


Abstract
The chicken anaemia virus-derived protein apoptin is a tumour-specific cell-killing agent. It is biologically active as a highly stable, multimeric complex, consisting of 30–40 monomers. In tumour cells, but negligibly in normal cells, apoptin is imported into the nucleus prior to the induction of apoptosis. Immunoelectron microscopic data we report here indicate that apoptin predominantly co-localises with heterochromatin and nucleoli within tumour cells. Apoptin’s preference for these DNA-dense nuclear bodies may be explained by our finding that apoptin cooperatively forms distinct superstructures with DNA in vitro. These superstructures do not grow beyond a diameter of ∼200 nm, containing up to 20 multimeric apoptin complexes and ∼3 kb of DNA. Furthermore, we show a single apoptin multimer to have eight independent, non-specific DNA-binding sites which preferentially bind strand ends, but which can also collaborate to bind longer stretches of DNA. Apoptin’s high affinity for naked, undecorated double- and single-stranded DNA and for DNA fibre ends suggests that it may also capture such DNA in superstructures in vivo. Since these forms of DNA are predominantly found in transcriptionally active, replicating and damaged DNA, apoptin could be triggering apoptosis by interfering with DNA transcription and synthesis.
INTRODUCTION
Apoptin is a protein expressed by chicken anaemia virus (CAV). This virus infects chicken thymocytes and lymphoblastoid cells, causing them to undergo apoptosis (1). Expression of apoptin alone reproduces CAV’s cell-killing effect (2). In CAV-infected transformed chicken cells in culture, apoptin migrates to the nucleus, where it is initially distributed in a fine granular pattern, but gradually becomes incorporated into larger aggregates and finally ends up in nuclear apoptotic bodies containing condensed DNA (2). Apoptin also induces apoptosis when overexpressed in a wide variety of human tumour cells, but it does not kill normal cells. In tumour cells, apoptin migrates to the nucleus, but it remains primarily in the cytoplasm in normal cells (3). In the tumour cell nucleus, the same morphological transition from granules to apoptotic bodies is observed as in the nucleus of a CAV-infected chicken cell.
The basic C-terminus of apoptin contains nuclear localisation signals (NLS), and their disruption greatly reduces both nuclear import and apoptosis induction, suggesting that apoptin’s localisation and activity are linked (4). However, nuclear localisation of apoptin does not seem to be sufficient for the induction of apoptosis, as direction of apoptin to the nucleus of normal cells by fusion to an SV40 large T antigen NLS does not affect cell fate (5). Apoptin was shown to be phosphorylated at Thr108 in tumour cells, a modification that is likely to be crucial for nuclear translocation and apoptotic activity, as a phosphorylation-mimicking mutant of apoptin (T108E) displays both of these hallmarks of apoptin’s tumour-specific behaviour, when expressed in normal cells (6).
Bacterially expressed, recombinant apoptin fused to maltose-binding protein (MBP–apoptin) is a good model for in vitro studies, as its microinjection into cultured cells produces essentially the same results as its expression within these cells (7). MBP–apoptin exists exclusively as a very stable, multimeric complex of 30–40 monomeric subunits which does not need to dissociate within the cell in order to be active (8).
Here, we discuss new data concerning apoptin’s biological activity. We found apoptin to co-localise with heterochromatin and nucleoli in tumour cells. Apoptin’s preference for DNA-dense nuclear bodies correlated with our in vitro studies, which showed the cooperative formation of distinct, higher-order nucleoprotein complexes or ‘superstructures’ containing up to 20 apoptin protein multimers and a single double-stranded (ds) DNA fibre. We established MBP–apoptin to have a high affinity for single-stranded (ss) and dsDNA, with little if any sequence specificity. If the formation of apoptin–DNA superstructures in vitro is analogous to the formation of nuclear apoptin-containing granules in vivo, the accumulation of apoptin in areas of cellular DNA may be the cause rather than the effect of nuclear apoptotic changes.
MATERIALS AND METHODS
Immunoelectron microscopy
Saos-2 cells, a human osteosarcoma-derived cell line, were transfected with pCMV-Vp3, expressing full-length apoptin (wt) (9). At 53 h after transfection, cells were fixed in 0.1% glutaraldehyde in 0.14 M cacodylate buffer pH 7.4 for 1 h at room temperature. After washing in phosphate buffer, cells were pelleted and embedded in 10% gelatin, cut into 1 mm3 cubes, cryoprotected in 2.3 M sucrose for 25 min and snap-frozen in liquid nitrogen. Ultra thin cryosections were prepared and incubated with the mouse monoclonal anti-apoptin antibody 111.3 (1:100 dilution) (3), followed by rabbit anti-mouse IgG (Dakopatts, Copenhagen, Denmark) and 10 nm protein A–gold. As a negative control, the primary antibody was omitted or incubated with an irrelevant isotype-matched antibody. After immunolabelling, sections were embedded and contrasted in methylcellulose/uranylacetate, and viewed with a Philips TEM 410 electron microscope.
Indirect immunofluorescence microscopy
Saos-2 cells were transfected with pCMV-Vp3 and fixed at 5 days post-transfection. Fixation sequence: 10 min formaldehyde, 5 min 100% methanol, 3 min 80% acetone. Subsequently, cells were stained with the 111.3 antibody and a fluorescein isothiocyanate (FITC)-labelled anti-mouse antibody. The apoptotic condition of individual cells was deduced from their nuclear morphology after staining with 4′,6-diamidino-2-phenylindole (DAPI) and examination using fluorescence microscopy.
Scanning force microscopy
The MBP–apoptin (wt, T108E) and H6-MBP expression in Escherichia coli was described previously (8). Purified MBP–apoptin was diluted in HPLC water (Sigma) to 2.5–100 µg/ml and filtered over 0.22 µm Ultrafree-MC spin filters (Millipore). pUC19 plasmid DNA was isolated and prepared for scanning force microscopy (SFM) analysis as described previously (10). pUC19 DNA was employed in its supercoiled, relaxed circular (DNase I-nicked) or linear form. DNase I-nicked pUC19 consisted mainly of circular DNA, but also contained a fraction of linearised molecules. Filtered protein (wt, T108E; 2.5–100 ng) was combined with pUC19 DNA (10–45 ng) in 10 µl of 5 mM HEPES pH 7.9, 3 mM KCl, 5.5 mM MgCl2. The mixture was incubated at 37°C for 15 min and then deposited on a disc of freshly cleaved mica (Ted Pella Inc.). After ∼60 s, the mica was gently rinsed with HPLC water. Excess water was removed and the disc was dried under a steady flow of filtered air (0.22 µm). Protein samples without DNA were prepared analogously. Images were acquired on a Nanoscope IIIa (Digital Instruments Inc.), operating in tapping mode in air with a type E scanner using silicon tips from Digital Instruments. Volumetric analyses were performed with the Image SXM 1.62 program, a modified version of NIH Image, provided by Dr Steve Barrett (Surface Science Research Centre, University of Liverpool, Liverpool, UK).
Cooperativity of MBP–apoptin/pUC19 binding. From a single MBP–apoptin (wt)/pUC19 DNA deposition, at a DNA to protein ratio of 3.8 kb per multimer, a total of 433 (series 1) or 437 (series 2) DNA molecules were selected from 18 images (4 × 4 µm). In series 1, higher-order complexes were only selected if the DNA was clearly distinguishable. Series 2 included higher-order complexes of the expected size but that did not contain any visible DNA. In both series, the DNA molecules were sorted into four categories: free, a primary complex with MBP–apoptin particle (1:1), a secondary complex (2:1) or a higher-order complex.
DNA wrapping in MBP–apoptin/pUC19 DNA. From several MBP–apoptin (wt)/pUC19 DNA depositions, at a DNA to protein ratio of 68 kb per multimer, we selected 49 pUC19 DNA molecules bound to a single MBP–apoptin multimer and determined the bending angle between the DNA ‘arms’ extending from the protein particle. For comparison, we collected a set of random bending angles by placing an isolated protein particle at random, regularly spaced positions on free pUC19 DNA molecules and measuring the bending angle.
Dynamic light scattering
All dynamic light scattering (DLS) measurements were recorded at room temperature on a DynaPro-MS/X (Protein Solutions Inc.). Data analysis was done with the Dynamics V5.25.44 program and the DynaLS V1.51 software package (Protein Solutions Inc.). Protein and DNA samples were filtered separately (0.22 µm) directly prior to the experiment. MBP–apoptin (wt, T108E) was diluted to 10–40 nM (multimers) in 20 mM HEPES pH 7.4, 10 mM NaCl, 1 mM EDTA. DNA (2.5–25 µg/ml) was diluted in the protein sample and the mixtures were injected directly into the DLS cuvette. Per experiment, between 10 and 20 measurements (10 s intervals) were collected. Per DNA species, between four and eight experiments were recorded and averaged. The correlation between the hydrodynamic diameter of the apoptin–DNA complex and DNA length was determined at a DNA to protein ratio of 400 bp per multimer. H6-MBP alone did not form complexes with DNA of any length. A solvent refractive index of 1.333 (aqueous buffer) was maintained throughout all experiments.
dsDNA species. (i) 12 bp, a stock solution (1 µg/µl) was prepared by combining complementary deoxyoligonucleotides (CTGGTAGATGGC, melting temperature 38°C). (ii) 81 bp PCR product of Vp3(29–52) open reading frame (ORF; nucleotides 87–155) with a 5′ BamHI overhang. (iii) 177 bp PCR product of the Vp1/Vp2 ORF overlap (nucleotides 852–1029 of the CAV genome). (iv) 396 bp PCR product of Vp3 ORF (nucleotides 1–366) with 5′ BamHI and 3′ SalI overhangs. (v) 651 bp PCR product of Vp2 ORF (nucleotides 380–1026 of the CAV genome). (vi) 820 bp BamHI–EcoRI fragment from pIC19R-CAV. (vii) 1.5 kb BamHI–EcoRI fragment from pIC19R-CAV. (viii) 2.6 kb linearised pIC19R following BamHI–EcoRI digestion of pIC19R-CAV. (ix) 3.7 kb PstI-linearised pET-23b (Novagen). (x) 6.6 kb BamHI-linearised pMal-p2E (NEB). (xi) 48.5 kb λ genomic DNA. All PCRs were carried out using VENT polymerase (NEB).
PCR-based assay for sequence-specific DNA binding
We generated a library of PCR fragments by amplifying an oligonucleotide that consisted of a stretch of 20 random nucleotides, flanked by two 15 nt primer sites, using VENT polymerase (NEB). DNA was incubated with MBP–apoptin (wt, 150 ng) at a DNA to protein ratio of ∼2.5 kb per multimer, in 10 mM HEPES pH 7.4, 50 mM NaCl, 1 mM EDTA at 4°C for 2 h. Under these conditions, at least 10 copies of each potential sequence are expected to be available (420). Subsequently, the protein was recovered using amylose resin (∼25 µl), and the retained DNA was extracted using Qiaquick spin columns and used in a new round of amplification. This procedure was repeated five times, during which the ionic strength was gradually increased to 150 mM NaCl. Finally, the retained DNA was cloned into appropriate cloning vectors, and positive clones (blue/white screening) were analysed by automated fluorescent sequencing.
Restriction fragment sorting
DNA templates. The DNA templates used were: (i) phage λ (cI857, strain 7) genomic DNA (48.5 kb) (Roche); (ii) pIC19R-CAV plasmid (5.0 kb), containing the CAV genome (11); and (iii) pcDNA3.1mychis(–)-MVp3 plasmid (7.1 kb) (8).
Fragment sorting assay. λ DNA was fully digested with RsaI, yielding 114 fragments of 6–2196 bp. pIC19R-CAV and pcDNA3.1mychis(–)-MVp3 were digested with AluI, yielding 26 (11–679 bp) and 33 fragments (6–706 bp), respectively. After deactivation of the enzyme, the digest was mixed with MBP–apoptin (wt, T108E) in 20 mM HEPES pH 7.4, 1 mM EDTA, at a DNA to protein ratio of ∼200 bp per multimer. After incubation on ice, for up to 3 h, the mixture was combined with ∼250 µl of suspended amylose. The resin was then eluted with an NaCl step gradient: 0–2 M NaCl, using five column volumes per step. Finally, the protein itself was eluted with 10 mM maltose. The DNA was recovered from each fraction, using Qiaquick spin filters (Qiagen), and subsequently analysed on 0.5–1.5% agarose gels (1× TBE, run at 4°C). Following electrophoresis, gels were stained with 5 µg/ml ethidium bromide (EB). If necessary, fractions were resolved at higher resolution on 4–10% TBE–PAGE (0.5 mm vertical gel, 4°C).
Fluorescence spectroscopy
Fluorescence emission and excitation spectra were recorded on a Perkin-Elmer LS-50B at room temperature. Spectra were averaged over five acquisitions. Settings: excitation wavelength = 517 nm, slit width = 10–15 nm. dsDNA species: 12 bp (CTGGTAGATGGC), 81 bp Vp3(29–52) and 177 bp Vp1/Vp2 ORF overlap. All protein and DNA stocks were dialysed extensively to 20 mM HEPES pH 7.4, 10 mM NaCl, 1 mM EDTA. Because DNA-dependent EB fluorescence is very sensitive to changes in ionic strength, the dialysis buffer was used as assay buffer. All DNA samples were diluted to 2.5 µg/ml and combined with EB (Bio-Rad) (bp/dye ≈5). MBP–apoptin (wt, T108E) was added to 300 µg/ml. The presence of MBP–apoptin alone did not affect the background fluorescence of unbound EB significantly. At all DNA to protein ratios tested, the exclusion of EB from dsDNA by MBP–apoptin reached its maximum level within the time required to record a single spectrum (∼30 s).
Quantitative ethidium bromide exclusion assay. dsDNA fragments (12, 81 and 177 bp) were diluted to 0.5–5 µg/ml and EB was added to a bp/dye of 5. The dsDNA–EB complex was then titrated by the stepwise addition of protein (wt, T108E; 2–200 µg/ml). All titrations were performed at three different dsDNA concentrations. At 10 mM NaCl, H6-MBP alone gave rise to <5% of the fluorescence decrease caused by MBP–apoptin. Fluorescence spectra were corrected for dilution effects and, when necessary, for background quenching by MBP.
Data analysis. DNA binding was deduced from the decrease in EB fluorescence at 600 nm (|ΔF600|) upon addition of protein. Titration curves ([protein] versus |ΔF600|) were plotted and analysed using Prism 3.00 (Graphpad Software Inc.). Curves were fitted by non-linear regression analysis, with either a Boltzmann sigmoidal or one-phase exponential association curve. The DNA-binding equilibrium was defined as: [protein]free + [DNA]free ↔ [protein – DNA]. The equilibrium dissociation constant was defined as: KD = ([protein]free[DNA]free)/[protein – DNA].
RESULTS
Apoptin is preferentially located in heterochromatin and nucleoli of tumour cells
In order to establish the location of apoptin within the ultrastructure of the cell prior to the onset of apoptosis, we analysed Saos-2 cells 2 days after transfection with apoptin (wt). At this time point, the majority of apoptin-expressing cells did not yet display visible apoptotic changes (3). Electron microscopy (EM) micrographs of coupes immunostained with an apoptin-specific antibody and gold-labelled protein A revealed numerous gold particles in heterochromatin and nucleoli (Fig. 1A). When detected by immunofluorescence microscopy, apoptin is known to be incorporated into small nuclear granules and larger bodies that have been identified as nucleoli, an example of which is depicted in Figure 1B (3). At present, apoptin’s localisation has not been confirmed by co-localisation with heterochromatin markers. Nevertheless, the electron-dense appearance of some of the areas stained by anti-apoptin antibody (Fig. 1A) correspond structurally to heterochromatin. Taken together, these results demonstrate that ectopically expressed apoptin targets particular nuclear structures that are rich in nucleic acid.
Figure 1.

Apoptin, expressed in human tumour cells, localises to heterochromatin and nucleoli. (A) Apoptin was detected in ultra thin cryosections of apoptin-expressing Saos-2 cells by immunogold staining. Indicated are: heterochromatin (HC), a nucleolus (NUC) and the nuclear membrane (NM); scale bar represents 200 nm. (B) Apoptin was detected in apoptin-expressing Saos-2 cells by indirect immunofluorescence microscopy. Top: Saos-2 cells, stained with anti-apoptin antibody 111.3. Bottom: nuclear morphology, as shown by DAPI staining. Similar results were published previously in Danen-van Oorschot et al. (3).
Apoptin and dsDNA assemble into superstructures in vitro
SFM analysis of purified MBP–apoptin in the presence of DNA indicated that the preference of apoptin for DNA-dense regions within the tumour cell nucleus could be caused by specific interactions between apoptin and DNA. MBP–apoptin multimers (Fig. 2A) assembled into higher-order nucleoprotein complexes, which we termed DNA·MBP–apoptin superstructures, upon the addition of DNA (nicked pUC19 plasmid, 2.7 kb), at a DNA to protein ratio of ∼300–600 bp per multimer. Similar superstructures formed with linear and supercoiled DNA (data not shown). Superstructures contained between 15 and 20 MBP–apoptin multimers each (Fig. 2B and C). MBP–apoptin multimers did not coalesce within the superstructures, but remained discernible as individual particles. The shape of the superstructures varied considerably: we observed elongated bodies, globular clusters, irregular aggregates and a small number of linear complexes with contiguous multimers arranged like beads on a string. However, it may well be that this variability does not reflect the solution state of the superstructures, as the samples had to be deposited on mica and dried prior to imaging.
Figure 2.

MBP–apoptin forms nucleoprotein superstructures with DNA with a hydrodynamic diameter of ∼100 nm. Analysis of MBP–apoptin complexes, deposited on mica, visualised by SFM. (A) MBP–apoptin alone, 2 × 2 µm surface area. (B) MBP–apoptin combined with pUC19 at a ratio of 320 bp per multimer. (C) As (B). Height is indicated in grey scale; bar is from 0.0 (black) to 1.5 nm (white). (D) The hydrodynamic diameter of MBP–apoptin–DNA superstructures in solution was determined as a function of DNA length by dynamic light scattering. MBP–apoptin (50 µg/ml, 20 nM multimers) was combined with 5 µg/ml dsDNA (7.5 µM bp, which corresponds to 350 bp/multimer) in 20 mM HEPES pH 7.4, 10 mM NaCl, 1 mM EDTA. Varying the DNA to protein ratio between 200 bp and 4 kb per multimer had no significant effect on the size of the complexes.
Indeed, DLS indicated the existence of a fairly uniform population of superstructures in solution. When small DNA fragments were used, correspondingly smaller superstructures were detected (Fig. 2D), but when MBP–apoptin was combined with linear DNA of at least 3 kb, we observed particle distributions corresponding to DNA·MBP–apoptin superstructures with a hydrodynamic diameter (DH) of 100 ± 5 nm (Fig. 2D). The apparent monodispersity of the DNA·MBP–apoptin superstructures in solution suggested that there was little variation in their overall shape. Assuming that the superstructures were roughly globular in solution, they had an average volume of ∼5.5 × 105 nm3, suggesting a stoichiometry of ∼20 MBP–apoptin multimers per superstructure. In general, the relationship between DNA length and multimer content observed in solution correlated well with the SFM images of immobilised DNA·MBP–apoptin. Complexes formed by MBP–apoptin T108E with DNA followed the same trend as those formed by MBP–apoptin wt, as determined by both SFM and DLS analysis (data not shown). Surprisingly, the superstructures did not grow beyond ∼120 nm in diameter, even when very long DNA fragments of 48.5 kb were used at a ratio of 400 bp of DNA per MBP–apoptin multimer. Apparently, under these conditions, the superstructures were sufficiently far apart on the stretches of dsDNA to diffuse as independent particles, as judged by light scattering. This conclusion was corroborated by our finding that MBP–apoptin formed only one superstructure per dsDNA fibre when combined with a 5.5 kb plasmid, at a DNA to protein ratio of 500 bp per multimer, as determined by SFM (data not shown).
DNA·MBP–apoptin superstructures assemble cooperatively
A striking feature of the SFM images was that hardly any superstructures were found to be in the process of assembly: we predominantly observed fully formed superstructures or apoptin multimers not bound to DNA. We also did not observe a significant effect on the size distribution of the superstructures as determined by DLS upon a 20-fold increase of the DNA to multimer ratio, for both MBP–apoptin wt and T108E. Even at a very large excess of DNA (∼11 kb of dsDNA per multimer), superstructures were by far the most abundant type of apoptin–DNA complex, as judged by SFM (Fig. 3A and B). These observations strongly suggested cooperative complex formation, in which binding of one or two apoptin multimers to DNA facilitates the quick incorporation of additional apoptin multimers.
Figure 3.

Formation of higher-order MBP–apoptin–DNA complexes is highly cooperative. (A) MBP–apoptin combined with nicked pUC19 at a ratio of 11 kb per multimer, or four plasmid molecules per protein particle. In theory, one molecule can accommodate 25 multimers. Height is indicated in grey scale; bar is from 0.0 (black) to 1.5 nm (white). (B) MBP–apoptin combined with nicked pUC19 at a ratio of 3.8 kb per multimer. (C) Distribution of binding states of pUC19 molecules as observed in 18 separate SFM images, which were comparable with (B). Primary complexes correspond to DNA molecules with a single bound apoptin multimer; secondary complexes carry two multimers. Series 1 contains only higher-order complexes where DNA was clearly visible. Series 2 includes higher-order complexes of the expected size but without distinguishable DNA due to slightly different imaging parameters. There is no significant difference between both series.
We counted the types of complexes formed when MBP–apoptin was incubated with 3.8 kb pUC19 dsDNA per multimer. We found that ∼70% of the DNA molecules did not bind MBP–apoptin multimers, close to 11% of DNA molecules contained primary complexes (a single bound multimer) and ∼18% contained superstructures (Fig. 3C). Little or no secondary complexes (two or more multimers per DNA molecule) were observed (≤0.5%). These observations allowed us to quantify the cooperative effect:
Given: Kn = [DNA·An – 1][A]/[DNA·An]1
where Kn is the dissociation constant of a single MBP–apoptin multimer dissociating from a DNA·MBP–apoptin complex containing ‘n’ MBP–apoptin multimers; [A] is the molar concentration of free MBP–apoptin multimers; [DNA·An] is the concentration of DNA·MBP–apoptin superstructures that each contain ‘n’ MBP–apoptin multimers, we can calculate the product of all dissociation constants K1, K2, …, Kn as:
Π(Ki) = [DNA][A]n/[DNA·An]2
where ‘n’ is the number of MBP–apoptin multimers. Lemma (2) defines the dissociation constant of a DNA·MBP–apoptin complex containing ‘n’ MBP–apoptin multimers dissociating into all its separate components. The level of cooperativity can be determined from the ratio ‘Q’:
Q = K1n/Π(Ki) = [DNA]n – 1 [DNA·An]/[DNA·A]n3
If binding is non-cooperative, by definition K1 = K2 = … = Kn and therefore ‘Q = 1’. A value ‘Q < 1’ would indicate anti-cooperative binding, whilst a value ‘Q > 1’ would indicate cooperative binding. Assuming that each DNA·MBP–apoptin superstructure contains about 20 MBP–apoptin multimers, the experimental results summarized in Figure 3C suggest that Q ≈ 2 × 1018, indicating a strong cooperative effect. Moreover, it implies that the geometric mean of all Kis (dissociation constants of all higher-order structures) is about eight times smaller than K1 (the dissociation constant of the primary complex), because 20√(2 × 1018) = 8. This result suggests that after the first MBP–apoptin multimer is bound, subsequent multimers bind approximately eight times more easily to a site directly adjacent to the growing superstructure.
Recombinant apoptin binds DNA in a sequence non-specific manner, but may have a preference for fibre ends
In order to establish whether apoptin has any sequence specificity, we performed a target detection assay (12). This assay selects sequences from a library of PCR fragments with a randomised internal target sequence of 20 bp. The library was incubated with MBP–apoptin multimers, the protein–DNA complexes were then captured on an amylose column and the retained DNA was amplified. This procedure was repeated several times under increasingly stringent conditions. The final library was expected to be enriched in DNA sequences that bind most tightly; however, we did not find any particular sequence motif.
We confirmed apoptin’s non-specific DNA binding in a selection assay on a library of unique, well-defined and blunt-ended dsDNA fragments (173 in total) of various sizes (6 bp up to 2.2 kb), that we obtained by fully digesting λ DNA with RsaI. We found that MBP–apoptin (wt and T108E) did not exhibit a significantly higher affinity towards any of the fragments sampled here, although fragments <250 bp were retained more efficiently than larger ones (Fig. 4).
Figure 4.
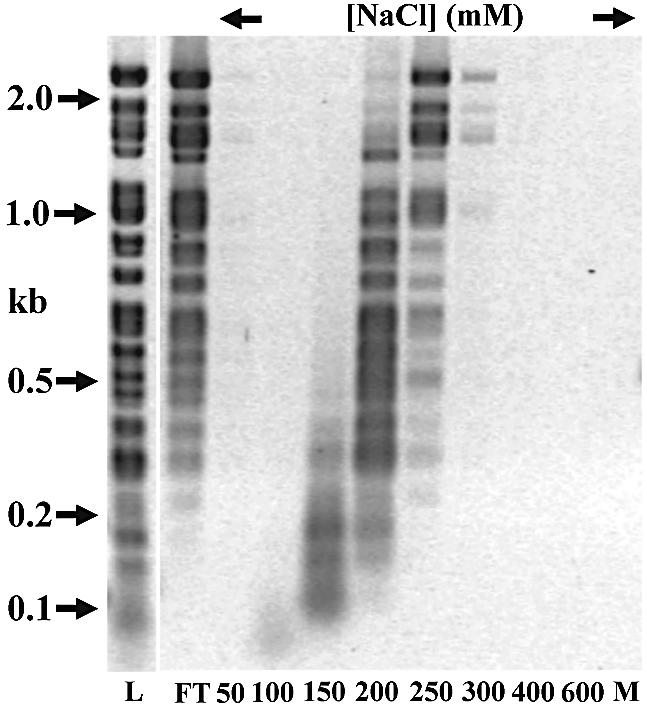
MBP–apoptin binding to DNA is sequence non-specific. MBP–apoptin was incubated with the λ/RsaI fragment pool in solution (10 mM NaCl). Protein–DNA complexes were recovered with amylose, and eluted with an NaCl step gradient. EB-stained 1.5% agarose gel, L = fragment pool, FT = flow-through, M = final wash, 2 M NaCl + 10 mM maltose. Any minor intensity differences in the overview gel (1.5% agarose) were not consistently reproducible upon adjusting the separation range of the gel to achieve higher resolution.
This increased affinity for shorter DNA fragments could be caused by a preferential binding to dsDNA fibre ends. Indeed, when we incubated MBP–apoptin (wt) with medium-sized linear DNA (1.2 kb) at a DNA to protein ratio of 61.5 kb per multimer, MBP–apoptin multimers were rarely observed at internal DNA positions by SFM. Instead, MBP–apoptin displayed a marked preference for binding to fibre ends (data not shown).
We found that MBP–apoptin also has affinity for ssDNA. ssDNA–cellulose (calf thymus) had a binding capacity of 2.2 ± 0.2 mg of MBP–apoptin per mg of immobilised ssDNA, whereas the corresponding dsDNA column had a capacity of 1.5 ± 0.2 mg of MBP–apoptin per mg of immobilised dsDNA. In similar experiments, we also observed binding of MBP–apoptin to E.coli 16S rRNA.
The apoptin protein multimer has multiple DNA-binding sites
By monitoring the exclusion of EB from DNA in response to the binding of MBP–apoptin, we further quantified the binding constants (13). Upon titration of a 12 bp dsDNA fragment, we established that MBP–apoptin (both wt and T108E) expelled ∼60% of bound EB at maximum binding. The titration curve was best fitted with a standard pseudo first-order association curve (Fig. 5A). MBP–apoptin bound 8 ± 2 12 bp fragments per multimer with an average equilibrium dissociation constant (KD) of 3 ± 1.5 nM per fragment (measured as the concentration of free MBP–apoptin at half-maximum EB quenching).
Figure 5.

Apoptin multimers contain about eight cooperative DNA-binding sites. (A) Titration of 12 bp dsDNA fragment (0.5 µg/ml, 65 nM fragment) with MBP–apoptin, |ΔF600| = reduction in ethidium fluorescence at 600 nm. (B) Titration of 177 bp blunt-ended dsDNA fragment (1 µg/ml, 8.7 nM fragment). Inset: emission spectra of 81 and 177 bp fragments (1 µg/ml) with and without 0.15 mg/ml MBP–apoptin.
Next, we titrated longer dsDNA fragments with MBP–apoptin multimers in order to test for cooperativity between the sites. We tested two different DNA substrates of 81 and 177 bp each, which were mixed sequence, well defined and blunt ended. MBP–apoptin (wt and T108E) displaced about the same amount of EB from these fragments as from the 12 bp fragment. The KD of the MBP–apoptin–DNA complex was 7.5 ± 1.5 nM for the 177 bp fragment and 3.3 ± 1.5 nM for the 81 bp fragment (Fig. 5B). The binding curves were sigmoidal, indicating cooperative binding of DNA to multiple binding sites on the MBP–apoptin multimer. Cooperative binding between MBP–apoptin and DNA seemed to be in disagreement with the observation that long DNA fragments binding multiple sites had a similar affinity as DNA dodecamers binding to a single site. This discrepancy is probably caused by preferential binding to DNA fibre ends.
Cooperative DNA binding through multiple binding sites could result in partial or complete wrapping of the DNA strand around the apoptin ‘core’. In the case of extensive wrapping around a particle of this size, a reduction of the apparent length of the DNA would result (14). In order to determine whether the DNA fibre was indeed wrapped around MBP–apoptin particles, we analysed complexes of MBP–apoptin (wt) with circular DNA (pUC19; lacking strand ends) at a DNA to protein ratio of 68 kb per multimer. Our analysis revealed that the DNA is considerably bent (66 ± 26°) by MBP–apoptin, but there was no clear indication for extensive DNA wrapping from DNA length measurements (data not shown).
DISCUSSION
Here, we show that ectopically expressed apoptin is localised in the DNA-dense nucleoli and heterochromatin of human tumour cells. Furthermore, we demonstrated that bacterially expressed apoptin multimers have a high intrinsic affinity for DNA and can form very distinct protein–DNA superstructures. Therefore, our in vitro studies suggest that direct interaction of apoptin protein with chromosomal DNA in vivo could result in aberrant protein–DNA structures, that might well trigger induction of apoptosis. Apoptin protein multimers bound DNA in a highly cooperative fashion, yet the size of these DNA·apoptin superstructures had a distinct ceiling: maximum-sized superstructures contained close to 20 apoptin multimers and 3 kb of DNA. Apparently, once a DNA·apoptin superstructure had started to form or had formed, it inhibited the formation of additional structures on the same DNA strand in its direct vicinity.
Immobilised ssDNA had a higher in vitro binding capacity per nucleotide for MBP–apoptin than dsDNA, which may indicate that ssDNA and dsDNA interact differently with apoptin multimers. Moreover, we found that apoptin multimers have a preference for the ends of dsDNA. We report that longer stretches of dsDNA preferentially coil around the apoptin core along a path formed by multiple DNA-binding sites. In addition, we observed significant bending of DNA by MBP–apoptin particles, although we could not demonstrate extensive wrapping. Altogether, apoptin’s interactions with DNA are expected to have an effect on the chromosomal organization within tumour cells, possibly by displacing histones (15). Disruption of the chromosomal superstructure could initiate apoptosis either directly, or indirectly through interference with transcription and/or replication processes (16,17).
Recently, Danen-van Oorschot et al. have shown that apoptin’s N-terminal (1–69) and C-terminal (80–121) domains induce cell death independently of each other, although both are less potent than full-length apoptin (5). To induce apoptosis, both domains need to be located in the nucleus. MBP-fused apoptin’s N- and C-terminal domains alone can bind DNA in vitro, but not as efficiently as full-length apoptin (S.R.Leliveld, M.H.M.Noteborn and J.P.Abrahams, unpublished results). Non-MBP-fused apoptin(1–80) forms superstructures, but these lack the size ceiling and are considerably more heterogeneous. These results suggest that DNA binding may well be essential for the activity of the isolated apoptin domains, and that the higher killing activity of full-length apoptin may well be due to its ability to bind DNA and assemble into superstructures. However, we cannot exclude the possibility that the killing mechanism of full-length apoptin is distinct from those of its separate domains, relying to a larger extent on DNA binding.
We report here that both MBP–apoptin wt and T108E mutant, which mimic constitutive phosphorylation, share similar DNA-binding properties. At first glance, this seems to disagree with apoptin’s tumour specificity, as apoptin is phosphorylated robustly both in vitro and in vivo in tumour cells but negligibly in normal cells. However, Rohn et al. (6) have reported that in human tumour cells, phosphorylated apoptin is enriched in the nucleus but dephosphorylated apoptin is present in the characteristic round subnuclear bodies, which were identified in this report as DNA-dense bodies. This might explain why both phosphorylated and non-phosphorylated apoptin shows affinity for DNA and/or RNA.
Alternatively, it may be that phosphorylation of apoptin is primarily involved in nuclear transport, based on the observation that the T108E gain-of-function point mutant conferred upon apoptin the ability to accumulate in the nucleus and kill normal cells. These data suggest that as soon as apoptin enters the nucleus, it would bind to DNA to induce apoptosis. However, transport of non-phosphorylated apoptin in normal cells did not result in the induction of apoptosis (5), which could mean that in normal cells, proteins are present that prevent non-phosphorylated apoptin from interacting with the chromatin or that other tumorigenic events might be of relevance. Tumour cells often contain altered chromatin structure, which associates with different patterns of histone acetylation and/or CpG methylation (18). Furthermore, in most if not all tumour cell lines, essential factors that control cell cycle progression have been rendered inoperable. Some of the effects of deficient cell cycle control on the integrity of cellular DNA have been described in detail for Saccharomyces cerevisiae Rad53 kinase. In its hyperphosphorylated state, Rad53 is mainly involved in stabilizing replisomes (replication forks) that have been transiently halted (19,20). In response to reversible inhibition of replication, e.g. by depletion of dNTPs, rad53 mutants accumulate replication intermediates and abnormal DNA structures, most of which contain large stretches of ssDNA. Because all eukaryotic cells employ essentially the same basic machinery for DNA replication, it is likely that the persistence of areas of aberrant DNA is mirrored in human tumour cells. If so, it is plausible that the formation of DNA·apoptin complexes in the tumour cell nucleus is facilitated by a combination of altered chromatin structure and exposed ssDNA. Furthermore, the tendency of MBP–apoptin to bind to DNA ends in vitro may also contribute, as tumour cells are likely to contain non-repaired strand breaks (21). In addition, unknown tumour-specific factors might also play an essential role in the establishment of apoptin–DNA superstructures under in vivo conditions.
In conclusion, we propose the following implications of apoptin’s DNA-binding properties. (i) The co-localisation of ectopically expressed apoptin with DNA-dense heterochromatin and nucleoli is likely to involve direct binding to cellular DNA. (ii) The multimeric state of recombinant apoptin allows it to bind DNA in a cooperative fashion and with high affinity, resulting in distinct apoptin–DNA superstructures. (iii) Apoptin’s accumulation in heterochromatin and nucleoli may reflect its preference for binding to a regularly arranged lattice (nucleosomes), which may stem from the architecture of the apoptin multimer, i.e. the configuration of its DNA-binding sites. Alternatively, the increased concentration of apoptin in DNA-dense nuclear structures may be caused by the local increase in DNA condensation. (iv) The formation of DNA·apoptin superstructure-like granules may be facilitated in tumour cells only, possibly aided by tumour-specific nuclear events
Acknowledgments
ACKNOWLEDGEMENT
We are grateful to J.J.M. Onderwater for technical assistance with immuno-EM.
REFERENCES
- 1.Jeurissen S., Wagenaar,F., Pol,J., Van der Eb,A. and Noteborn,M. (1992) Chicken anaemia virus causes apoptosis of thymocytes after in vivo infection and of cell lines after in vitro infection. J. Virol., 66, 7383–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noteborn M.H., Todd,D., Verschueren,C., de Gauw,H., Curran,W., Veldkamp,S., Douglas,A., McNulty,M., Van der Eb,A. and Koch,G. (1994) A single chicken anaemia virus protein induces apoptosis. J. Virol., 68, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danen-van Oorschot A.A., Fischer,D., Grimbergen,J., Klein,B., Zhuang,S.-M., Falkenburg,J., Backendorf,C., Quax,P., Van der Eb,A. and Noteborn,M. (1997) Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc. Natl Acad. Sci. USA, 94, 5843–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang S.-M., Landegent,J.E., Verschueren,C.A., Falkenburg,J.F., van Ormondt,H., Van der Eb,A.J. and Noteborn,M.H. (1995) Apoptin, a protein encoded by chicken anaemia virus, induces cell death in various human hematologic malignant cells in vitro. Leukemia, 9 (Suppl. 1), S118–S120. [PubMed] [Google Scholar]
- 5.Danen-van Oorschot A.A., Zhang,Y.-H., Leliveld,S.R., Rohn,J.L., Seelen,M.C., Bolk,M.W., van Zon,A., Erkeland,S.J., Abrahams,J.P., Mumberg,D. and Noteborn,M.H. (2003) Importance of nuclear localization of apoptin for tumor-specific induction of apoptosis. J. Biol. Chem., 278, epub, ahead of print. [DOI] [PubMed] [Google Scholar]
- 6.Rohn J., Zhang,Y., Aalbers,R., Otto,N., den Hertog,J., Henriquez,N., van de Velde,J., Kuppen,P., Mumberg,D., Donner,P. and Noteborn,M. (2002) A tumour-specific kinase activity regulates the viral death protein Apoptin. J. Biol. Chem., 277, 50820–50827. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y.-H., Leliveld,S.R., Kooistra,K., Molenaar,C., Rohn,J.L., Tanke,H.J., Abrahams,J.P. and Noteborn,M.H.M. (2003) Recombinant apoptin multimers kill tumour cells but are non-toxic and epitope-shielded in a normal-cell-specific fashion. Exp. Cell Res., in press. [DOI] [PubMed] [Google Scholar]
- 8.Leliveld S.R., Zhang,Y.-H., Rohn,J.L., Noteborn,M. and Abrahams,J.P. (2003) Apoptin induces tumour-specific apoptosis as a globular multimer. J. Biol. Chem., 278, 9042–9051. [DOI] [PubMed] [Google Scholar]
- 9.Zhuang S.-M., Shvarts,A., Jochemsen,A.G., van Oorschot,A.A., Van der Eb,A.J. and Noteborn,M.H. (1995) Differential sensitivity to Ad E1B 21kD and Bcl-2 proteins of apoptin-induced versus p53-induced apoptosis. Carcinogenesis, 16, 2939–2944. [DOI] [PubMed] [Google Scholar]
- 10.Dame R.T., Wyman,C. and Goosen,N. (2000) H-NS mediated compaction of DNA visualised by atomic force microscopy. Nucleic Acids Res., 28, 3504–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noteborn M.H., de Boer,G., van Roozelaar,D., Karreman,C., Kranenburg,O., Vos,J., Jeurissen,S., Hoeben,R., Zantema,A. and Koch,G. (1991) Characterization of cloned chicken anaemia virus DNA that contains all elements for the infectious replication cycle. J. Virol., 65, 3131–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thiessen H.-J. and Bach,C. (1990) Target detection assay (TDA): a versatile procedure to determine DNA binding sites as demonstrated on SP1 protein. Nucleic Acids Res., 18, 3203–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boger D., Fink,B., Brunette,S., Tse,W. and Hedrick,M. (2001) A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J. Am. Chem. Soc., 123, 5878–5891. [DOI] [PubMed] [Google Scholar]
- 14.Verhoeven E., Wyman,C., Moolenaar,G., Hoeijmakers,J. and Goosen,N. (2001) Architecture of nucleotide excision repair complexes: DNA is wrapped by UvrB before and after damage recognition. EMBO J., 20, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polach K. and Widom,J. (1996) A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol., 258, 800–812. [DOI] [PubMed] [Google Scholar]
- 16.Schickling O., Stegh,A., Byrd,J. and Peter,M. (2001) Nuclear localization of DEDD leads to caspase-6 activation through its death effector domain and inhibition of RNA polymerase I-dependent transcription. Cell Death Differ., 8, 1157–1168. [DOI] [PubMed] [Google Scholar]
- 17.Xu X., Hamhouyia,F., Thomas,S., Burke,T., Girvan,A., McGregor,W., Trent,J., Miller,D. and Bates,P. (2001) Inhibition of DNA replication and induction of S phase cell cycle arrest by G-rich oligonucleotides. J. Biol. Chem., 276, 43221–43230. [DOI] [PubMed] [Google Scholar]
- 18.Rountree M., Bachman,K., Herman,J. and Baylin,S. (2001) DNA methylation, chromatin inheritance and cancer. Oncogene, 20, 3156–3165. [DOI] [PubMed] [Google Scholar]
- 19.Lopes M., Cotta-Ramusino,A., Liberi,G., Plevani,P., Muzi-Falconi,M., Newlon,C. and Foiani,M. (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature, 412, 557–561. [DOI] [PubMed] [Google Scholar]
- 20.Sogo J., Lopes,M. and Foiani,M. (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science, 297, 599–602. [DOI] [PubMed] [Google Scholar]
- 21.Tong W., Hande,M., Lansdorp,P. and Wang,Z. (2001) DNA strand break-sensing molecule poly(ADP-ribose) polymerase cooperates with p53 in telomere function, chromosome stability and tumour suppression. Mol. Cell. Biol., 21, 4046–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]