

Abstract
In Caenorhabditis elegans adults, the single Rho GTPase orthologue, RHO-1, stimulates neurotransmitter release at synapses. We show that one of the pathways acting upstream of RHO-1 in acetylcholine (ACh)-releasing motor neurons depends on Gα12 (GPA-12), which acts via the single C. elegans RGS RhoGEF (RHGF-1). Activated GPA-12 has the same effect as activated RHO-1, inducing the accumulation of diacylglycerol and the neuromodulator UNC-13 at release sites, and increased ACh release. We showed previously that RHO-1 stimulates ACh release by two separate pathways—one that requires UNC-13 and a second that does not. We show here that a non-DAG-binding-UNC-13 mutant that partially blocks increased ACh release by activated RHO-1 completely blocks increased ACh release by activated GPA-12. Thus, the upstream GPA-12/RHGF-1 pathway stimulates only a subset of RHO-1 downstream effectors, suggesting that either the RHO-1 effectors require different levels of activated RHO-1 for activation or there are two distinct pools of RHO-1 within C. elegans neurons.
Keywords: C. elegans , G protein, mUNC13, neurotransmitter, Rho GTPase
Introduction
Rho family GTPases regulate numerous processes in eukaryotic cells, including organisation of the cytoskeleton, cell morphology and motility, gene expression, and neurotransmitter release (Jaffe and Hall, 2005; McMullan et al, 2006). They function as molecular switches and are active when GTP is bound and are inactive when GDP is bound. They are activated by specific guanine-nucleotide-exchange factors (GEFs), which stimulate the exchange of GTP for GDP (Schmidt and Hall, 2002). More than 85 RhoGEFs have been identified (Jaffe and Hall, 2005), all of which contain a Dbl homology (DH) domain that is responsible for the GDP–GTP exchange activity; most also contain a Pleckstrin homology (PH) domain and a variety of interaction domains that are implicated in signal transduction.
We have recently shown that the single Caenorhabditis elegans Rho orthologue (RHO-1) acts in adult animals to regulate neurotransmitter release (McMullan et al, 2006). Expression of a constitutively active RHO-1 (G14V) in C. elegans cholinergic motor neurons stimulates acetylcholine (ACh) release; conversely, inhibition of endogenous RHO-1 via the Rho inhibitor C3 transferase reduces ACh release. RHO-1 regulates ACh release by at least two separate pathways. In one, presynaptic RHO-1 increases ACh release by stimulating the accumulation of diacylglycerol (DAG) and the DAG-binding protein UNC-13 at sites of neurotransmitter release; this pathway is blocked by UNC-13 mutants unable to bind DAG. A second UNC-13-independent pathway is revealed by a RHO-1 mutant unable to increase DAG levels; this RHO-1 mutant protein retains the ability to stimulate ACh release by a mechanism that is not blocked by the non-DAG-binding-UNC-13 mutant.
The heterotrimeric G proteins Gq, G12, and G13 have been shown to mediate signals from G protein-coupled receptors (GPCRs) to Rho GTPases. These G proteins are believed to activate directly RhoGEFs that contain a Regulator of G protein Signalling (RGS) domain: p115 RhoGEF, for example, binds activated Gα12 or Gα13 through its RGS domain, and Gα13 binding stimulates p115 RhoGEF activity (Hart et al, 1998; Kozasa et al, 1998). Mutations in either Drosophila Gα12 or p115 RhoGEF homologues (concertina and DRhoGEF2) disrupt gastrulation, suggesting that these proteins may act in the same signalling pathway (Parks and Wieschaus, 1991; Barrett et al, 1997; Hacker and Perrimon, 1998). In C. elegans, the single Gα12 protein (GPA-12) has been implicated in the control of pharyngeal pumping (van der Linden et al, 2003). Both GPA-12 and the single RGS RhoGEF, RHGF-1, are coexpressed in the C. elegans nervous system, although GPA-12 is more strongly expressed in cells where RHGF-1 is absent, including the hypodermis, muscle, intestinal cells, and pharynx (van der Linden et al, 2003; Yau et al, 2003).
To understand how RHO-1 is regulated in the adult C.elegans nervous system, we have examined the role of the GPA-12/RHGF-1 signalling pathway in neurotransmitter release in motor neurons. We show that GPA-12 stimulates ACh release by a pathway that depends on RHGF-1, RHO-1, and UNC-13.
Results
GPA-12 stimulates ACh release in a RHO-1-dependent manner
To test for a role of GPA-12 in the control of ACh release, we used a heat shock-inducible, constitutively active Gα12 transgene (hs∷GPA-12 (Q205L)) constructed by van der Linden et al (2003) (Figure 1A). ACh release was quantified by measuring the time course of paralysis induced by the ACh esterase inhibitor aldicarb (Nonet et al, 1993; Nguyen et al, 1995; Miller et al, 1996; Nurrish et al, 1999). Aldicarb blocks removal of endogenously released ACh, resulting in hypercontraction of the muscles and paralysis. Increases in ACh release cause animals to become paralysed faster with aldicarb treatment; conversely, animals with a decrease in ACh release become paralysed more slowly and continue to move at time points when wild-type animals are completely paralysed. Heat shock-induced expression of constitutively active GPA-12 (Q205L) in adults increased aldicarb sensitivity both immediately and 24 h post-heat shock compared to controls, suggesting that GPA-12 increased ACh release (Figure 1B). Even without heat shock, the hs∷GPA-12 (Q205L) animals were slightly hypersensitive to aldicarb, probably due to a leakiness of the heat shock promoter (Figure 1B). Expression of either constitutively active GPA-12 (Q205L) or constitutively active RHO-1 (G14V) (McMullan et al, 2006) mutants resulted in aldicarb hypersensitivity, consistent with a role for GPA-12 in activating RHO-1.
Figure 1.
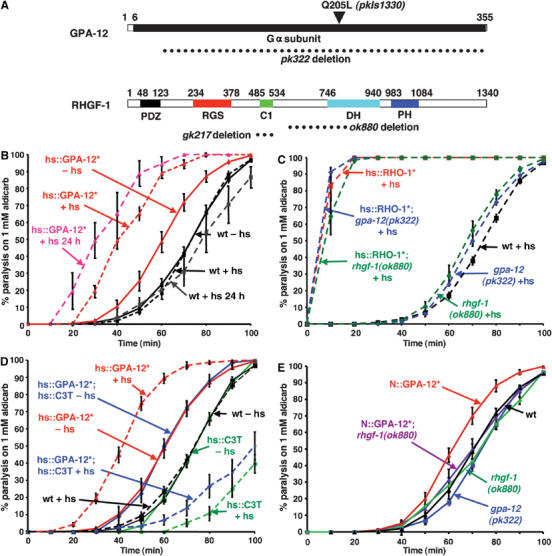
A constitutively active GPA-12 (Q205L) acts within motor neurons to increase ACh release upstream of RHO-1. (A) In C. elegans GPA-12 is the single Gα12 orthologue, and RHGF-1 is the single RGS-containing RhoGEF, see text for abbreviations. To investigate the role of GPA-12 and RHGF-1 on ACh release, we tested pk322 and ok880 deletion mutants, respectively (deletions indicated by dotted lines): pk322 removes almost the entire coding region of gpa-12 and is likely a null mutant, ok880 is an in-frame deletion that removes the N-terminal part of the DH RhoGEF domain (residues 599–804). Another mutation rhgf-1(gk217) is an in-frame deletion that removes the RHGF-1 C1 domain. The pkIs1330 integrated transgene (van der Linden et al, 2003) expresses the constitutively active GPA-12 (Q205L) mutant from a heat shock promoter. (B–D) Levels of ACh release were assessed by testing for rates of paralysis on the ACh esterase inhibitor aldicarb, animals with increased rates of ACh release paralyse faster on aldicarb. Dashed lines indicate heat shocked animals. Mean % of paralysed animals are shown. Error bars indicate s.e.m., all assays were performed at least five times. (B) The hs∷ GPA-12 (Q205L) transgene (hs∷GPA-12*) paralysed faster on aldicarb than wild-type animals (wt) even in the absence of heat shock (−hs). Rates of paralysis were increased both immediately (+hs) and 24 h (+hs 24 h) post-heat shock. (C) gpa-12(pk322) and rhgf-1(ok880) mutant animals were slightly aldicarb hypersensitive immediately after heat shock. Neither altered aldicarb hypersensitivity caused by heat shock expression of constitutively active RHO-1 (G14V). (D) Aldicarb hypersensitivity caused by heat shock expression of GPA-12 (Q205L) (hs∷GPA-12*+hs) was blocked by inhibition of endogenous RHO-1 by the specific Rho inhibitor C3 transferase expressed from a heat shock promoter (hs∷GPA-12*; hs∷C3T+hs). (E) Expression of the constitutively active GPA-12 (Q205L) in cholinergic neurons using the unc-17 promoter (N∷GPA-12*) caused aldicarb hypersensitivity that was suppressed by the rhgf-1(ok880) mutation (N∷GPA-12*;rhgf-1(ok880)). Mutations in both gpa-12(pk322) and rhgf-1(ok880) did not change the response to aldicarb.
Two results suggested that GPA-12 was acting upstream of RHO-1. First, aldicarb hypersensitivity of animals expressing constitutively active RHO-1 (G14V) was not altered by the presence of the null gpa-12(pk322) mutation (Figure 1A and C). Second, inhibition of endogenous RHO-1 by C3 transferase almost completely suppressed the increase in aldicarb hypersensitivity caused by expression of the constitutively active GPA-12 (Q205L) (Figure 1D). The gpa-12 (pk322) mutation alone had no effect on the sensitivity to aldicarb in the absence of heat shock (Figure 1E) and was very slightly hypersensitive to aldicarb in the presence of heat shock (Figure 1C), suggesting that GPA-12 signalling is not required for standard rates of ACh release under laboratory conditions.
To test whether GPA-12 had a role in motor neuron development, we labelled cholinergic motor neurons using GFP expressed from the acr-2 promoter. We observed no gross morphological differences in neuronal development between heat shocked wild-type and hs∷GPA-12 (Q205L) animals both immediately and 24 h after heat shock (Figure 2A–C).
Figure 2.
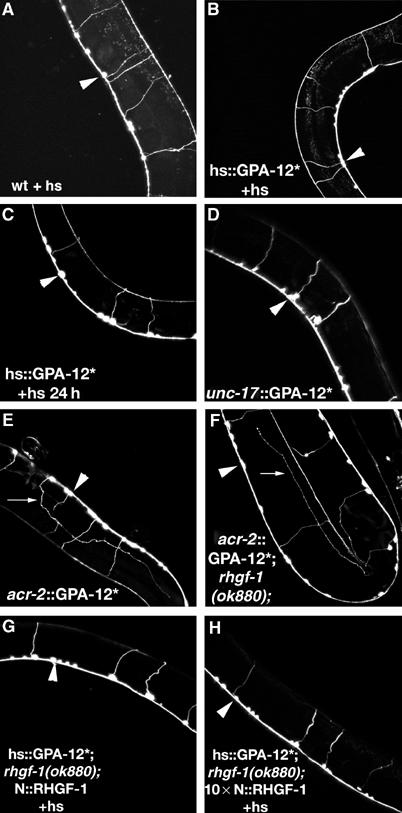
Expression of GPA-12 (Q205L) from the acr-2 promoter changes gross neuronal morphology. (A–G) Soluble GFP was expressed in the cholinergic motor neurons from the acr-2 promoter. In most cases, the anterior side of the animal is towards the upper left of the picture, in (B) anterior is top, and in (F) anterior is the leftmost part of the animal. Arrowheads indicate the ventral nerve cord. Motor neuron morphology was the same immediately after heat shock in wild-type (A) and in hs∷GPA-12 (Q205L) animals immediately (B), and 24 h after heat shock (C). GPA-12 (Q205L) expressed in cholinergic cells from the unc-17 promoter (unc-17∷GPA-12*) did not alter gross morphology of the motor neurons (D). Expression of GPA-12 (Q205L) from the p.acr-2 promoter (acr-2∷GPA-12*) did cause a pathfinding defect (indicated by an arrow) (E) and this was not suppressed by the rhgf-1(ok880) mutation (F). Expression of rhgf-1 cDNA from the acr-2 promoter injected at 10 ng/μl (G) or 100 ng/μl (H) in hs∷GPA-12 (Q205L); rhgf-1(ok880) animals did not alter gross neuronal morphology.
Gα12 acts presynaptically to stimulate ACh release
Although the hs∷GPA-12 (Q205L) transgene is expressed in the cholinergic neurons, it is also expressed in many other cells, including muscle cells. We tested whether the transgene was acting presynaptically in two ways. First, we expressed the constitutively active GPA-12 (Q205L) specifically in cholinergic cells using the promoter for the unc-17 vesicular ACh transporter (p.unc-17) and showed it caused aldicarb hypersensitivity, albeit with a weaker effect than the hs∷GPA-12 (Q205L) transgene (Figure 1E). Secondly, we tested the sensitivity of the muscles to ACh release in heat shock-induced transgenic animals expressing GPA-12 (Q205L). The drug levamisole activates the nicotinic ACh receptors in the muscle causing animals to become paralysed at a rate dependent on muscle sensitivity to ACh (Nurrish et al, 1999). The heat shocked animals showed no significant increase in levamisole sensitivity compared to controls, suggesting that the heat shock expression of GPA-12 (Q205L) did not alter muscle response to ACh (Supplementary Figure 1A and B). Thus, GPA-12, like RHO-1, can act presynaptically to increase ACh release.
We also expressed constitutively active GPA-12 (Q205L) specifically in the cholinergic motor neurons using the acr-2 promoter. This transgene, however, caused neuronal pathfinding defects (Figure 2E) and, therefore, could not be used to assay for aldicarb sensitivity. By contrast, expression of GPA-12 (Q205L) from the unc-17 promoter did not cause a detectable neurite pathfinding defect (Figure 2D).
RHGF-1 is required for GPA-12-mediated ACh release
RHGF-1 is the single RhoGEF containing a G protein-regulated RGS domain in C. elegans. RHGF-1 binds to GPA-12 (Yau et al, 2003), and is thus a strong candidate for mediating GPA-12's ability to stimulate RHO-1. We obtained a full-length SL1 spliced cDNA for rhgf-1, which encodes a protein that contains five conserved domains-PDZ (PSD-95/Dlg/ZO-1), RGS, C1 (PKC homology domain 1), DH, and PH (Figure 1A). We received a mutant strain with a deletion in rhgf-1(ok880), from which we isolated cDNA and confirmed the presence of an in-frame deletion that removes amino acids 599–804 (Figure 1A). The predicted truncated protein lacks the first 58 residues of the DH domain, which is required for RhoGEF activity in mammalian RhoGEFs (Kristelly et al, 2004). A second allele rhgf-1(gk217) has an in-frame deletion that removes the C1 domain (Figure 1A), leaving the other conserved domains. Strains carrying this mutation appear to have at least one other closely linked mutation that decreases the rate of paralysis induced by aldicarb (see Materials and methods), and so we were unable to test this allele. The responses of rhgf-1(ok880) mutants in the aldicarb and levamisole assays were indistinguishable from those of wild type in the absence of heat shock, but were very slightly aldicarb hypersensitive in the presence of heat shock (Figure 1C and E and Supplementary Figure 1A and B). However, rhgf-1(ok880) significantly reduced aldicarb hypersensitivity caused by the hs∷GPA-12 (Q205L) transgene (Figure 3A and B), and it completely suppressed the aldicarb hypersensitivity of animals expressing GPA-12 (Q205L) in cholinergic neurons (from the unc-17 promoter) (Figure 1E), suggesting that GPA-12 (Q205L)-mediated increases in ACh release require RHGF-1.
Figure 3.
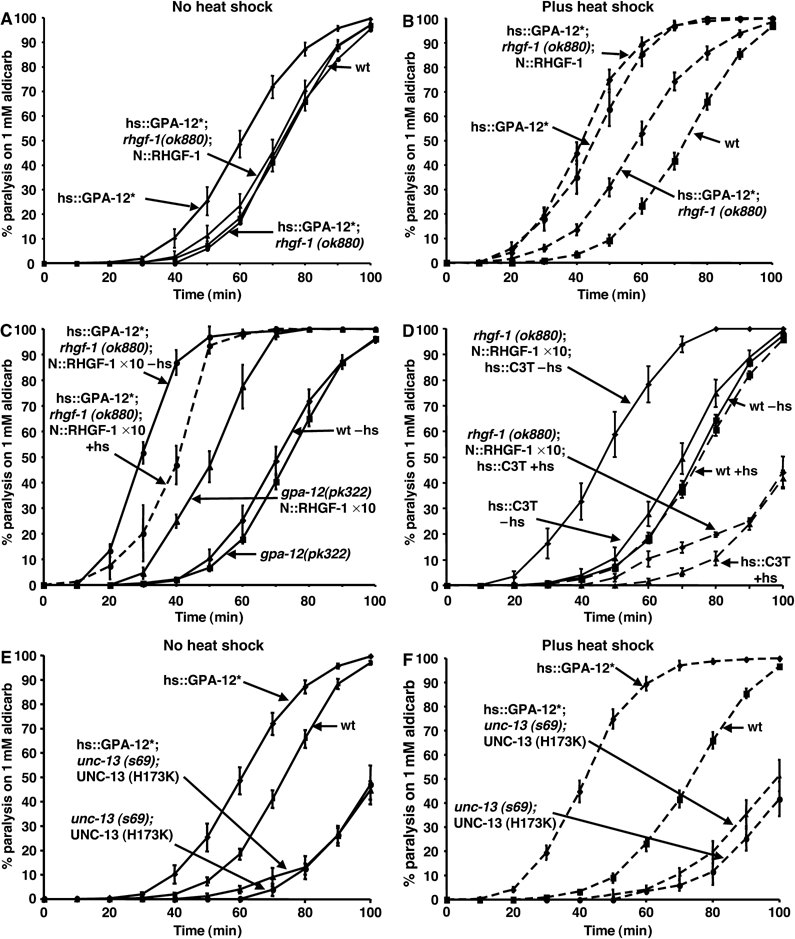
GPA-12 (Q205L) stimulation of ACh release requires RHGF-1, DAG, and UNC-13. (A, B) The RGS RhoGEF mutation (rhgf-1(ok880)) completely suppressed aldicarb hypersensitivity caused by the hs∷GPA-12 (Q205L) transgene (hs∷GPA-12*;rhgf-1(ok880)) in the absence of heat shock (A) and strongly suppresses in the presence of heat shock (B). In hs∷GPA-12 (Q205L); rhgf-1(ok880) animals, expression of RHGF-1 from the cholinergic motor neuron-specific acr-2 promoter (N∷RHGF-1) at low copy number (injected at 10 ng/μl, hs∷GPA-12*;rhgf-1(ok880);N∷RHGF-1) had no effect on the response of animals to aldicarb in the absence of heat shock (A), but restored aldicarb hypersensitivity caused by heat-shock-induced expression of GPA-12 (Q205L) (B). (C) A transgene with a 10-fold increase in p.acr-2∷RHGF-1 (injected at 100 ng/μl N∷RHGF-1 × 10) caused aldicarb hypersensitivity in hs∷GPA-12*;rhgf-1(ok880) animals without heat shock (−hs). Aldicarb hypersensitivity was decreased slightly upon heat-shock-induced expression of GPA-12 (Q205L) (+hs) and much more strongly by removal of all GPA-12 using the gpa-12(pk322) mutation. (D) Inhibition of endogenous RHO-1 by heat shock expression of C3 transferase (hs∷C3T +hs) blocked aldicarb hypersensitivity caused by N∷RHGF-1 × 10 (compare rhgf-1(ok880);N∷RHGF-1 × 10;hsC3T in the absence (−hs) or presence (+hs) of C3 transferase expression). (E, F) Replacement of endogenous UNC-13 by a mutant UNC-13S unable to bind diacylglycerol (DAG) (unc-13(s69); UNC-13S (H173K)) caused aldicarb resistance (E) that is not altered by heat shock expression of constitutively active GPA-12 (Q205L) (F).
RHGF-1 acts within the cholinergic motor neurons upstream of RHO-1
To determine the site of action of RHGF-1, we rescued the rhgf-1(ok880) mutants using full-length rhgf-1 cDNA expressed from the cholinergic-motor-neuron-specific acr-2 promoter (N∷RHGF-1). This transgene restored aldicarb hypersensitivity induced by heat shock of hs∷GPA-12 (Q205L) in rhgf-1(ok880) animals (Figure 3B), but not in the absence of heat shock (Figure 3A). Transgenic animals are created by injecting DNA into the gonads of C. elegans and looking for stable transmission of the DNA in the progeny (Mello and Fire, 1995). N∷RHGF-1 transgenics were created by injecting either 10 ng/μl (N∷RHGF-1) or 100 ng/μl (N∷RHGF-1 × 10). The N∷RHGF-1 × 10 transgene caused aldicarb hypersensitivity in hs∷GPA-12 (Q205L); rhgf-1(ok880) mutants in the absence of heat shock (Figure 3C). Surprisingly, aldicarb hypersensitivity was slightly decreased upon heat shock-induced expression of the constitutively active GPA-12 (Q205L) (Figure 3C). Removal of all GPA-12 activity using null gpa-12(pk322) animals also reduced the aldicarb hypersensitivity associated with the N∷RHGF-1 × 10 transgene, although not back to wild-type levels (Figure 3C). Inhibition of endogenous RHO-1 by heat shock expression of C3 transferase almost completely suppressed the aldicarb hypersensitivity of N∷RHGF-1 × 10 to levels observed with expression of C3 transferase alone, consistent with a role for RHGF-1 upstream of RHO-1 (Figure 3D). Gross neuronal morphology of transgenic animals expressing both low levels and high levels of RHGF-1 were normal (Figure 2G and H).
GPA-12 increases ACh release at existing synapses
GPA-12 could be stimulating an increase in ACh release through either an increase in the number of ACh-releasing synapses, an increase in the release of ACh from existing synapses, or both. To test the first possibility, we measured the number of synapses in hs∷GPA-12 (Q205L) animals using CFP-labelled synaptobrevin (SNB-1∷CFP) expressed specifically in cholinergic motor neurons using the acr-2 promoter. Synaptobrevin is an integral membrane protein enriched in synaptic vesicles and thus acts as a marker for release sites. We observed no significant difference in the number of SNB-1∷CFP puncta before heat shock or immediately and 24 h post-heat shock in either wild-type (2.48±0.11 versus 2.51±0.10 and 2.38±0.11 puncta per 10 μm, all errors are s.e.m.) or hs∷GPA-12 (Q205L) animals (2.49±0.09 versus 2.56±0.11 and 2.3±0.11 puncta per 10 μm) (Figure 4A and B). Thus, heat shock induction of GPA-12 (Q205L) in adults did not increase the number of release sites in cholinergic motor neurons, suggesting that constitutively active GPA-12 (Q205L) increases ACh release at pre-existing release sites. Mutation of rhgf-1, either in the presence or absence of heat shock-induced GPA-12 (Q205L) also failed to change the number of SNB-1∷CFP puncta (2.58±0.10 versus 2.54±0.09 puncta per 10 μm, Figure 4B), suggesting that the rhgf-1(ok880) mutation does not suppress GPA-12 (Q205L) by reducing the number of ACh release sites. We also observed no change in SNB-1∷CFP puncta numbers in gpa-12(pk322) mutants (2.5±0.1 puncta per 10 μm, Figure 4B). The rhgf-1(ok880) mutants did have a decrease in the number of release sites in the dorsal cord (2.27±0.06 puncta per 10 μm, Figure 4), but this just failed to reach a statistically significant difference from the non-heat-shocked wild type (P=0.051 in a two-tailed t-test). In all mutant and transgenic animals tested, there was no significant difference from wild type in average size (non-heat-shocked wild type puncta size 0.7 μm2±0.05) (Figure 4C) or fluorescence (Figure 4D) of the SNB-1 puncta. The only notable difference was a much increased variation in SNB-1 puncta size in hs∷GPA-12 (Q205L) animals immediately, but not 24 h, post-heat shock.
Figure 4.

hs∷GPA-12 (Q205L) does not increase the number, size, or fluorescence of SNB-1∷CFP puncta. (A) Dorsal cord neuromuscular junctions were labelled by expressing a CFP-tagged version of synaptobrevin (SNB-1∷CFP, which is enriched in synaptic vesicles) in cholinergic motor neurons (using the acr-2 promoter). Animals with an integrated array expressing the constitutively active GPA-12 (Q205L) from a heat shock promoter (hs∷GPA-12 (Q205L)) either without heat shock (top panel) or with heat shock (lower panel) had the same density of SNB-1∷CFP puncta. (B–D) Numbers (B), average size (C), and average fluorescence (D) of SNB-1∷CFP puncta in dorsal nerve cords were measured using Image J for animals with the indicated genotypes and heat shock conditions (see Materials and methods). Bars are means±s.e.m., numbers of animals counted are given in brackets. Statistical analysis was performed using a two-tailed t-test and no significant differences were detected (P>0.05).
GPA-12 increases UNC-13 levels at release sites via RHGF-1
We have previously shown that RHO-1 acts via two pathways in motor neurons to increase ACh release: one dependent on the DAG-binding neuromodulator UNC-13 and the other UNC-13 independent (McMullan et al, 2006). In the UNC-13 dependent pathway, RHO-1 increases the short form of UNC-13 (UNC-13S) at release sites via a spatially restricted increase in DAG levels. The enrichment of UNC-13 at release sites has been shown to correlate with increased neurotransmitter release (Betz et al, 1998; Lackner et al, 1999; Nurrish et al, 1999). Expression of constitutively active RHO-1 (G14V) in cholinergic motor neurons causes an UNC-13S∷YFP fusion protein to localise to punctate structures that colocalise with the SNB-1∷CFP marker for synaptic vesicles (McMullan et al, 2006). Both immediately and 24 h after heat shock expression of constitutively active GPA-12 (Q205L), the time points we observed aldicarb hypersensitivity, UNC-13S∷YFP in the dorsal nerve cord was localised into punctate structures, whereas it was not so localised in non-heat-shocked animals (1.84±0.11 pre-heat shock versus 2.29±0.12 and 2.2±0.14 versus puncta per 10 μm, using two-tailed t-test P=0.036 between before and immediately after heat shock) (Figure 5A–D and I), as we previously observed for RHO-1 (G14V) (McMullan et al, 2006). These UNC-13S∷YFP puncta colocalise with the release-site marker SNB-1∷CFP, confirming that GPA-12 (Q205L) causes UNC13S to localize to sites of neurotransmitter release (Figure 5H). In the rhgf-1(ok880) mutant animals, UNC-13S∷YFP fails to become punctate in response to heat-shock-induced GPA-12 (Q205L) expression (1.97±0.09 versus 1.86±0.11 puncta per 10 μm) (Figure 5E, F and I). Thus, rhgf-1(ok880) blocks both GPA-12 (Q205L)-mediated increases in ACh release and relocalisation of UNC-13S∷YFP, suggesting that GPA-12 (Q205L) stimulates ACh release via changes in UNC-13 localisation in a RHGF-1-dependent manner. Neither the gpa-12(pk322) nor the rhgf-1(ok880) mutant altered UNC-13∷YFP puncta density in the dorsal cord (Figure 5I).
Figure 5.

GPA-12 (Q205L) regulates the distribution of UNC-13S∷YFP in the dorsal nerve cord. UNC-13∷YFP and SNB-1∷CFP transgenes and image analysis were as described previously (McMullan et al, 2006). In (A–G), digital images were converted from greyscale into a 32-colour look-up table (Image J) to visualize pixel intensities. (A–F) UNC-13S∷YFP (expressed from the internal unc-13S promoter) was diffusely distributed in the dorsal nerve cord axons of untreated (A) and heat shocked (B) wild-type animals as well as non-heat-shocked animals containing the hs∷GPA-12 (Q205L) array (hs∷GPA-12*) (C). UNC-13S∷YFP became more punctate (as indicated by arrows) in the dorsal cords of hs∷GPA-12 (Q205L) (hs∷GPA-12*) animals immediately after heat shock (D). However, a mutation in the single RGS RhoGEF, rhgf-1(ok880) (E, F), blocked this increase, compare (D) with (F). (G) Unlike the wild-type protein, UNC-13S (H173K)∷GFP, which is predicted not to bind to DAG, remains diffusely distributed in heat shocked hs∷GPA-12 (Q205L) (hs∷GPA-12*) animals. (H) CFP-tagged synaptobrevin expressed from the acr-2 promoter (top), and YFP-tagged UNC-13S expressed from the unc-13s promoter (middle) were simultaneously visualized in the dorsal cord of heat shocked hs∷GPA-12 (Q205L) animal. In the merged image (bottom), the SNB-1∷CFP puncta colocalise with UNC-13S∷YFP puncta. (I) Numbers of either UNC-13S∷YFP or non-DAG-binding UNC-13S (H173K)∷GFP puncta in dorsal nerve cords were counted in the indicated genotypes and heat shock conditions (see Materials and methods). Bars are means±s.e.m., numbers of animals counted are given in brackets. Significance was determined by two-tailed t-test. *Significant difference (P<0.05).
The GPA-12/RHGF-1 pathway activates a subset of RHO-1 effectors
Replacement of wild-type UNC-13 by the non-DAG-binding UNC-13S (H173K) mutant causes resistance to aldicarb (Lackner et al, 1999) (Figure 3E). Constitutively active RHO-1 (G14V) causes aldicarb hypersensitivity, an effect that is only partially suppressed by UNC-13S (H173K), demonstrating the existence of both UNC-13-dependent and UNC-13-independent pathways downstream of RHO-1 (McMullan et al, 2006). In contrast to constitutively active RHO-1, the aldicarb hypersensitivity of animals expressing constitutively active GPA-12 (Q205L) was completely suppressed by UNC-13S (H173K) (Figure 3E and F). Thus, GPA-12-mediated aldicarb hypersensitivity requires binding of UNC-13 to DAG, whereas its downstream effector RHO-1 can act via both UNC-13-dependent and UNC-13-independent pathways. Heat shock expression of the constitutively active GPA-12 (Q205L) did not significantly alter the localisation of the non-DAG-binding UNC-13S (H173K)∷GFP (1.33±0.19 versus 1.53±0.14 puncta per 10 μm. P=0.55, two-tailed student's t-test) (Figure 5 G and I), suggesting that GPA-12 (Q205L) alters UNC-13S localization via a spatially restricted increase in DAG levels at sites of neurotransmitter release. There appeared to be a difference in numbers of UNC-13S (H173K)∷GFP puncta between heat shocked wild-type animals and heat shocked hs∷GPA-12 (Q205L) transgenic animals (1.16±0.19 versus 1.53±0.14) (Figure 5I); this was not statistically significant (P=0.07 two-tailed t-test), however.
Constitutively active GPA-12 (Q205L) causes defects in development and pharyngeal pumping independent of RHGF-1
Previous work has shown that the constitutively active GPA-12 (Q205L) causes developmental growth arrest, which is secondary to decreased pharyngeal pumping. It is possible that the aldicarb hypersensitivity and changes in UNC-13 localization caused by GPA-12 (Q205L) is also secondary to reduced pharyngeal pumping. However, both the growth arrest and decreased pharyngeal pumping associated with GPA-12 (Q205L) were suppressed by mutations in the tpa-1 gene, whereas aldicarb hypersensitivity was not suppressed by tpa-1 (Supplementary Figure 2). We have repeated the experiments of van der Linden et al (2003) and confirmed the ability of tpa-1 mutations to suppress growth and pharyngeal pumping defects caused by GPA-12 (Q205L) expression (Figure 6A and B). The rhgf-1(ok880) mutation, which suppressed GPA-12 (Q205L) aldicarb hypersensitivity, failed to suppress the pharyngeal pumping and growth defects caused by expression of GPA-12 (Q205L) (Figure 6A and B). Thus, the hs∷GPA-12 (Q205L);rhgf-1(ok880) animals have reduced pharyngeal pumping but an almost normal response to aldicarb, indicating that changes in aldicarb sensitivity associated with expression of constitutively active GPA-12 (Q205L) appear to be independent of changes in pharyngeal pumping. The rhgf-1(ok880) mutation also failed to suppress the neuronal pathfinding defect caused by expression of the constitutively active GPA-12 (Q205L) from the acr-2 promoter (Figure 2F).
Figure 6.
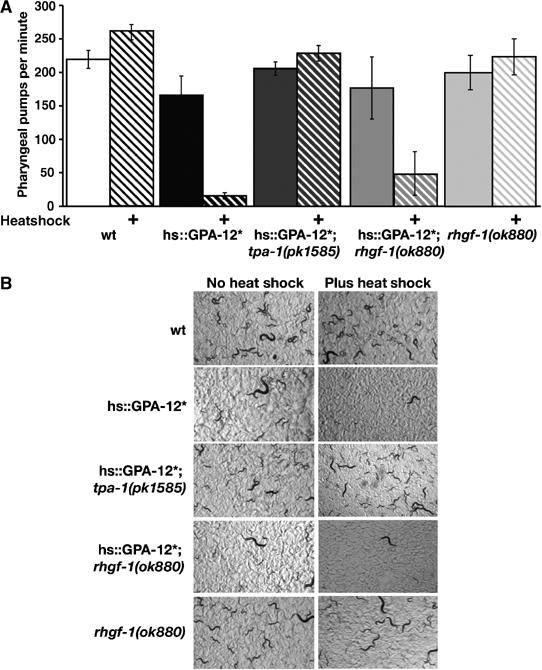
TPA-1 and RHGF-1 define at least two different GPA-12 signalling pathways regulating distinct behaviours. (A) Heat shock expression of the constitutively active GPA-12 (Q205L) severely reduced pharyngeal pumping, and this was suppressed by a PKC mutation tpa-1(pk1585) (van der Linden et al, 2003), whereas the rhgf-1(ok880) mutation has no effect. (B) Twenty L1-stage animals were placed on plates, immediately heat shocked, and photographed 4 days later. Both wild-type and rhgf-1(ok880) animals produced adults both with and without heat shock. However, heat shock expression of the constitutively active GPA-12 (Q205L) results in a developmental arrest and few adults are observed. This arrest is suppressed by tpa-1(pk1585), but not by rhgf-1(ok880).
Discussion
We show that activation of the C. elegans Gα12 orthologue (GPA-12) in adult animals causes aldicarb hypersensitivity, an effect consistent with an increase of ACh release at neuromuscular junctions. In mammals, Gα12 activates an RGS RhoGEF, which in turn activates RhoA. We believe this pathway is conserved in C. elegans as both a mutation in rhgf-1 (which encodes the RGS RhoGEF RHGF-1) and inhibition of RHO-1 strongly suppresses the ability of GPA-12 to cause aldicarb hypersensitivity. RHGF-1 overexpression also causes aldicarb hypersensitivity that is strongly suppressed by inhibition of endogenous RHO-1. Thus, both GPA-12 and RHGF-1 require RHO-1 to increase ACh release. A mutation in gpa-12 does somewhat suppress the effects of RHGF-1 overexpression, which might either indicate a requirement for some basal GPA-12 activation of RHGF-1, even when overexpressed, that some functions of GPA-12 act in parallel to RHGF-1, or both. As the constitutively active GPA-12 (Q205L) mutant is not fully suppressed by rhgf-1(ok880), this may also represent some RHGF-1-independent signalling by GPA-12, although we do not know if rhgf-1(ok880) represents a null mutation. Nonetheless, we believe the genetic data support a model in which the Gα12/RGS RhoGEF/RhoA signalling pathway present in mammals is conserved in the control of ACh release in C. elegans.
Work from several groups has previously shown that three other C. elegans Gα subunits, EGL-30 (Gαq), GOA-1 (Gαo), and GSA-1 (Gαs), act in motor neurons to regulate ACh release (Mendel et al, 1995; Segalat et al, 1995; Brundage et al, 1996; Lackner et al, 1999; Schade et al, 2005) (Figure 7). Several lines of evidence suggest that the GPA-12/RHGF-1/RHO-1 pathway also acts within C. elegans motor neurons. Expression of RHGF-1 specifically in cholinergic motor neurons causes a dose-dependent effect; rescue of the GPA-12 (Q205L) induced aldicarb hypersensitivity at low levels and constitutive aldicarb hypersensitivity at high levels. It is interesting that heat-shock-induced expression of GPA-12 (Q205L) slightly decreases aldicarb hypersensitivity caused by RHGF-1 overexpression. Perhaps, feedback controls inhibit ACh release above a certain threshold of GPA-12/RHGF-1 signalling. Expression of GPA-12 (Q205L) from a cholinergic cell-specific-promoter (p.unc17) also causes aldicarb hypersensitivity, suggesting GPA-12 can act within cholinergic motor neurons, although unc-17 expression has also been observed in a subset of non-neural intestinal cells (J Rand, personal communication, 2006). Heat shock expression of constitutively active GPA-12 (Q205L) gives a much greater aldicarb hypersensitivity than expression driven from the unc-17 promoter. This could reflect either differences in levels of expression, or a role for GPA-12 in other cells in increasing ACh release, such as in interneurons or neuropeptide-releasing cells. As GPA-12 and RHGF-1 are both expressed in motor neurons and physically interact (Yau et al, 2003), our data best fits a model in which one site of action of the GPA-12/RHGF-1/RHO-1 pathway is at the C. elegans neuromuscular junction (Figure 7). Thus, at least four Gα proteins act to regulate neurotransmitter release in cholinergic motor neurons. Of these, three (Gαq, Gαo, and Gα12) converge to regulate DAG levels, and Gαs appears to act downstream of DAG. Each Gα protein is likely to be coupled to multiple GPCRs, suggesting that locomotion is controlled by multiple extracellular signals produced in response to changes in the internal and external environment.
Figure 7.

GPA-12 is a fourth G-protein pathway acting through RHGF-1 and RHO-1 to regulate ACh release in C. elegans cholinergic motor neurons. Our data suggest that GPA-12 (Gα12) acts within motor neurons to stimulate ACh release as do EGL-30 (Gαq) and GSA-1 (Gαs) (underlined), whereas GOA-1 (Gαo) acts to inhibit release (shaded). EGL-30, GOA-1, and GPA-12 pathways converge to regulate levels of diacylglycerol (DAG), GSA-1 appears to act downstream of DAG. DAG recruits UNC-13 to release sites and the accumulation of UNC-13 at release sites correlates with increases in ACh release. RHO-1 is also able to increase ACh release by inhibiting DGK-1 and by a second presynaptic pathway independent of DGK-1 inhibition and of DAG binding to UNC-13. The UNC-13-independent pathway is not activated by GPA-12, suggesting two distinct pools of RHO-1 may exist. This model is derived from data from the following papers (Mendel et al, 1995; Segalat et al, 1995; Brundage et al, 1996; Lackner et al, 1999; Miller et al, 1999; Nurrish et al, 1999; Richmond et al, 2001; Chase et al, 2004; Schade et al, 2005; McMullan et al, 2006).
GPA-12 stimulates ACh release via RHGF-1, RHO-1, and UNC-13 relocalisation to pre-existing release sites most probably through a spatially restricted increase in DAG levels. Unlike a constitutively active RHO-1 (G14V) mutant, a constitutively active GPA-12 (Q205L) acts in an entirely DAG- and UNC-13-dependent manner, as shown by the ability of the non-DAG-binding UNC-13S (H173K) mutant to block GPA-12 (Q205L) stimulation of ACh release. Two explanations, not mutually exclusive, may explain the difference between the expression of constitutively active RHO-1 and constitutively active GPA-12. In the first, GPA-12 (Q205L) causes a much lower amount of active RHO-1 to be formed than expression of RHO-1 (G14V). If the RHO-1 effector that triggers the UNC-13-dependent pathway has a higher affinity for active RHO-1 than effectors that trigger the UNC-13-independent pathway, then GPA-12 activation will preferentially activate the UNC-13-dependent pathway. In the second, the cholinergic motor neurons contain two distinct RHO-1 pools (Figure 7). One pool of RHO-1 is activated by GPA-12 via RHGF-1 and causes increases in the levels of both DAG and UNC-13 at release sites and thus an increase in ACh release. The second pool of RHO-1 is not activated by GPA-12 and its stimulation increases ACh release via an UNC-13-independent pathway; activation of this pool of RHO-1 may also activate the UNC-13-dependent pathway as well but we cannot test this possibility without knowing the upstream activators of this second RHO-1 pool. Selection of RHO-1 signalling outputs by different upstream activators has been previously reported: the scaffold protein CNK-1 links RhoGEFs and RhoA to activation of the Janus kinase (JNK), while decreasing RhoA stimulated stress fibre formation and activation of the serum response factor (SRF) (Jaffe et al, 2005). The use of scaffold proteins appears to be an important mechanism for channelling intracellular signals along specific pathways to ensure the appropriate cellular responses (Smith and Scott, 2002; Marinissen and Gutkind, 2005).
The failure of GPA-12 (Q205L) to activate the UNC-13-independent pathway downstream of RHO-1 implies other pathways, in addition to GPA-12, act upstream of RHO-1. The existence of other inputs to RHO-1 is also supported by the differing phenotypes of the null gpa-12(pk322) (no defect in ACh release) and that induced by the inhibition of endogenous RHO-1 by expression of C3 transferase (strong reduction in ACh release). Thus, under standard laboratory conditions, there is likely to be little signalling through the GPA-12/RHGF-1/RHO-1 pathway, and RHO-1 is likely to be activated by some other RhoGEF. Candidates for other upstream activators of RHO-1 are UNC-73 isoforms containing a RhoGEF domain. Loss of UNC-73 RhoGEF activity in neurons causes lethargic locomotion, similar to that caused by inhibiting RHO-1 via C3 transferase, although this change in locomotion has been reported to be independent of changes in ACh release (Steven et al, 2005). A large scale RNAi screen for regulators of ACh release has also implicated the gei-18 RhoGEF in this regulation (Sieburth et al, 2005).
The hs∷GPA-12 (Q205L) transgene causes increased aldicarb hypersensitivity both immediately and 24 h after heat shock. As we do not know the stability of the expressed GPA-12 (Q205L) protein, it is possible that the GPA-12 pathway is still active 24 h after heat shock or that this pathway causes other long-lasting changes that result in aldicarb hypersensitivity even after levels of GPA-12 (Q205L) protein have dropped to pre-heat shock levels. However, at both time points after heat shock, the recruitment of UNC-13 to release sites remains significantly higher than in non-heat-shocked animals, plus the numbers and size of cholinergic motor neuron release sites (as measured using the SNB-1∷CFP reporter) remain unaltered. Thus, we have no evidence to suggest any difference in signalling immediately and 24 h after heat shock. Interestingly, immediately after heat shock, the variation in the size of SNB-1 puncta is increased, although the average is not significantly changed. This could suggest a relocation of synaptic vesicles from some synapses to others, or possibly a change between the ratio of SNB-1 in synaptic vesicles and on the plasma membrane. However, this increased variation is not observed 24 h after heat shock and thus does not correlate with either aldicarb hypersensitivity or the increase in UNC-13S at release sites.
The constitutively active GPA-12 (Q205L) causes a developmental growth arrest due to strong inhibition of pharyngeal pumping, an effect that is suppressed by mutations in the tpa-1 PKC homologue (van der Linden et al, 2003). We also observe a motor neuron pathfinding defect associated with expression of the constitutively active GPA-12 (Q205L) from the acr-2 promoter. The rhgf-1(ok880) mutation fails to suppress the developmental arrest, reduction in pharyngeal pumping, and pathfinding defect but does suppress GPA-12 (Q205L)-mediated aldicarb hypersensitivity, indicating that changes in feeding are not responsible for the increased sensitivity to aldicarb. A mutation in tpa-1 fails to prevent GPA-12 (Q205L)-mediated aldicarb hypersensitivity, suggesting that GPA-12 acts in at least two independent signalling pathways, one that regulates pharyngeal pumping in a TPA-1-dependent manner and the second that regulates ACh release in an RHGF-1-dependent manner. Consistent with this suggestion, GPA-12 is expressed in many cell types where RHGF-1 is absent, including the pharynx (van der Linden et al, 2003; Yau et al, 2003). The neuronal pathfinding defect caused by p.acr-2∷GPA-12 (Q205L) is detected in neurons expressing p.acr-2∷GFP, suggesting that GPA-12 is acting cell-autonomously. It will be interesting to test if this pathfinding defect is suppressed by mutations in tpa-1, or whether it represents a third GPA-12 effector pathway.
The rhgf-1(ok880) mutant completely suppresses the aldicarb hypersensitivity due to cholinergic expression of the constitutively active GPA-12 (Q205L), but it does not completely suppress the increase induced by heat shock expression of the same GPA-12 mutant. This may reflect differences in the levels and/or places of GPA-12 (Q205L) expression from the two promoters. Alternatively, GPA-12 may interact with downstream effectors other than RHGF-1 when expressed at high levels or in noncholinergic cells. The rhgf-1(ok880) mutation removes a large part of the DH domain and is thus predicted to have no RhoGEF activity based on mammalian studies; however, numerous other domains are retained, and it is possible that high levels of GPA-12 can signal through these domains. A null rhgf-1 mutation might completely suppress aldicarb hypersensitivity induced by heat shock expression of GPA-12 (Q205L). Mutants with a deletion in the single Drosophila orthologue DRhoGEF2 fail to gastrulate (Barrett et al, 1997; Hacker and Perrimon, 1998), whereas rhgf-1(ok880) mutants are viable, suggesting a different requirement for these proteins in Drosophila and C. elegans or that domains other than the RhoGEF DH domain are required for viability. It will be useful to test if a null rhgf-1 mutant is lethal. However, in the case of gpa-12, it is clear that the null gpa-12(pk322) mutant is viable (van der Linden et al, 2003), whereas mutations in the Drosophila Gα12 homologue concertina, which acts upstream of DRhoGEF2, are not (Parks and Wieschaus, 1991). Thus, we believe that the Gα12/RGS RhoGEF/RhoA signalling pathways in C. elegans and Drosophila are used for different purposes in development.
Materials and methods
Strains
The N2 (wild type) strain, NL594 gpa-12(pk322), NL4258 pkIs1330[hs∷GPA-12 (Q205L)]; tpa-1(pk1585) dpy-20(e1282), RB976 rhgf-1(ok880), and VC430 rhgf-1(gk217) were obtained from the Caenorhabditis Genetics Center (University of Minnesota). rhgf-1(ok880) was backcrossed through N2 four times to give QT313 rhgf-1(ok880)*4. VC430 was backcrossed four times with selection for the rhgf-1(gk217) to give the QT263 strain. QT263 has a recessive aldicarb-resistance phenotype (ric); however, transgenes that rescue rhgf-1(ok880) failed to rescue the ric phenotype and we suspect that a closely linked gene to rhgf-1(gk217) is responsible. Thus, the rhgf-1(gk217) mutation was not analysed. QT199 pkIs1330 was obtained by backcrossing NL4258 twice with N2 and isolating animals with a growth arrest phenotype when heat shocked (i.e. had lost tpa-1(pk1401)). QT307 pkIs1330, rhgf-1(ok880), was generated by standard genetic crosses and the presence of the pkIs1330 and the rhgf-1(ok880) deletion were confirmed by PCR. For aldicarb assays, UNC-13S constructs were expressed in unc-13(s69) mutants. All strains were cultivated at 20°C unless otherwise stated and were maintained as described previously (Brenner, 1974).
Transgenes and germline transformation
Plasmids (listed as QT#, SJN, or EJH) were constructed by standard techniques, and verified by DNA sequencing. Transgenic strains (listed as QT) were isolated by microinjection of 100 ng/μl of plasmid, unless otherwise described below, together with p.acr-2∷gfp (KP#307), p.ttx-3∷gfp (a gift of O Hobert), or p.unc-122∷gfp (a gift of P Sengupta) as a marker. Unless otherwise stated, all injections were performed into N2 animals. Plasmids and transgenic strains were constructed as follows.
GPA-12 transgenes. We obtained the wild-type gpa-12 cDNA from yk336c8 (Yuji Kohara, National Institute of Genetics, Mishima, Japan). Using PCR, the NotI sequence plus 1 bp (GCGGCCGCC) was inserted immediately at the 3′ of the ATG start codon and the mutation Q205L was created along with an adjacent silent change to create a BglII site. This cDNA, encoding the constitutively active GPA-12 (Q205L) mutant, was then placed either under the control of the unc-17 promoter (McMullan et al, 2006) (EJH15) or the acr-2 promoter (EJH14). They were injected at 10 ng/μl into N2 to give nzEx230 and nzEx124, respectively.
RHGF-1 transgenes. The 3′ end of the RHGF-1 (F13e6.6) cDNA was derived from yk877c7 (a gift of Yuji Kohara, National Institute of Genetics, Mishima, Japan). The SL1 containing 5′ end was obtained using total N2 C. elegans RNA transcribed into cDNA using the SMARTRACE kit (Stratagene) as per the manufacturer's instructions. The oligos ggggggcggccgcaagcagtggtatcaacgcagagt (SMART oligo) and atgcgatctggataccaagcc were used in a first round PCR, followed by a second round PCR using oligos cccccgcggccgcggtttaattacccaagtttgag (SL1) and tgtggagaatatttcgaacgcccaacg, this PCR product was inserted into pBS using NotI and EcoRI to give SJN290. This 5′ end was then joined with the rest of the cDNA sequence from yk877c7, and a NotI site plus 1 bp (GCGGCCGCC) was inserted immediately 5′ of the STOP codon to give a full-length RHGF-1 cDNA (SJN305). The full-length RHGF-1 cDNA (NheI–KpnI fragment of SJN305) was then inserted behind the p.acr-2 promoter (Nurrish et al (1999), which is expressed in the neurons VA, DA, VB, DB, IL1, RMD, and PVQ) to give EJH4. SMART RACE was also used on total RNA from RB976 rhgf-1(ok880) using the oligos TGATGGTGTGGCCAAAGTAA and GCATTCAAGTCAAAGGGCAT in the first round PCR, and CGTAGTTTGCGCACTCACAT and TGTAGGGATGCTATCTGGGG in the second round to amplify rhgf-1 cDNA around the ok880 deletion.
RHGF-1 expressed from the p.acr-2 promoter (EJH4) was coinjected with p.acr-2∷GFP (KP#307) into QT307 at either high (100 ng/μl QT365 pkIs1330; rhgf-1(ok880); nzEx121) or low (10 ng/μl QT430 pkIs1330; rhgf-1(ok880); nzEx166) concentrations.
C3 transferase transgenes. N-terminally tagged GFP∷C3 transferase was expressed from a heat shock promoter (QT#99) (McMullan et al, 2006). nzEx4 contains an extrachromosomal version of QT#99 injected at 1 ng/μl. QT457 pkIs1330; nzEx4 was constructed by standard genetic cross.
UNC-13 transgenes. For UNC-13 and SNB-1 visualization: rol-6, KP#291 (which encodes a full length unc-13S∷YFP fusion gene which restores locomotion to animals carrying unc-13 alleles) and KP#282 (which encodes an SNB-1∷CFP fusion expressed from the acr-2 promoter (Nurrish et al, 1999)) were either integrated in a wild-type background (nuIs59) or exists as an extrachromosomal array after injection into QT307 pkIs1330; rhgf-1(ok880) to give QT435 pkIs1330; rhgf-1(ok880); nzEx189. KP#273 encodes UNC-13S∷GFP, which contains a point mutation resulting in the amino-acid change H173K. nzEx195 contains an extrachromosomal array of KP#273 injected into QT467 unc-13(s69); pkIs1330 to give QT469 unc-13(s69); pkIs1330; nzEx196.
Induction of heat shock-inducible transgenes
Expression from the heat shock promoter was achieved using two rounds of heat shock at 33°C for 60 min, separated by 30 min at 20°C, followed by recovery at 20°C for 30 min. Animals were then tested immediately or 24 h after heat shock.
Microscopy
Strains expressing p.acr-2∷GFP (KP#307), p.unc13S∷UNC13S∷YFP, p.acr-2∷SNB-1∷CFP (nzIs59), or p.unc-13S∷UNC13S(H173K)∷GFP (integrated to give nzIs25) were imaged by mounting on agarose pads and viewed on a Biorad 2100 upright multiphoton confocal microscope with a Nikon × 60 objective. Images (8 bit) were obtained using LaserSharp software (Biorad). Digital images were processed to give maximum intensity projections of a z-series using Image J (NIH). Images were blinded with respect to genotype and treatment and thresholded to highlight objects containing more than five pixels with an intensity greater than 100 (pixels have a value between 0 and 255 based on fluorescent intensity), these were defined as puncta. Highlighted objects outside the dorsal cord, usually auto fluorescence in the gut, were deleted from the image. The UNC-13S∷YFP and SNB-1∷CFP puncta numbers and size were then counted using Image J. The average puncta intensity was the average pixel value within the all the puncta minus the average value across the whole image (nonthreasholded background value) using Image J. Error bars for all measurements indicate the standard error of the mean. The following strains were used for microscopy; KP1683 [nuIs59], QT368 [nuIs59;pkIs1330], QT435 [pkIs1330; rhgf-1(ok880); nzEx189], QT365 [pkIs1330; rhgf-1(ok880); nzEx121], QT430 [pkIs1330; rhgf-1(ok880); nzEx166], QT419 [pkIs1330; nzIs25], QT377 nzEx124, QT461 nzEx124; rhgf-1(ok880), QT603 gpa-12(pk322); nzIs59, and QT604 rhgf-1(ok880); nzIs59.
Analysis of locomotion and sensitivity to drug treatment
Sensitivity to 1 mM aldicarb (Greyhound Chromatography) or 100 μM levamisole (Sigma) was determined by analysing the onset of paralysis as described previously (Nurrish et al, 1999). For each experiment, at least 20 animals were tested and each experiment was repeated at least five times. Error bars indicate the s.e.m.
Embryonic arrest assays
Eggs were obtained by bleaching 20 gravid adult animals using 50% bleach and 1 M NaOH. Eggs were left to hatch overnight at 20°C and 20 of the resulting L1 animals were transferred to clean seeded plates, and then if required heat shocked twice starting within 1 h of transfer. Plates were incubated at 20°C for 4 days, and representative pictures were taken.
Pharyngeal pumping assays
Young adults were transferred onto clean seeded plates, left for 20 min, and pharyngeal pumps were recorded using the Etho programme (Jim Thomas, University of Washington, USA). If an animal moved off the bacterial food during the assay, it was voided and started again. All animals were assayed within 30 min after heat shock. At least five animals per genotype were assayed.
Statistical analysis
In all cases, statistical analysis was performed using an unpaired two-tailed t-test. P-values of less than 0.05 were considered statistically significant and are indicated on figures by *.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank other members of the Nurrish Lab for advice and discussion, Giovanni Lesa and Martin Raff for comments on the manuscript. Strains were obtained from the C. elegans Genetics Center (CGC), which is supported by the National Institute of Health National Center for Research Resources. The ok880 and gk217 deletions were provided by the C. elegans knockout consortium. The gpa-12 and part of the rhgf-1 cDNA were provided by Yuji Kohara.
References
- Barrett K, Leptin M, Settleman J (1997) The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell 91: 905–915 [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N (1998) Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21: 123–136 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, Sternberg PW, Simon MI (1996) Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron 16: 999–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR (2004) Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci 7: 1096–1103 [DOI] [PubMed] [Google Scholar]
- Hacker U, Perrimon N (1998) DRhoGEF2 encodes a member of the Dbl family of oncogenes and controls cell shape changes during gastrulation in Drosophila. Genes Dev 12: 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280: 2112–2114 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A, Schmidt A (2005) Association of CNK1 with Rho guanine nucleotide exchange factors controls signaling specificity downstream of Rho. Curr Biol 15: 405–412 [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC (1998) p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280: 2109–2111 [DOI] [PubMed] [Google Scholar]
- Kristelly R, Gao G, Tesmer JJ (2004) Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J Biol Chem 279: 47352–47362 [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM (1999) Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24: 335–346 [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS (2005) Scaffold proteins dictate Rho GTPase-signaling specificity. Trends Biochem Sci 30: 423–426 [DOI] [PubMed] [Google Scholar]
- McMullan R, Hiley E, Morrison P, Nurrish SJ (2006) Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev 20: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C, Fire A (1995) DNA transformation. Methods Cell Biol 48: 451–482 [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RH, Sternberg PW (1995) Participation of the protein Go in multiple aspects of behavior in C. elegans. Science 267: 1652–1655 [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA 93: 12593–12598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB (1999) Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron 24: 323–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB (1995) Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB (1993) Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73: 1291–1305 [DOI] [PubMed] [Google Scholar]
- Nurrish S, Segalat L, Kaplan JM (1999) Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron 24: 231–242 [DOI] [PubMed] [Google Scholar]
- Parks S, Wieschaus E (1991) The Drosophila gastrulation gene concertina encodes a G alpha-like protein. Cell 64: 447–458 [DOI] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM (2001) An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412: 338–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade MA, Reynolds NK, Dollins CM, Miller KG (2005) Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the G alpha(s) pathway and define a third major branch of the synaptic signaling network. Genetics 169: 631–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Hall A (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 16: 1587–1609 [DOI] [PubMed] [Google Scholar]
- Segalat L, Elkes DA, Kaplan JM (1995) Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science 267: 1648–1651 [DOI] [PubMed] [Google Scholar]
- Sieburth D, Ch'ng Q, Dybbs M, Tavazoie M, Kennedy S, Wang D, Dupuy D, Rual JF, Hill DE, Vidal M, Ruvkun G, Kaplan JM (2005) Systematic analysis of genes required for synapse structure and function. Nature 436: 510–517 [DOI] [PubMed] [Google Scholar]
- Smith FD, Scott JD (2002) Signaling complexes: junctions on the intracellular information super highway. Curr Biol 12: R32–R40 [DOI] [PubMed] [Google Scholar]
- Steven R, Zhang L, Culotti J, Pawson T (2005) The UNC-73/Trio RhoGEF-2 domain is required in separate isoforms for the regulation of pharynx pumping and normal neurotransmission in C. elegans. Genes Dev 19: 2016–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden AM, Moorman C, Cuppen E, Korswagen HC, Plasterk RH (2003) Hyperactivation of the G12-mediated signaling pathway in Caenorhabditis elegans induces a developmental growth arrest via protein kinase C. Curr Biol 13: 516–521 [DOI] [PubMed] [Google Scholar]
- Yau DM, Yokoyama N, Goshima Y, Siddiqui ZK, Siddiqui SS, Kozasa T (2003) Identification and molecular characterization of the G alpha12-Rho guanine nucleotide exchange factor pathway in Caenorhabditis elegans. Proc Natl Acad Sci USA 100: 14748–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2