 and β2AR by anti-TREK-1 antibodies from solubilized synaptosomal proteins prepared from wild-type (WT) but not from TREK-1 KO (TREK-1−/−) mice.
If AKAP150 did not change maximal TREK-1 inhibition by Gs-coupled receptors, there was a clear difference in the kinetics of inhibition. Inhibition was twice as fast in the presence of AKAP150. For β2AR, the calculated value of the time constant of inhibition τ is 26.9±5 s (n=10) in control conditions versus 13.6±3 s (n=9) in the presence of AKAP150 (Figure 6C). For 5HT4sR, a similar acceleration of inhibition kinetics was observed: τ=4.2±0.7 s (n=6) in the presence of AKAP150 compared to 7.8±1.4 s (n=6) in control conditions (not shown). These results suggest that the anchoring of PKA to close proximity of TREK-1 by AKAP150 is essential for a faster inhibition of TREK-1 by Gs-coupled receptors.
AKAP150 forms regulation complexes comprising TREK-1 and membrane receptors
We have previously shown that atrial cardiomyocytes express an endogenous TREK current inhibited by isoproterenol, a β-adrenergic agonist (Terrenoire et al, 2001). On the other hand, AKAP150 has been shown to interact with β2AR both in vitro and in vivo (Gao et al, 1997; Liu et al, 2004). In transfected MDCK cells, β2AR and TREK-1 labelings did not overlap (Figure 5E), but in the presence of AKAP150, the stainings showed a perfect overlay, supporting the hypothesis that AKAP150 can bring TREK-1 and β2AR into close proximity. Evidence of such a promiscuous association was also found in brain membranes. AKAP150 and β2AR were specifically co-immunoprecipitated with TREK-1 from synaptosomal proteins prepared from WT mice but not from TREK-1−/− mice (Figure 6F).
In neurons, AKAP150 is located at the cell body periphery and in the dendrites where it concentrates in post-synaptic membranes (Carr et al, 1992; Colledge and Scott, 1999). In cultured hippocampal neurons, no macroscopic TREK-1 currents could be recorded. When overexpressed, TREK-1 was detected in the same dendritic dense bodies as AKAP150 (Figure 7A). A background current was recorded upon TREK-1 transfection that could not be stimulated by AA corresponding to AKAP150/TREK-1 channels (Figure 7B). In the axon that did not contain AKAP150 aggregates, TREK-1 immunoreactivity was faint and diffuse. In post-synaptic dense bodies, AKAP150 is known to interact with the PDZ domain-containing proteins PSD95 and SAP97 that recruit PDZ-interacting proteins including G-coupled receptors (Colledge et al, 2000). The colocalization of AKAP150 and TREK-1 in these sites suggests that TREK-1 may form protein complexes not only with β2AR but also with PDZ-interacting G-protein-coupled receptors in neurons expressing AKAP150 and PSD95/SAP97.
Figure 7.

Expression of TREK-1 in hippocampal neurons. (A) Immunolocalization of TREK-1 (green labeling) and AKAP150 (red labeling) in a transfected hippocampal neuron. Overlapping green and red labelings are in yellow. The open arrowhead shows the axon. Plain arrowhead shows a dendrite. This region of the dendrite is enlarged in the lower panels. Both TREK-1 and AKAP150 are present in the postsynaptic dense bodies that are indicated by plain arrowheads in the right panel. (B) In primary cultured neurons, no TREK-1 macroscopic currents were recorded. When overexpressed in these neurons, TREK-1 produced an active background K+ channel that could not be stimulated by AA (10 μM).
Discussion
In this report, we show that AKAP150 is a partner protein of TREK channels. The binding of AKAP150 to the post-M4 regulatory domain of TREK-1 radically transforms the electrophysiological properties and regulation of this channel (Figure 8). This post-M4 regulatory domain contains the structural elements (E306 surrounded by a positively charged cluster) that are necessary for the mechano-, pH and PUFA sensitivities of the TREK channels (Maingret et al, 1999; Honoré et al, 2002; Chemin et al, 2005).
Figure 8.
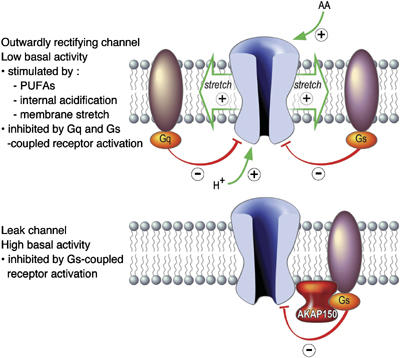
AKAP150 has a dual effect on TREK channels as direct molecular switch and regulatory microdomain scaffold. AKAP150 transforms the voltage-dependent and AA-, pH- and stretch-stimulated TREK channels into leak channels. These leak channels are no longer inhibited by Gq-coupled receptors, but still under the negative control of Gs-coupled receptors. This inhibition is faster, owing to the organization of a regulatory microdomain by AKAP150.
Expressed alone, TREK-1 and TREK-2 channels produce outwardly rectifying currents relatively inactive at rest but strongly activated by a wide variety of physical and chemical stimuli such as membrane stretch, osmotic swelling, internal acidification and bioactive lipids such as PUFAs (Patel et al, 1998; Maingret et al, 1999, 2000a; Honoré et al, 2002; Chemin et al, 2005; Lopes et al, 2005) (Figure 8). These different regulations have been related to altered functions in TREK-1−/− mice. For example, animals without TREK-1 have lost the neuroprotection induced by PUFAs against ischemia (Heurteaux et al, 2004). This is probably because ischemia, by increasing AA levels and intracellular loads of protons and by causing cell swelling, activates TREK-1, thus protecting neurons by increasing resting membrane potential and limiting Ca2+ influx via voltage-gated Ca2+ channels (Lauritzen et al, 2000). Also, TREK-1−/− mice that lack this particular K+ channel in small sensory neurons associated with nociception display altered pain response to mechanical stimuli and osmotic changes (Alloui et al, 2006).
When TREK-1 is expressed with AKAP150, its electrophysiological properties change radically. The TREK-1/AKAP150 channel becomes an active leak channel, which is no longer sensitive to internal acidification, mechanical stretch and AA (Figure 8). In neurons, TREK-1 and AKAP150 colocalize in dendrites where the formation of TREK-1/AKAP150 complexes is expected to occur (Figure 7). TREK-1 and AKAP150 are well expressed in brain regions such as cortex, hippocampus and striatum, where a TREK-1/AKAP150 channel is expected to form and produce a leak conductance that will be important for cell excitability. Dendritic excitability in cortex pyramidal neurons is known to be shaped by interaction between nonselective cationic currents and leak K+ currents (Day et al, 2005).
Because AKAP150 and its human ortholog AKAP79 belong to a family of anchoring proteins that coordinate multienzyme signaling complexes, TREK-1 channels bound to AKAP150 and switched in the active leak mode become part of larger protein complexes including PKA and G-coupled receptors. AKAP150 provides a mechanism to address PKA and other enzymes to the postsynaptic membrane to regulate synaptic activity and plasticity (for a review, see Bauman et al, 2004). In the nervous system, the function of AKAP79/150 is to bring PKC, PKA and PP2B into close proximity to a variety of membrane receptors including excitatory NMDA and AMPA glutamate receptors (Colledge et al, 2000), mGluR5 metabotropic glutamate receptor (Cho et al, 2002) and inhibitory GABAA receptors (Brandon et al, 2003). AKAP150 also associates with other types of ion channels such as the inwardly rectifying K+ channel Kir2.1 (Dart and Leyland, 2001) and the KCNKQ2 M-type K+ channel (Hoshi et al, 2003). The recruitment of PKA to TREK-1 via the TREK-1/AKAP150 complex is probably at the origin of the acceleration of channel inhibition following Gs-coupled receptor activation. At the same time, TREK-1, which is inhibited by the PKC activator PMA and by Gq-coupled receptors when expressed alone (Fink et al, 1996; Chemin et al, 2003), becomes resistant to these effectors in the presence of AKAP150. This decrease of efficacy is probably due to the fact that interaction of AKAP150 with the regulatory post-M4 domain of TREK-1 masks the serine residue (S300) that is normally phosphorylated by PKC (Murbartian et al, 2005). This interaction probably also prevents access of inhibitory lipids DAG and PA to this key region for TREK-1 function (Chemin et al, 2003).
As previously indicated, one important function of AKAP150 is to associate with neurotransmitter receptors. This paper provides direct evidence of a TREK-1/AKAP150/membrane receptor complex formation. TREK-1, AKAP150 and β2AR can be co-precipitated from brain tissue (Figure 6F) and have been shown to colocalize when coexpressed in transfected cells (Figure 6E). The function in neurons of this ternary complex containing TREK-1 and β2AR has yet to be deciphered. AKAP150 is also known to interact with the C-ter SH3 and GK regions of PSD95 and SAP97 (Colledge et al, 2000). A variety of receptors including receptors for serotonin (5HT2a, 5HT2c) and noradrenaline (β1AR) contain PDZ-interacting sites that can themselves interact with the PDZ domain-containing proteins PSD95 and SAP97 (Bockaert et al, 2004). Therefore, via AKAP150 and their binding to PSD95/SAP97, TREK channels may well form several other types of signaling complexes with neurotransmitter receptors. One particular TREK channel might be integrated into several different types of scaffolds depending on neuron type and, within neurons, depending on compartment type (soma, dendrites, etc.). It is to be expected that this organization of one particular type of K+ channel in several different types of assemblies will have important implications in normal as well as in pathological brain functions.
Materials and methods
Proteomics
Mouse brain synaptosomes from WT C57bl6 and TREK-1−/− mice (Heurteaux et al, 2004) were prepared using a one-step preparation method based on the known isopycnic densities of various cellular compartments (Hibino et al, 2002). The synaptic proteins were solubilized in PBS (Eurobio) containing 1% Triton X-100, 2 mM EDTA and a mixture of protease inhibitors (Roche Diagnostics, Basel, Switzerland), then centrifuged at 20 000 g for 30 min at 4°C. Solubilized proteins (10 mg) were incubated overnight with affinity-purified anti-TREK-1 antibodies (Maingret et al, 2000a) covalently linked to protein A-Sepharose 4B fast flow beads (Sigma) (100 μg of IgGs bound to 50 μl of beads). The beads were briefly washed twice with PBS and 0.1% Triton X-100 in SigmaPrep spin columns (Sigma). Proteins were eluted in Laemmli sample buffer (Bio-Rad), then separated by SDS–PAGE. Each lane was cut in 10 fragments and the precipitated proteins were identified via direct nanoLC-ESI-MS/MS analysis of trypsin-digested gel fragments.
Molecular biology
AKAP150 was cloned from a mouse brain cDNA library and inserted into pCMV-Myc (Clontech, Palo Alto, CA, USA), pBud.CE4.1 (pBud) (Invitrogen, Carlsbad, CA, USA) and pIRES2 HcRed. This vector was derived from pIRES2 EGFP (Clontech) by substituting HcRed coding sequence to EGFP. Mutants of TREK-1 and TASK-1/CtTREK-1 chimera have been described previously (Patel et al, 1998). Mouse β2AR and hamster 5HT2bR were amplified by RT–PCR and subcloned into pBud.CE4.1.
Cell culture and electrophysiology
MDCK cells were grown in minimal essential medium (MEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified incubator with 5% CO2 at 37°C. MDCK cells were transfected with Lipofectamin 2000 (Invitrogen). For electrophysiological recordings, COS cells were cultured and transfected using DEAE-dextran as previously described (Patel et al, 1998). For whole-cell experiments, the bath solution (EXT) contained 150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 3 mM MgCl2 and 10 mM HEPES at pH 7.4 with NaOH and the pipette solution (INT) contained 155 mM KCl, 3 mM MgCl2, 5 mM EGTA and 10 mM HEPES at pH 7.2 with KOH. For recordings in symmetrical conditions, EXT contained 155 mM KCl, 1 mM CaCl2, 3 mM (or 0 mM) MgCl2 and 10 mM HEPES at pH 7.4 with KOH. For inside-out patch recordings, the bath solution was INT and pipettes were filled with EXT. Recordings were carried out at room temperature (21–22°C) using a Multi Clamp 700A computer-controlled patch-clamp amplifier (Axon Instruments, USA). The Pclamp software was used to analyze recorded data. Mechanical stimulation was applied through an open loop pressure generating system. AA, CPT-cAMP and PMA were all purchased from Sigma-Aldrich Chimie.
Defolliculated Xenopus oocytes were injected with plasmids encoding TREK-1 (0.5 ng), AKAP150 (0.3 ng) and β2AR, mGluR5, 5HT2bR or 5HT4sR (0.3 ng). They were used for electrophysiological studies 3–4 days following injection. In a 0.3 ml perfusion chamber, a single oocyte was impaled with two standard microelectrodes (1–2.5 MΩ resistance) filled with 3 M KCl and maintained under voltage clamp with a Dagan TEV 200 amplifier, in standard ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 2 mM MgCl2, 5 mM HEPES, pH 7.4 with NaOH). Stimulation of the preparation, data acquisition and analysis were performed using pClamp software (Axon Instruments, USA). Drugs were applied externally by addition to the superfusate. All experiments were performed at room temperature (21–22°C). Noradrenaline (NA), 5-hydroxy-tryptamine (5-HT) and L-glutamic acid were purchased from Sigma-Aldrich Chimie.
Immunocytochemistry
Transfected cells on coverslips were fixed with PBS containing 4% paraformaldehyde (15 min at 21–22°C), then permeabilized with PBS and 0.1% Triton X-100 (PBST) and blocked for 1 h with 5% horse serum (HS) in PBST. Primary and secondary antibodies were diluted in PBST and 5% HS and incubated for 1 h at 21–22°C. Three 5-min washes with PBST were carried out between each incubation step and at the end of the procedure. Coverslips were mounted in Dako® Fluorescent Mounting medium (Dako Corporation, Carpinteria, CA, USA). The following antibodies were used: rabbit anti-TREK-1 polyclonal antibodies (Maingret et al, 2000a), mouse monoclonal antibody 9E10 against the myc epitope (Roche Diagnostics, Mannheim, Germany), rat monoclonal antibody 3F10 against hemagglutinin (HA) epitope (Roche Diagnostics), goat anti-rabbit IgGs conjugated to Alexa Fluor® 488 (Molecular Probes Europe BV, Leiden, The Netherlands), donkey anti-mouse IgGs conjugated to Alexa Fluor® 594 (Molecular Probes) and donkey anti-rat IgGs conjugated to Texas Red (Jackson Immunoresearch). Microscopy analysis and data acquisition were carried out with an Axioplan 2 Imaging Microscope (Carl Zeiss, Le Pecq, France).
Immunoprecipitation and Western blot analysis
Mouse brain synaptosomes and MDCK cells were homogenized in PBS containing saponin (0.5% w/v), Triton X-100 (0.5% w/v) and protease inhibitors (Roche Diagnostics, Basel, Switzerland). Lysates were clarified by centrifugation at 20 000 g for 30 min. Anti-TREK-1 antibodies were immobilized on protein A-Sepharose 4B fast flow (Sigma, Saint Louis, MO, USA) for immunoprecipitation from MDCK cells or on TrueBlotTM anti-rabbit Ig IP beads (eBioscience, San Diego, CA) for the mouse brain lysate. Immunoprecipitated proteins were separated on 10% SDS polyacrylamide gel and blotted onto nitrocellulose membrane (Hybond-C extra, Amersham Biosciences, Freiburg, Germany). Detection was carried out using mouse monoclonal antibody 9E10 against the myc epitope (Roche Diagnostics), goat polyclonal antibodies C-20 against AKAP150 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit polyclonal antibodies M-20 against β2AR (Santa Cruz Biotech).
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to I Lauritzen, A Patel and E Honoré for mutant TREK-1 plasmids and discussions and A Dupuy and JP Pin for 5HT4sR and mGluR5 vectors. We thank M Jodar for excellent technical assistance with cell culture and oocyte preparation, and I Haddad for mass spectrometry. S Thümmler was the recipient of a Postdoctoral Fellowship of the Deutsche Forschungsgemeinschaft (DFG, Germany) and S Feliciangeli was supported by the Association de Recherche sur le Cancer (ARC, France). F Lesage is the recipient of a ‘contrat d'interface' INSERM/CHU, service de neurologie, Hôpital Pasteur, Nice. This work was supported by the Ligue Nationale Contre le Cancer (Equipe labellisée) and by the Japan–France Integrated Action Program SAKURA (06980UF).
References
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M (2006) TREK-1, a K+ channel involved in polymodal pain perception. EMBO J 25: 2368–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E (2002) Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277: 33598–33603 [DOI] [PubMed] [Google Scholar]
- Bang H, Kim Y, Kim D (2000) TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem 275: 17412–17419 [DOI] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD (2004) Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology 46: 299–310 [DOI] [PubMed] [Google Scholar]
- Bockaert J, Fagni L, Dumuis A, Marin P (2004) GPCR interacting proteins (GIP). Pharmacol Ther 103: 203–221 [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Colledge M, Kittler JT, Brandon JM, Scott JD, Moss SJ (2003) A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABA(A) receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol Cell Neurosci 22: 87–97 [DOI] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD (1992) Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP79. J Biol Chem 267: 16816–16823 [PubMed] [Google Scholar]
- Chemin J, Girard C, Duprat F, Lesage F, Romey G, Lazdunski M (2003) Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J 22: 5403–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E (2005) A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J 24: 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Brown MW, Bashir ZI (2002) Mechanisms and physiological role of enhancement of mGlu5 receptor function by group II mGlu receptor activation in rat perirhinal cortex. J Physiol 540: 895–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000) Targeting of PKA to glutamate receptors through a MAGUK–AKAP complex. Neuron 27: 107–119 [DOI] [PubMed] [Google Scholar]
- Colledge M, Scott JD (1999) AKAPs: from structure to function. Trends Cell Biol 9: 216–221 [DOI] [PubMed] [Google Scholar]
- Dart C, Leyland ML (2001) Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J Biol Chem 276: 20499–20505 [DOI] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ (2005) Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci 25: 8776–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac S, Franco M, Bendahhou S, Warth R, Knauer S, Barhanin J, Lazdunski M, Lesage F (2004) ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GTP exchange factor for ARF6. EMBO Rep 5: 1171–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M (1997) TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16: 5464–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M (2000) The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol Pharmacol 57: 906–912 [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854–6862 [PMC free article] [PubMed] [Google Scholar]
- Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M (1998) A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 17: 3297–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185–196 [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S (2005) International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57: 527–540 [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, Lazdunski M (2004) TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J 23: 2684–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Pironkova R, Onwumere O, Vologodskaia M, Hudspeth AJ, Lesage F (2002) RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca2+ channels. Neuron 34: 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honoré E, Maingret F, Lazdunski M, Patel AJ (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO J 21: 2968–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6: 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D (2005) Physiology and pharmacology of two-pore domain potassium channels. Curr Pharm Des 11: 2717–2736 [DOI] [PubMed] [Google Scholar]
- Kim Y, Gnatenco C, Bang H, Kim D (2001) Localization of TREK-2 K+ channel domains that regulate channel kinetics and sensitivity to pressure, fatty acids and pHi. Pflugers Arch 442: 952–960 [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Blondeau N, Heurteaux C, Widmann C, Romey G, Lazdunski M (2000) Polyunsaturated fatty acids are potent neuroprotectors. EMBO J 19: 1784–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J (1996a) TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15: 1004–1011 [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Reyes R, Fink M, Duprat F, Guillemare E, Lazdunski M (1996b) Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J 15: 6400–6407 [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M (2000) Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801 [DOI] [PubMed] [Google Scholar]
- Lesage F, Terrenoire C, Romey G, Lazdunski M (2000) Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem 275: 28398–28405 [DOI] [PubMed] [Google Scholar]
- Levitan IB (2006) Signaling protein complexes associated with neuronal ion channels. Nat Neurosci 9: 305–310 [DOI] [PubMed] [Google Scholar]
- Liu G, Shi J, Yang L, Cao L, Park SM, Cui J, Marx SO (2004) Assembly of a Ca2+-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. EMBO J 23: 2196–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CM, Rohacs T, Czirjak G, Balla T, Enyedi P, Logothetis DE (2005) PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol 564: 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E (1999) Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274: 26691–26696 [DOI] [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E (2000a) TREK-1 is a heat-activated background K+ channel. EMBO J 19: 2483–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E (2000b) Lysophospholipids open the two-pore domain mechano-gated K+ channels TREK-1 and TRAAK. J Biol Chem 275: 10128–10133 [DOI] [PubMed] [Google Scholar]
- Maingret F, Honoré E, Lazdunski M, Patel AJ (2002) Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K+ channel. Biochem Biophys Res Commun 292: 339–346 [DOI] [PubMed] [Google Scholar]
- Murbartian J, Lei Q, Sando JJ, Bayliss DA (2005) Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J Biol Chem 280: 30175–30184 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E (2001) Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 24: 339–346 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci 2: 422–426 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17: 4283–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C, Lauritzen I, Lesage F, Romey G, Lazdunski M (2001) A TREK-1-like potassium channel in atrial cells inhibited by beta-adrenergic stimulation and activated by volatile anesthetics. Circ Res 89: 336–342 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information