

Abstract
The ability of several different influenza A virus strains to infect and replicate in primary, differentiated airway epithelial cell cultures from Syrian golden hamsters was investigated. All virus strains tested replicated equivalently in the cultures and displayed a preference for infecting nonciliated cells. This tropism correlated with the expression of both α2,3- and α2.,6-linked sialic acid on the nonciliated cells. In contrast, the ciliated cells did not have detectable α2,6-linked sialic acid and expressed only low amounts of α2,3-linked sialic acid. In contrast to clinical isolates, laboratory strains of influenza A virus infected a limited number of ciliated cells at late times post infection. The presence of α2,3- and α2,6-linked sialic acid residues on the same cell type suggests , Syrian golden hamsters and differentiated airway epithelial cell cultures derived from hamsters may provide a system for studying the reassortment of influenza A virus strains which utilize different forms of sialic acid as a primary virus receptor.
Keywords: influenza, sialic acid, respiratory epithelial cells, airway epithelial, ciliated cells, Clara cells, goblet cell, virus receptors, virus tropism
Introduction
Influenza virus infection is estimated to be responsible for approximately 36,000 deaths and 100,000 hospitalizations annually in the United States alone (Thompson et al., 2004; Thompson et al., 2003). These numbers would increase considerably in the event of an influenza pandemic, which would result from the introduction of an influenza A virus strain encoding a hemagglutinin (HA) gene that was not present in viruses circulating recently in the human population (Barnett et al., 2005; Bartlett and Hayden, 2005; Wilson et al., 2005). Pandemic influenza A virus strains are generated through one of two mechanisms: reassortment or adaptation. Since the influenza A virus genome consists of eight distinct RNA segments (Lamb and Krug, 2001), infection of one cell with two influenza A virus strains can lead to the exchange of genetic material, a process called reassortment, resulting in a new virus strain possessing biological properties derived from either parental viruses (Horimoto and Kawaoka, 2005). The presence of influenza A virus strains bearing various combinations of the 16 HA and 9 neuraminidase (NA) serotypes in a range of animal reservoirs provides a pool of virus strains that could be the source of the next influenza pandemic (Fouchier et al., 2005; Rohm et al., 1996; WHO, 1980) . The influenza pandemics of 1957 and 1968 resulted from reassortment of human and avian influenza A viruses (Webster et al., 1997). In contrast, the influenza pandemic of 1918 is believed to have been caused by an avian influenza A virus which acquired the ability to infect and efficiently spread in the human population through adaptation, not reassortment (Reid et al., 2004; Taubenberger et al., 2005; Tumpey et al., 2005).
The primary, but certainly not the only, hurdle influenza A virus must overcome when crossing a species barrier is believed to be utilization of host cell receptors (Ito, 2000). Influenza A virus uses sialic acid (SA) residues present at the terminus of oligosaccharide modifications of host cell glycoproteins as attachment receptors (Chu and Whittaker, 2004). SA residues are usually linked to the penultimate carbohydrate group via α2,3-, α2,6- or α2,8-linkages, and most influenza A virus strains characterized to date utilize either α2,3- or α2,6-SA as a primary receptor (Glaser et al., 2005; Matrosovich et al., 2000). Avian influenza A virus strains preferentially bind to α2,3-SA since this form of SA is abundant in the gastrointestinal tract of various avian species (Gambaryan et al., 2002; Gambaryan et al., 2004; Kim et al., 2005; Matrosovich et al., 2000). However, while the human respiratory tract contains both α2,3- and α2,6-SA (Matrosovich et al., 2004; Shinya et al., 2006; Slepushkin et al., 2001; van Riel et al., 2006; Zhang et al., 2005), most human influenza A virus strains show a binding preference for α2,6-SA as a receptor (Baum and Paulson, 1990; Gagneux et al., 2003; Matrosovich et al., 2000; Matrosovich et al., 2004).
In mammals, the primary tissues infected by influenza A virus are the trachea as well as the upper and lower bronchial tubes (Zambon, 2001). In rare cases, virus infection of the alveolar epithelial cells occurs, leading to viral pneumonia. Several animal models have been used to study influenza A virus pathogenesis, most notably the laboratory mouse (Lu et al., 1999; Novak et al., 1993). However, most human influenza A virus strains must be adapted for efficient replication in mice (Brown et al., 2001; Lu et al., 1999). Human influenza A virus clinical isolates replicate and cause disease in ferrets (Govorkova et al., 2005; Herlocher et al., 2001; Maher and DeStefano, 2004; Reuman et al., 1989; Sweet et al., 2002; Zitzow et al., 2002), Syrian golden hamsters (Ali et al., 1982; Daly et al., 2003; Murphy et al., 1997; Potter and Jennings, 1976; Renis, 1977; Snyder et al., 1989; Stein-Streilein and Guffee, 1986), cotton rats (Ottolini et al., 2005) and non-human primates (Rimmelzwaan et al., 2001), but with the possible exception of ferrets, none of these animal models is used extensively.
Since Syrian golden hamsters can be infected with influenza A virus and infected hamsters can transmit virus to uninfected animals, we characterized the ability of influenza A virus strains to infect primary, differentiated tracheal epithelial cells (TECs) derived from Syrian golden hamsters. Our results demonstrate that Syrian golden hamsters and hamster TECs, represent an attractive system for studying infection of nonciliated airway epithelial cells, investigating the reassortment of influenza A virus strains which have dissimilar receptor preferences and generating influenza A virus strains with altered or expanded receptor utilization.
Results
Hamster TECs support influenza A virus replication
The ability of influenza A viruses to replicate in well-differentiated hamster TEC cultures grown in Transwell tissue culture inserts was determined by infecting the cultures with recombinant influenza A virus strains A/WSN/33 (rWSN;H1N1) and A/Udorn/72 (rUdorn;H3N2), and the clinical isolates A/California/7/2004 (H3N2), A/Memphis/14/96 (H1N1), A/Memphis/5/98 (H3N2). Infectious virus titers in the supernatants of virus-infected cells were determined by plaque assay at various times post infection. Hamster TEC cultures are routinely infected between days 10 and 14 after initiation of an air liquid interface (ALI), a time when the culture is fully differentiated into a polarized, heterogeneous cell population (Rowe et al., 2004). Both recombinant strains and clinical isolates displayed similar patterns of replication, consisting of a peak in viral titer at 1 to 2 days post infection, and then a decline to undetectable amounts by 4 to 5 days post infection (Fig. 1A, B). Virus was detected in the apical but not the basolateral supernatants over the time course of infection, (Rowe and Pekosz, 2006), and proteases produced by the cells were responsible for cleavage of the HA protein into its active subunits (Rowe et al., 2004). Thus, laboratory strains and clinical isolates of influenza A virus replicate to equivalent infectious virus titers in hamster TECs.
Figure 1.

Influenza A virus replicates in hamster TEC cultures. (A) Recombinant viruses rWSN and rUdorn and (B) clinical isolates A/California/7/2004, A/Memphis/14/96, and A/Memphis/5/98 were used to infect hamster TEC cultures at day 10 ALI with an MOI of 3. Infectious virus titers were determined at the indicated days. Virus titers from the apical chamber are shown as no infectious virus was detected in the basolateral chamber. The titer of the input virus is graphed at time = 0 and the 1 hr timepoint represents the initial inoculum that was remaining after washing. The horizontal, dashed line indicates the limit of detection, 2000 PFU/ml.
The cell tropism associated with influenza A virus infection was determined by immunostaining virus-infected hamster TEC cultures for expression of the influenza A virus HA protein and the ciliated cell marker β–tubulin IV after infection with rUdorn. Viral antigen expression correlated with the peak of virus titer, with antigen positive cells being abundant at day 1 and decreasing in subsequent days post infection (Fig. 2). Viral antigen appeared to predominate in the nonciliated cell population, particularly on day one post infection, but limited numbers of viral antigen-positive, ciliated cells could be detected at day 2 post infection. The decreased numbers of cell nuclei (visualized by TO-PRO-3 fluorescence) over time (Fig. 2, lower panels) suggested that virus infection induced cell death within the culture, most likely in the virus-infected cell population. However, virus infection did not result in death of all the cells in the culture, as demonstrated by the presence of ciliated cells at all times post infection (Fig. 2, upper panels). This data demonstrates that the nonciliated cells in the hamster TEC cultures, are the primary cell type infected by rUdorn virus and that some subset of cells in the cultures are unable to support influenza A virus replication.
Figure 2.
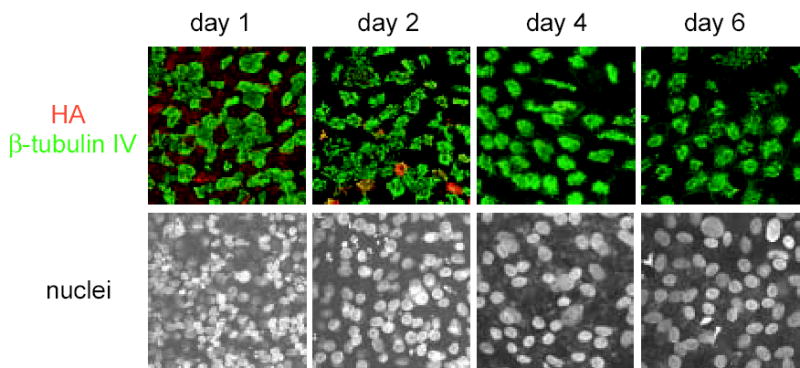
Influenza A virus infection of hamster TECs is self-limiting and specific for a subset of cells. Hamster TECs were infected at day 10 ALI with rUdorn at an MOI of 3, and monitored over 6 days for expression of viral antigen (HA, red) and β-tubulin IV (green). Cell nuclei were visualized with TO-PRO-3 fluorescence (bottom panel). All images were taken at a 63X magnification.
Expression of α2,3- and α2,6-linked SA in hamster TECs
Since influenza viral antigen expression was detected predominantly in the nonciliated cell population, we determined whether virus receptor expression could be responsible for the cell type-specific infection pattern. The expression of α2,3- and α2,6-linked SAs, the primary receptors for influenza A virus entry, was determined using lectins that recognize either α2,3- (Maackia amurensis agglutinin, MAA) or α2,6- (Sambucus nigra agglutinin, SNA) SA. Both α2,3- and α2,6-SA moieties were expressed (Fig. 3A), and quantitation of the number of cells per field expressing each linkage gave a ratio of 1.5:1 of α2,3- to α2,6-SA expressing cells (Fig. 3B). We next determined the distribution of α2,3- and α2,6-SA on specific cell types within the hamster TEC culture. α2,6-SA was localized to nonciliated cells, as demonstrated by the lack of co-localization of SNA with the ciliated cell marker protein β-tubulin IV (Fig. 3C, upper panels). α2,3-SA had a broader distribution than α2,6-SA, and was detected on both ciliated and nonciliated cells (Fig. 3C lower panels). It is important to note however, that the population of cells that expressed the highest levels of α2,3-SA were not β-tubulin IV positive (Fig. 3C lower panels, white asterisks). Thus, the nonciliated cell population expressed both α2,3- and α2,6- linked SAs while the ciliated cells only express α2,3-SA but to a lower level than that observed on nonciliated cells.
Figure 3.
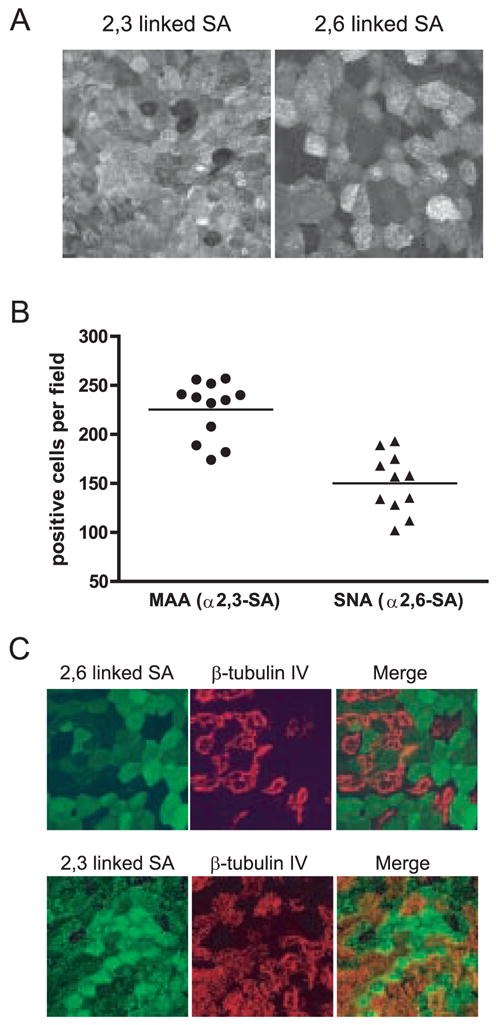
The α2,3- and α2,6-linked forms of SA are expressed in hamster TEC cultures. (A) Representative image showing the distribution of α2,3- or α2,6-SA. (B) Quantitation of the number of cells per field expressing α2,3- or α2,6-SA. Each point represents an individual field. (C) The expression of α2,3- or α2,6-SA (green) compared to the ciliated cell marker β-tubulin IV (red). There is a low level α2,3-SA expressed in ciliated cells as compared to nonciliated cells (white asterisks indicate nonciliated cells expressing high amounts of α2,3-SA) but α2,6-SA appears to be expressed exclusively in nonciliated cells. All images were taken at a 63X magnification.
rUdorn viral antigen was detected primarily in nonciliated cells at day one post infection (Fig. 2), and this correlates with the high level of α2,3- and α2,6-linked SA expression in the same population of cells. To conduct a more thorough analysis of virus strain specific differences in cell tropism, hamster TECs were infected with rWSN (H1N1) or rUdorn (H3N2), and viral antigen expression was analyzed during the first replication cycle (6 hrs post infection), as well as 1 and 2 days post infection. The rWSN virus has been passaged extensively in mice and embryonated eggs, conditions that have been shown to result in a preference for α2,3-SA as an entry receptor (Gambaryan et al., 1999; Gambaryan et al., 1997; Matrosovich et al., 2000) and it encodes amino acids at positions 190 and 225 of the HA protein that dictate preferred binding to α2,3-SA (Glaser et al., 2005; Stevens et al., 2006) . The rUdorn virus has been passaged in MDCK cells and MDCK cell passage is believed to maintain the α2,6-linked SA receptor preference of human influenza A virus strains (Gambaryan et al., 1999; Gambaryan et al., 1997; Matrosovich et al., 2000). The amino acids present at positions 226 and 228 of the HA protein predict a preference for binding to α2,6 SA (Connor et al., 1994; Gambaryan et al., 2005; Gambaryan et al., 1999; Gambaryan et al., 1997; Govorkova et al., 1999; Ito et al., 1997; Rogers and Paulson, 1983; Rogers et al., 1983; Stevens et al., 2006), however, binding of rUdorn HA to α2,3 and α2,6-linked SA was detected using ganglioside binding assays (Ryan-Poirier et al., 1998).
At six hours post infection, rUdorn HA was expressed predominantly in the nonciliated cell population (Fig. 4A a–c), and this same pattern is seen at day one post infection (Fig. 4A e–g). At day two post infection, few viral antigen positive cells are detected (Fig. 4A i–k, white asterisks in k denote antigen positive cells), suggesting that most of the virus infected cells have succumbed to infection. Interestingly, almost all the antigen positive cells detected at day two post infection are ciliated. The numbers of viral antigen positive cells were quantified on days 1 and 2 post infection and data indicate that there are limited numbers (less than 5%) of infected, ciliated cells on both days post infection (Fig. 4B). However, the majority of infected cells are not ciliated and the numbers of infected, nonciliated cells drop dramatically between days 1 and 2 post infection. The numbers of ciliated cells in the infected cultures did not drop as significantly as the total number of nuclei, indicating that virus infection was eliminating nonciliated cells, consistent with the cell tropism data (Figure 4C). This data indicates that the primary cell type infected by rUdorn is the nonciliated cell, with some infected ciliated cells detected at days 1 and 2 post infection. Since only α2,3-SA is detected in ciliated cells, the rUdorn virus is most likely utilizing this form of sialic acid to enter ciliated cells.
Figure 4.
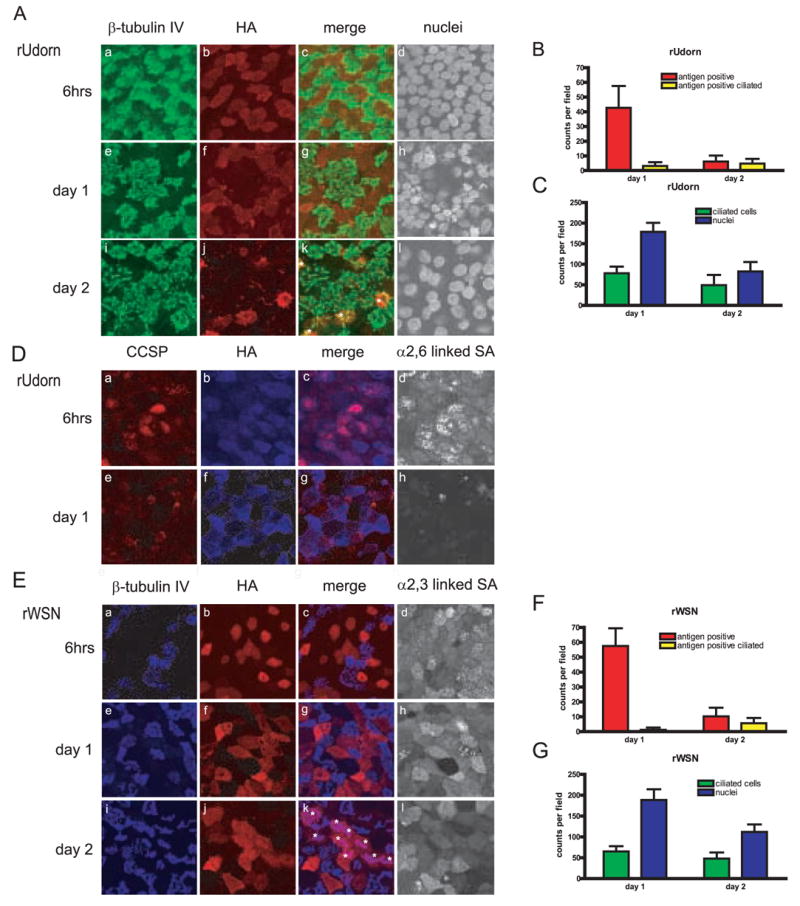
Influenza A virus infection targets nonciliated cells in hamster TEC cultures. Cultures were infected with an MOI of 3, and were analyzed for antigen expression at the indicated times post infection (A) rUdorn HA expression (red) compared to β–tubulin IV (green) (D) Colocalization of rUdorn HA (blue) with the secretory cell protein CCSP (red) and α2,6-SA (E) rWSN HA expression (red) compared to β–tubulin IV (blue) and α2,3-SA. White asterisks in panel k indicate rWSN-infected ciliated cells at day two post infection. The same field is shown in each of the four panels for each row. Quantitation of antigen positive cells (B,F) and ciliated cells and nuclei (C,G) at days one and two post infection. Ten to fourteen fields were counted for each virus per day and the data is expressed as a percentage of the total cells (nuclei) present in each field. There are approximately 150–250 nuclei per field in uninfected cultures (Rowe et al., 2004)). All images were taken at a 63X magnification.
The Clara cell secretory protein (CCSP) was used as a marker for one type of nonciliated cell present in the hamster TEC cultures (Singh and Katyal, 2000). CCSP co-localized with rUdorn HA at six hours post infection (Fig. 4D a–c), indicating that Clara cells are at least one type of nonciliated cell infected by influenza A virus. At day one post infection, rUdorn HA expression was clearly detected (Fig. 4D f), but the CCSP staining appeared slightly diminished, likely due to the cytopathic effects of virus replication on this cell type or to reduced translation of host cell proteins in virus-infected cells (Fig. 4D e and g). Expression of α,2,6-SA was evident at six hours post infection, but difficult to detect at day one post infection (Figure 4D, panels d,h) most likely due to the combined effect of death of the nonciliated cells and reduced SA expression due to the activity of the viral NA protein.
Viral antigen expression in rWSN-infected cells was similar to that of rUdorn at six hours and day one post infection, with HA detected predominantly in nonciliated cells (Fig. 4E a–c and e–g). At day two post infection, rWSN HA was detected in a limited number of ciliated cells, however antigen positive nonciliated cells are also present (Fig. 4E i–k, ciliated cells marked with asterisks). The numbers of antigen positive cells were quantified and the data again show that the nonciliated cells are the primary cell type infected by rWSN, with infected ciliated cells low in number (less than 5%) at days 1 and 2 post infection (Figure 4F). The reduction in number of nuclei per field was much greater than the reduction of ciliated cells per field, indicating the nonciliated cells were preferentially eliminated in virus-infected monolayers (Figure 4G). When the expression of α2,3-SA – the SA form expressed on ciliated cells – was assessed, the ciliated cells infected at day 2 showed reduced amounts of α2,3-SA when compared to nonciliated cells at six hours or 1 day after infection (Figure 4E, panels d,h and l). This data suggests that rWSN infects primarily nonciliated cells, with limited numbers of ciliated cells infected at days 1 and 2 post infection. Taken together, the data in figure 4 indicates the expression of α2,3- and α2,6-SA on nonciliated cells correlates very closely with the predominant viral antigen expression at early and late times during infection with both rUdorn and rWSN viruses.
In addition to rWSN and rUdorn, clinical isolates A/California/7/2004, A/Memphis/14/96, and A/Memphis/5/98 were also examined for HA expression in the HamsterTEC cultures at the same timepoints (Figure 5). The A/Memphis/14/96 and A/Memphis/5/98 viruses have been documented to bind preferentially to α2,6-SA (Matrosovich et al., 2003; Matrosovich et al., 2004). HA expression of all three clinical isolates displayed the same antigen pattern - expression of HA exclusively in the non-ciliated cells during the first round of virus replication (Fig. 5A a–c, e–g, i–k) and day 1, with little to no antigen detected at day 2.. The number of antigen positive cells, ciliated cells, and nuclei at day 1 and day 2 were quantified for each clinical isolate (Fig 5., B–G). While the number of ciliated cells stay relatively constant through two days post infection, nuclei counts decrease, suggesting again that the nonciliated cell population is infected and eliminated after infection with influenza A virus (Figure 4 C, E and G). However, in contrast to the laboratory strains, antigen positive ciliated cells were not detected at any time point with the three clinical isolates (Fig.5, B, D and F). Thus, it appears that the ability of rWSN and rUdorn to infect a limited number of ciliated cells is not shared by the clinical isolates of the influenza A virus strains analyzed, and may be related to altered SA receptor usage (Deom et al., 1986; Ohuchi et al., 1997; Ward, 1997), or that tissue culture passage has created minor populations able to bind to alternate receptors in the case of rUdorn. Clinical human isolates of influenza A virus, however, seem to maintain a more strict receptor specificity for the α2,6-SA expressing nonciliated cell population.
Figure 5.
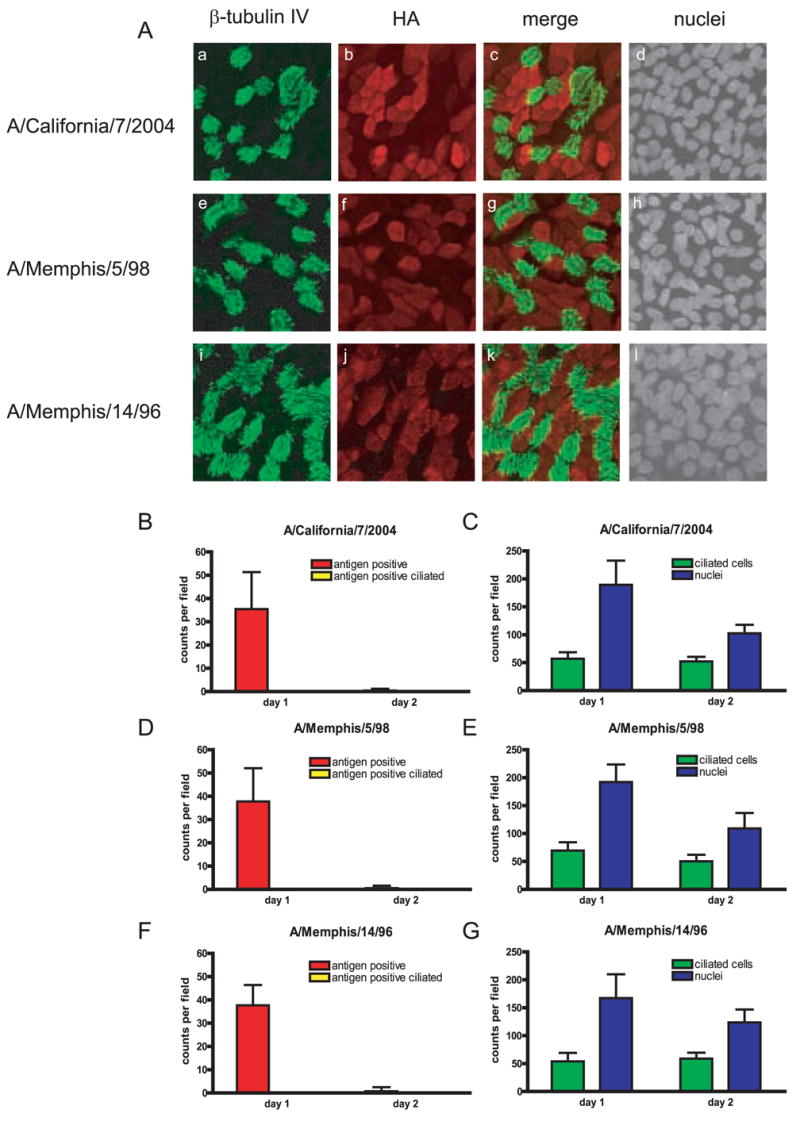
Influenza A virus clinical isolates exclusively target nonciliated cells in hamster TEC cultures. Cultures were infected with an MOI of 3, and were analyzed for antigen expression at indicated times post infection. (A) clinical isolates A/California/7/2004, A/Memphis/5/98. and A/Memphis/14/96 at nine hours post infection. HA expression (red) compared to β–tubulin IV (green). The same field is shown in each of the four panels for each row. (B–G) Quantitation of antigen positive cells (B,D and F) and ciliated cells and nuclei (C,E and G) at days one and two post infection. Ten to fourteen fields were counted for each virus per day and the data is expressed as a percentage of the total cells (nuclei) present in each field. There are approximately 150–250 nuclei per field in un-infected cultures (Rowe et al., 2004)). All images were taken at a 63X magnification.
Discussion
Receptor utilization is an important determinant of viral pathogenesis and cell tropism. For influenza A virus, receptor utilization is particularly important as it is thought to be one of the key factors enabling avian influenza A viruses to replicate in and spread between humans (Parrish and Kawaoka, 2005). The ongoing outbreak of H5N1 virus occurring in poultry throughout Asia and parts of Europe has led to over one hundred cases of human infection, thus increasing the likelihood that a variant of H5N1 displaying an expanded receptor tropism for α2,6-SA may emerge (Bartlett and Hayden, 2005; Holmes et al., 2005). In order to better understand the process that can lead to the emergence of a pandemic influenza virus strain, appropriate animal and tissue culture models must be developed. Our data with Syrian golden hamster airway epithelial cell cultures provides such a model system.
Hamster TECs are susceptible to lab adapted as well as clinical isolates of influenza A virus (this study and (Rowe and Pekosz, 2006), and Syrian golden hamsters have been shown to be an animal model for influenza A virus infection (Ali et al., 1982; Potter and Jennings, 1976; Renis, 1977; Stein-Streilein and Guffee, 1986) . Murine tracheal tissues and TEC cultures contain α2,3-SA on ciliated cells and little to no detectable α2,6-SA (CMN and AP unpublished observations) perhaps explaining the poor ability of human influenza A virus strains to replicate and cause disease in mice. Since hamster airway epithelial cells possess both α2,3- and α2,6-SA receptors, they are suitable hosts for comparative studies between viruses displaying different SA receptor specificities, avian and human influenza viruses for example. Syrian golden hamsters have been shown to support the replication of equine influenza A virus strains (Daly et al., 2003) and these influenza A virus strains have a preference for utilizing α2,3-SA as a receptor (Medeiros et al., 2004) due to the prevalence of α2,3-SA in the equine respiratory tract (Lin et al., 2001).
Since both forms of SA were present in the same cell type, primarily nonciliated cells, hamster TEC cultures may facilitate studies on the reassortment between influenza A viruses which utilize different forms of SA (avian and human influenza viruses for example) as well as the adaptation of α2,3-SA utilizing viruses to α2,6-SA usage. The expression of α2,3-SA in ciliated cells was reduced when compared to expression in nonciliated cells and ciliated cells were less susceptible to infection with rUdorn and rWSN. This may indicate that the absolute level of SA on the plasma membrane may also dictate the susceptibility of a cell to influenza A virus infection, but definitive statements on this await more careful and accurate quantification of SA levels in specific cell types. The effect of viral neuraminidase activity and specificity on the amount of available SA may also have a profound effect on cell susceptibility to virus infection.
The expanded cell tropism of rUdorn in hamster TEC cultures when compared to primary influenza A virus isolates may reflect an the ability of the Udorn HA protein to recognize both α2,3- and α2,6-SA (Ryan-Poirier et al., 1998). Curiously, the Udorn HA protein possess amino acid residues at positions 193, 226 and 228 that are found in H3 subtype viruses that bind preferentially to α2,6-SA (Medeiros et al., 2001; Medeiros et al., 2004; Stevens et al., 2006; van Riel et al., 2006). Position 186 of H3 subtype HA proteins has been implicated in altered receptor specifity with substitutions of G186V or S186I resulting in increased α2,3-SA binding (Gambaryan et al., 1999; Widjaja et al., 2006), however, the rUdorn strain encodes a S at this amino acid. This indicates that other, as yet unidentified amino acids may alter SA receptor recognition in H3 subtype HA proteins or that the assays used to detect α2,3- and α2,6-SA binding are not reflective of the cell tropism documented in this study. It is important to note that while receptor utilization and expression are important determinants of influenza A virus cell tropism in the respiratory tract (Matrosovich et al., 2004) (Shinya et al., 2006; van Riel et al., 2006; Zhang et al., 2005; Zhang et al., 2002) , it has been demonstrated that interferon and interferon signaling are also important factors that limit influenza A virus tropism in vivo and in vitro (Garcia-Sastre et al., 1998a; Garcia-Sastre et al., 1998b).
The expression of α2,3- and α2,6-SA in the human respiratory tract has been investigated extensively (Matrosovich et al., 2004; Slepushkin et al., 2001; Zhang et al., 2005). Most studies support the model that α2,3-SA is present primarily, if not exclusively, in the ciliated cells. The expression of α2,6-SA has been documented to be either exclusively in the nonciliated cells (Matrosovich et al., 2004) or present in both ciliated and nonciliated cells (Ibricevic et al., ; Zhang et al., 2005). The amount of SA on each cell type has yet to quantified precisely. Highly pathogenic H5N1 influenza A virus strains have limited ability to attach to and replicate in the upper respiratory tract as compared to the lower respiratory tract – again the result (at least in part) of altered expression of α2,3- and α2,6-SA (Shinya et al., 2006; van Riel et al., 2006). The SA receptor pattern on hamster TEC cultures differs from that described for humans, an important factor to consider when interpreting hamster pathogenesis experiments. However, there are two points to consider when deciding between hamsters or mice for influenza pathogenesis studies. First, the hamster model may be better suited to pathogenesis experiments with human influenza A virus strains when compared to the mouse model since α2,6-SA is not expressed in the mouse respiratory tract, but is present in the upper and lower respiratory tract of the hamster (CMN and AP unpublished observations). Second, humans and hamsters have significant numbers of mucous-secreting goblet cells (Atherton et al., 2003; Rowe et al., 2004) while mice possess virtually undetectable numbers (Look et al., 2001; Shahzeidi et al., 2003; Walter et al., 2002). Since hamster and human goblet cells also express SA receptors, studies on the effects of influenza on this cell type are best performed in the hamster model.
It has been suggested that replication of influenza A virus in nonciliated cells is important for efficient virus replication and/or transmission in humans (Matrosovich et al., 2004). This may also explain the ability of hamsters to support the replication and transmission of human influenza A virus strains (Ali et al., 1982; Daly et al., 2003) and suggests that studying transmission of human and avian influenza A viruses in hamsters may help shed light on viral and host factors that are important for virus transmission. Our data demonstrates that primary, differentiated hamster TEC cultures can serve as a useful model of influenza A virus pathogenesis and cell tropism, and provides a relevant cell culture system for studying influenza virus strains that have different SA receptor specificities.
Materials and Methods
Reagents and antibodies
The components in TEC basic media (TEC basic), proliferation media (TEC plus), and maintenance media (TEC MM) have been described previously (Rowe et al., 2004). Rabbit anti-hamster Clara cell secretory protein (CCSP; 1:500 immunofluorescence) was kindly provided by Gurmukh Singh (VA medical center, Pittsburgh, PA). Other primary antibodies were purchased and used as follows: mouse anti-β Tubulin IV (1:100 immunofluorescence; BioGenex, San Ramon, CA), goat anti-H3 hemagglutinin (HA-H3 subtype) Aichi/2/68 sera (1:500 immunofluorescence; NIH/NIAD reference reagent V314-591-157), goat anti-H1 hemagglutinin (HA-H0 subtype) A/PR8/34 sera (1:500 immunofluorescence; NIH/NIAD reference reagent V314-511-157). The influenza A virus HA antibodies showed minimal reactivity to mock infected hamster TEC cultures (data not shown). Secondary antibodies were used as follows: donkey anti-mouse IgG conjugated to fluorescein isothiocyanate (1:250 immunofluorescence), donkey anti-goat IgG conjugated to rhodamine red (1:250 immunofluorescence), and goat anti-mouse IgG conjugated to rhodamine red (1:250 immunofluorescence) were purchased from Jackson Immunoresearch (Westgrove, PA); goat anti-mouse IgG conjugated to Alexa Fluor 647 (1:500 immunofluorescence), donkey anti-mouse IgG conjugated to Alexa Fluor 647 (1:500 immunofluorescence), goat anti-rabbit IgG conjugated to Alexa Fluor 594 (1:500 immunofluorescence) and TO-PRO-3 nuclear stain (1:500) were purchased from Molecular Probes (Eugene, OR); anti-digoxigenin-fluoroscein, FAb fragment (1:50) was purchased from Roche applied science (Indianapolis, IN).
Cell lines
Madin-Darby canine kidney cells (MDCK American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle medium (DMEM, Sigma, St. Louis, MO) containing 10% fetal bovine serum (Atlanta Biologics, Atlanta, GA), 100U/ml of penicillin, and 100ug/ml of streptomycin (Invitrogen, Grand Island, NY) and maintained at 37°C in a humidified environment containing 5% CO2.
Viruses
Recombinant influenza A viruses A/WSN/33 (rWSN) (Neumann et al., 1999; Takeda et al., 2002) and A/Udorn/72 (rUdorn) (Takeda et al., 2002), and the influenza A virus clinical isolates A/California/7/2004 (Centers for Disease Control, Atlanta GA), A/Memphis/14/96 (H1N1), A/Memphis/5/98 (H3N2) (St. Jude Children’s Hospital, Memphis, TN) were used in this study. The latter two virus strains have been characterized with respect to their cell tropism in human TEC cultures (Matrosovich et al., 2004) and receptor specificity (Matrosovich et al., 2003) . Virus stocks were generated by infecting MDCK cells and infectious virus was quantified by plaque assay using MDCK cells as previously described (Paterson and Lamb, 1993). The H3 amino acid numbering system is used when referring to amino acid positions in HA.
Hamster TEC cultures
Hamster TEC isolation and culture conditions were performed as previously described (Rowe et al., 2004). In all experiments, hamster TEC cultures were seeded and maintained in 0.4μM pore, 0.33cm2 Transwell-Clear support membrane (Corning Costar, Corning NY) (Rowe et al., 2004).
Infection of Hamster TECs
Hamster TEC cultures were infected between days 10 and 14 after ALI. Hamster TECs were infected via the apical chamber with 1x106 plaque forming units (pfu) of virus diluted in warm DMEM with penicillin/streptomycin in a total volume of 100μl. If all the cells in the culture were susceptible to influenza virus infection, this would correspond to a multiplicity of infection (MOI) of approximately 3. Since the cultures are psuedostratified, not all of the cells in the culture will be exposed to the apically administered virus inoculum. Also, not all cell types express influenza A virus receptors. Cells were incubated with virus at 37°C for 1 hour, inoculum removed, and cells washed three times with 200μL of warm DMEM with penicillin/streptomycin. After washing, 100μL of DMEM with penicillin/streptomycin and 500μL of TEC maintenance media (MM) (Rowe et al., 2004) was placed in the apical and basolateral chambers, respectively. Apical and basolateral supernatants were collected at the indicated times post infection and stored at −70°C. All growth curves and immunostaining on hamster TEC cultures were reproduced at least three times.
Immunofluorescence confocal microscopy
At indicated times post infection, hamster TECs were washed three times with phosphate buffered saline (PBS, GIBCO Inc., Carlsbad, CA), and fixed in PBS containing 2% paraformaldehyde for 15min at room temperature. Cells were washed three times and permeabilized with PBS containing 0.2% Triton-X 100 and 0.1% sodium citrate for ten minutes at room temperature. Cells were washed with PBS and incubated in PBS containing 3% normal goat or normal donkey serum and 0.5% bovine serum albumin (blocking buffer) for 30 minutes at room temperature. Cells were washed, and incubated with blocking buffer containing primary antibody for one hour at room temperature, washed again, and incubated with blocking buffer containing secondary antibodies and TO-PRO-3 for 45 minutes. The wash solution for all steps is PBS with 0.2% Tween-20. Transwell-Clear membranes were mounted using 10uL of Molecular Probes ProLong antifade (Molecular Probes), and slides were imaged using a Ziess LSM 510 Meta confocal microscope. All images were obtained with a 63x oil objective. All images presented are a flattened composite of 5–10 micron Z-stack images collected with LSM software.
Detection of lectin binding
The DIG glycan differentiation kit (Roche, Indianapolis, IN) was used. SA linked to the penultimate galactose or N-acetylgalactosamine via an α2,3 linkage was detected with digoxigenin-conjugated Maackia amurensis agglutinin (MAA) lectin while α2,6 linked SA was detected with digoxigenin-conjugated Sambucus nigra agglutinin (SNA) lectin. Samples were stained according to the manufacturer’s protocol using anti-digoxigenin FITC. Lectin-positive cells in ten to twelve fields were counted to determine the number of cells expressing each linkage. There are between 150–250 total cells per field, as determined by TO-PRO-3 staining. Lectin staining on hamster TEC cultures was reproduced in three separate experiments.
Acknowledgments
We acknowledge the Molecular Microbiology Imaging facility for technical support and all the members of the Pekosz laboratory for insightful discussions and comments. We thank Nancy Cox and Robert Webster for providing influenza A virus strains. This work was supported by the Whitaker Foundation Young Investigator award (A.P) and Department of Health and Human Services, Public Health Services grant AI053629 (A.P.).
References
- Ali MJ, Teh CZ, Jennings R, Potter CW. Transmissibility of influenza viruses in hamsters. Arch Virol. 1982;72(3):187–97. doi: 10.1007/BF01348964. [DOI] [PubMed] [Google Scholar]
- Atherton HC, Jones G, Danahay H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L730–739. doi: 10.1152/ajplung.00089.2003. [DOI] [PubMed] [Google Scholar]
- Barnett DJ, Balicer RD, Lucey DR, Everly GS, Omer SB, Steinhoff MC, Grotto I. A Systematic Analytic Approach to Pandemic Influenza Preparedness Planning. PLoS Medicine. 2005;2(12) doi: 10.1371/journal.pmed.0020359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JG, Hayden FG. Influenza A (H5N1): Will It Be the Next Pandemic Influenza? Are We Ready? Ann Intern Med. 2005;143(6):460–462. doi: 10.7326/0003-4819-143-6-200509200-00011. [DOI] [PubMed] [Google Scholar]
- Brown EG, Liu H, Kit LC, Baird S, Nesrallah M. Pattern of mutation in the genome of influenza A virus on adaptation to increased virulence in the mouse lung: identification of functional themes. Proc Natl Acad Sci U S A. 2001;98(12):6883–8. doi: 10.1073/pnas.111165798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VC, Whittaker GR. Influenza virus entry and infection require host cell N-linked glycoprotein. PNAS. 2004;101(52):18153–18158. doi: 10.1073/pnas.0405172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RJ, Kawaoka Y, Webster RG, Paulson JC. Receptor Specificity in Human, Avian, and Equine H2 and H3 Influenza Virus Isolates. Virology. 1994;205(1):17–23. doi: 10.1006/viro.1994.1615. [DOI] [PubMed] [Google Scholar]
- Daly JM, Yates RJ, Browse G, Swann Z, Newton JR, Jessett D, Davis-Poynter N, Mumford JA. Comparison of hamster and pony challenge models for evaluation of effect of antigenic drift on cross protection afforded by equine influenza vaccines. Equine Vet J. 2003;35(5):458–62. doi: 10.2746/042516403775600433. [DOI] [PubMed] [Google Scholar]
- Deom CM, Caton AJ, Schulze IT. Host Cell-Mediated Selection of a Mutant Influenza A Virus That Has Lost a Complex Oligosaccharide from the Tip of the Hemagglutinin. PNAS. 1986;83(11):3771–3775. doi: 10.1073/pnas.83.11.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RAM, Munster V, Wallensten A, Bestebroer TM, Herfst S, Smith D, Rimmelzwaan GF, Olsen B, Osterhaus ADME. Characterization of a Novel Influenza A Virus Hemagglutinin Subtype (H16) Obtained from Black-Headed Gulls. J Virol. 2005;79(5):2814–2822. doi: 10.1128/JVI.79.5.2814-2822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan A, Webster R, Matrosovich M. Differences between influenza virus receptors on target cells of duck and chicken. Archives of Virology. 2002;147(6):1197–1208. doi: 10.1007/s00705-002-0796-4. [DOI] [PubMed] [Google Scholar]
- Gambaryan AS, Karasin AI, Tuzikov AB, Chinarev AA, Pazynina GV, Bovin NV, Matrosovich MN, Olsen CW, Klimov AI. Receptor-binding properties of swine influenza viruses isolated and propagated in MDCK cells. Virus Research. 2005;114(1–2):15–22. doi: 10.1016/j.virusres.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Gambaryan AS, Robertson JS, Matrosovich MN. Effects of Egg-Adaptation on the Receptor-Binding Properties of Human Influenza A and B Viruses. Virology. 1999;258(2):232–239. doi: 10.1006/viro.1999.9732. [DOI] [PubMed] [Google Scholar]
- Gambaryan AS, Tuzikov AB, Pazynina GV, Webster RG, Matrosovich MN, Bovin NV. H5N1 chicken influenza viruses display a high binding affinity for Neu5Ac[alpha]2-3Gal[beta]1-4(6-HSO3)GlcNAc-containing receptors. Virology. 2004;326(2):310–316. doi: 10.1016/j.virol.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Gambaryan AS, Tuzikov AB, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Bovin NV, Matrosovich MN. Specification of Receptor-Binding Phenotypes of Influenza Virus Isolates from Different Hosts Using Synthetic Sialylglycopolymers: Non-Egg-Adapted Human H1 and H3 Influenza A and Influenza B Viruses Share a Common High Binding Affinity for 6′-Sialyl(N-acetyllactosamine) Virology. 1997;232(2):345–350. doi: 10.1006/viro.1997.8572. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. The role of interferon in influenza virus tissue tropism. J Virol. 1998a;72(11):8550–8. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998b;252(2):324–30. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Glaser L, Stevens J, Zamarin D, Wilson IA, Garcia-Sastre A, Tumpey TM, Basler CF, Taubenberger JK, Palese P. A Single Amino Acid Substitution in 1918 Influenza Virus Hemagglutinin Changes Receptor Binding Specificity. J Virol. 2005;79(17):11533–11536. doi: 10.1128/JVI.79.17.11533-11536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorkova EA, Matrosovich MN, Tuzikov AB, Bovin NV, Gerdil C, Fanget B, Webster RG. Selection of receptor-binding variants of human influenza A and B viruses in baby hamster kidney cells. Virology. 1999;262(1):31–8. doi: 10.1006/viro.1999.9892. [DOI] [PubMed] [Google Scholar]
- Govorkova EA, Rehg JE, Krauss S, Yen H-L, Guan Y, Peiris M, Nguyen TD, Hanh TH, Puthavathana P, Long HT, Buranathai C, Lim W, Webster RG, Hoffmann E. Lethality to Ferrets of H5N1 Influenza Viruses Isolated from Humans and Poultry in 2004. J Virol. 2005;79(4):2191–2198. doi: 10.1128/JVI.79.4.2191-2198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlocher ML, Elias S, Truscon R, Harrison S, Mindell D, Simon C, Monto AS. Ferrets as a transmission model for influenza: sequence changes in HA1 of type A (H3N2) virus. J Infect Dis. 2001;184(5):542–6. doi: 10.1086/322801. [DOI] [PubMed] [Google Scholar]
- Holmes EC, Taubenberger JK, Grenfell BT. Heading Off an Influenza Pandemic. Science. 2005;309(5737):989. doi: 10.1126/science.1117128. [DOI] [PubMed] [Google Scholar]
- Horimoto T, Kawaoka Y. Influenza: Lessons from Past Pandemics, Warnings from Current Incidents. Nature Reviews Microbiology Nat Rev Micro. 2005;3(8):591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, HoltzmaN MJ, Brody SL. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol. 2006 doi: 10.1128/JVI.02677-05. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T. Interspecies transmission and receptor recognition of influenza A viruses. Microbiol Immunol. 2000;44(6):423–30. doi: 10.1111/j.1348-0421.2000.tb02516.x. [DOI] [PubMed] [Google Scholar]
- Ito T, Suzuki Y, Takada A, Kawamoto A, Otsuki K, Masuda H, Yamada M, Suzuki T, Kida H, Kawaoka Y. Differences in sialic acid-galactose linkages in the chicken egg amnion and allantois influence human influenza virus receptor specificity and variant selection. J Virol. 1997;71(4):3357–3362. doi: 10.1128/jvi.71.4.3357-3362.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Ryu SY, Seo SH. Cells in the respiratory and intestinal tracts of chickens have different proportions of both human and avian influenza virus receptors. J Microbiol. 2005;43(4):366–9. [PubMed] [Google Scholar]
- Lamb RA, Krug RM. Orthomyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4th ed. . Lippincott, Williams & Wilkins; Philadelphia: 2001. pp. 1487–532. [Google Scholar]
- Lin C, Holland RE, Jr, Williams NM, Chambers TM. Cultures of equine respiratory epithelial cells and organ explants as tools for the study of equine influenza virus infection. Arch Virol. 2001;146(11):2239–47. doi: 10.1007/s007050170034. [DOI] [PubMed] [Google Scholar]
- Look DC, Walter MJ, Williamson MR, Pang L, You Y, Sreshta JN, Johnson JE, Zander DS, Brody SL. Effects of paramyxoviral infection on airway epithelial cell Foxj1 expression, ciliogenesis, and mucociliary function. Am J Pathol. 2001;159(6):2055–69. doi: 10.1016/S0002-9440(10)63057-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. A Mouse Model for the Evaluation of Pathogenesis and Immunity to Influenza A (H5N1) Viruses Isolated from Humans. J Virol. 1999;73(7):5903–5911. doi: 10.1128/jvi.73.7.5903-5911.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher JA, DeStefano J. The ferret: an animal model to study influenza virus. Lab Anim (NY) 2004;33(9):50–3. doi: 10.1038/laban1004-50. [DOI] [PubMed] [Google Scholar]
- Matrosovich M, Matrosovich T, Carr J, Roberts NA, Klenk H-D. Overexpression of the {alpha}-2,6-Sialyltransferase in MDCK Cells Increases Influenza Virus Sensitivity to Neuraminidase Inhibitors. J Virol. 2003;77(15):8418–8425. doi: 10.1128/JVI.77.15.8418-8425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrosovich M, Tuzikov A, Bovin N, Gambaryan A, Klimov A, Castrucci MR, Donatelli I, Kawaoka Y. Early Alterations of the Receptor-Binding Properties of H1, H2, and H3 Avian Influenza Virus Hemagglutinins after Their Introduction into Mammals. J Virol. 2000;74(18):8502–8512. doi: 10.1128/jvi.74.18.8502-8512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A. 2004;101(13):4620–4. doi: 10.1073/pnas.0308001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, Escriou N, Naffakh N, Manuguerra JC, van der Werf S. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology. 2001;289(1):74–85. doi: 10.1006/viro.2001.1121. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Naffakh N, Manuguerra JC, Werf Svd. Binding of the hemagglutinin from human or equine influenza H3 viruses to the receptor is altered by substitutions at residue 193. Archives of Virology. 2004;149(8):1663–1671. doi: 10.1007/s00705-003-0287-2. [DOI] [PubMed] [Google Scholar]
- Murphy BR, Park EJ, Gottlieb P, Subbarao K. An influenza A live attenuated reassortant virus possessing three temperature-sensitive mutations in the PB2 polymerase gene rapidly loses temperature sensitivity following replication in hamsters. Vaccine. 1997;15(12–13):1372–8. doi: 10.1016/s0264-410x(97)00031-5. [DOI] [PubMed] [Google Scholar]
- Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A. 1999;96 (16):9345–50. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak M, Moldoveanu Z, Schafer DP, Mestecky J, Compans RW. Murine model for evaluation of protective immunity to influenza virus. Vaccine. 1993;11 (1):55–60. doi: 10.1016/0264-410x(93)90339-y. [DOI] [PubMed] [Google Scholar]
- Ohuchi M, Ohuchi R, Feldmann A, Klenk HD. Regulation of receptor binding affinity of influenza virus hemagglutinin by its carbohydrate moiety. J Virol. 1997;71(11):8377–8384. doi: 10.1128/jvi.71.11.8377-8384.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolini MG, Blanco JCG, Eichelberger MC, Porter DD, Pletneva L, Richardson JY, Prince GA. The cotton rat provides a useful small-animal model for the study of influenza virus pathogenesis. J Gen Virol. 2005;86 (10):2823–2830. doi: 10.1099/vir.0.81145-0. [DOI] [PubMed] [Google Scholar]
- Parrish CR, Kawaoka Y. The Origins of New Pandemic Viruses: The Acquisition of New Host Ranges by Canine Parvovirus and Influenza A Viruses. Annual Review of Microbiology. 2005;59(1):553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- Paterson RG, Lamb RA. Molecular Virology: A Practical Approach. Oxford University Press; Oxford, U.K: 1993. The molecular biology of influenza viruses and paramyxoviruses; pp. 35–73. [Google Scholar]
- Potter CW, Jennings R. The hamster as a model system for the study of influenza vaccines. Postgrad Med J. 1976;52(608):345–51. doi: 10.1136/pgmj.52.608.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AH, Fanning TG, Janczewski TA, Lourens RM, Taubenberger JK. Novel Origin of the 1918 Pandemic Influenza Virus Nucleoprotein Gene. J Virol. 2004;78(22):12462–12470. doi: 10.1128/JVI.78.22.12462-12470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renis HE. Influenza virus infection of hamsters. A model for evaluating antiviral drugs. Arch Virol. 1977;54(1–2):85–93. doi: 10.1007/BF01314381. [DOI] [PubMed] [Google Scholar]
- Reuman PD, Keely S, Schiff GM. Assessment of signs of influenza illness in the ferret model. J Virol Methods. 1989;24(1–2):27–34. doi: 10.1016/0166-0934(89)90004-9. [DOI] [PubMed] [Google Scholar]
- Rimmelzwaan GF, Kuiken T, van Amerongen G, Bestebroer TM, Fouchier RAM, Osterhaus ADME. Pathogenesis of Influenza A (H5N1) Virus Infection in a Primate Model. J Virol. 2001;75(14):6687–6691. doi: 10.1128/JVI.75.14.6687-6691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GN, Paulson JC. Receptor determinants of human and animal influenza virus isolates: differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology. 1983;127(2):361–73. doi: 10.1016/0042-6822(83)90150-2. [DOI] [PubMed] [Google Scholar]
- Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature. 1983;304(5921):76–78. doi: 10.1038/304076a0. [DOI] [PubMed] [Google Scholar]
- Rohm C, Zhou N, Suss J, Mackenzie J, Webster RG. Characterization of a Novel Influenza Hemagglutinin, H15: Criteria for Determination of Influenza A Subtypes. Virology. 1996;217(2):508–516. doi: 10.1006/viro.1996.0145. [DOI] [PubMed] [Google Scholar]
- Rowe RK, Brody SL, Pekosz A. Differentiated Cultures of Primary Hamster Trachealairway Epithelial Cells. In Vitro Cell Dev Biol Anim. 2004;40(10):303–311. doi: 10.1290/0408056.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RK, Pekosz A. Bidirectional Virus Secretion and Nonciliated Cell Tropism following Andes Virus Infection of Primary Airway Epithelial Cell Cultures. J Virol. 2006;80(3):1087–1097. doi: 10.1128/JVI.80.3.1087-1097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan-Poirier K, Suzuki Y, Bean WJ, Kobasa D, Takada A, Ito T, Kawaoka Y. Changes in H3 influenza A virus receptor specificity during replication in humans. Virus Res. 1998;56(2):169–76. doi: 10.1016/s0168-1702(98)00067-7. [DOI] [PubMed] [Google Scholar]
- Shahzeidi S, Aujla PK, Nickola TJ, Chen Y, Alimam MZ, Rose MC. Temporal analysis of goblet cells and mucin gene expression in murine models of allergic asthma. Exp Lung Res. 2003;29(8):549–65. doi: 10.1080/01902140390240159. [DOI] [PubMed] [Google Scholar]
- Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: Influenza virus receptors in the human airway. Nature. 2006;440(7083):435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- Singh G, Katyal SL. Clara Cell Proteins. Ann NY Acad Sci. 2000;923(1):43–58. doi: 10.1111/j.1749-6632.2000.tb05518.x. [DOI] [PubMed] [Google Scholar]
- Slepushkin VA, Staber PD, Wang G, McCray J, Paul B, Davidson BL. Infection of Human Airway Epithelia with H1N1, H2N2, and H3N2 Influenza A Virus Strains. Molecular Therapy. 2001;3(3):395–402. doi: 10.1006/mthe.2001.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MH, London WT, Maassab HF, Murphy BR. Attenuation and phenotypic stability of influenza B/Texas/1/84 cold-adapted reassortant virus: studies in hamsters and chimpanzees. J Infect Dis. 1989;160(4):604–10. doi: 10.1093/infdis/160.4.604. [DOI] [PubMed] [Google Scholar]
- Stein-Streilein J, Guffee J. In vivo treatment of mice and hamsters with antibodies to asialo GM1 increases morbidity and mortality to pulmonary influenza infection. J Immunol. 1986;136(4):1435–1441. [PubMed] [Google Scholar]
- Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. Glycan Microarray Analysis of the Hemagglutinins from Modern and Pandemic Influenza Viruses Reveals Different Receptor Specificities. Journal of Molecular Biology. 2006;355(5):1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sweet C, Jakeman KJ, Bush K, Wagaman PC, Mckown LA, Streeter AJ, Desai-Krieger D, Chand P, Babu YS. Oral Administration of Cyclopentane Neuraminidase Inhibitors Protects Ferrets against Influenza Virus Infection. Antimicrob Agents Chemother. 2002;46(4):996–1004. doi: 10.1128/AAC.46.4.996-1004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Pekosz A, Shuck K, Pinto LH, Lamb RA. Influenza A virus M2 ion channel activity is essential for efficient replication in tissue culture. J Virol. 2002;76(3):1391–9. doi: 10.1128/JVI.76.3.1391-1399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437(7060):889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. Influenza-Associated Hospitalizations in the United States. JAMA. 2004;292(11):1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. Jama. 2003;289(2):179–86. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. Characterization of the Reconstructed 1918 Spanish Influenza Pandemic Virus. Science. 2005;310(5745):77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RAM, Osterhaus ADME, Kuiken T. H5N1 Virus Attachment to Lower Respiratory Tract. Science. 2006;312(5772):399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest. 2002;110(2):165–75. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AC. Virulence of influenza A virus for mouse lung. Virus Genes. 1997;14(3):187–94. doi: 10.1023/a:1007979709403. [DOI] [PubMed] [Google Scholar]
- Webster RG, Shortridge KF, Kawaoka Y. Influenza: interspecies transmission and emergence of new pandemics. FEMS Immunology and Medical Microbiology. 1997;18(4):275–279. doi: 10.1111/j.1574-695X.1997.tb01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull World Health Organ. 1980;58(4):585–91. [PMC free article] [PubMed] [Google Scholar]
- Widjaja L, Ilyushina N, Webster RG, Webby RJ. Molecular changes associated with adaptation of human influenza A virus in embryonated chicken eggs. Virology. 2006;350(1):137–145. doi: 10.1016/j.virol.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Wilson N, Baker M, Crampton P, Mansoor O. The potential impact of the next influenza pandemic on a national primary care medical workforce. Hum Resour Health. 2005;3(1):7. doi: 10.1186/1478-4491-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon MC. The pathogenesis of influenza in humans. Rev Med Virol. 2001;11(4):227–41. doi: 10.1002/rmv.319. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ. Infection of Ciliated Cells by Human Parainfluenza Virus Type 3 in an In Vitro Model of Human Airway Epithelium. J Virol. 2005;79(2):1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory Syncytial Virus Infection of Human Airway Epithelial Cells Is Polarized, Specific to Ciliated Cells, and without Obvious Cytopathology. J Virol. 2002;76(11):5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitzow LA, Rowe T, Morken T, Shieh W-J, Zaki S, Katz JM. Pathogenesis of Avian Influenza A (H5N1) Viruses in Ferrets. J Virol. 2002;76(9):4420–4429. doi: 10.1128/JVI.76.9.4420-4429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]