

Abstract
Autophagy, the process by which cells recycle cytoplasm and dispose of excess or defective organelles, has entered the research spotlight largely owing to the discovery of the protein components that drive this process. Identifying the autophagy genes in yeast and finding orthologs in other organisms reveals the conservation of the mechanism of autophagy in eukaryotes and allows the use of molecular genetics and biology in different model systems to study this process. By mostly morphological studies, autophagy has been linked to disease processes. Whether autophagy protects from or causes disease is unclear. Here, we summarize current knowledge about the role of autophagy in disease and health.
Cellular homeostasis requires a constant balance between biosynthetic and catabolic processes. Eukaryotic cells primarily use two distinct mechanisms for large-scale degradation, the proteasome and autophagy; but only autophagy has the capacity to degrade entire organelles. The three types of autophagy are macroautophagy, microautophagy, and chaperone-mediated autophagy (1). Here, we will focus on macroautophagy, hereafter called autophagy, which plays an important physiological role in human health. In autophagy, a double- or multi-membrane–bound structure, called the autophagosome or autophagic vacuole, is formed de novo to sequester cytoplasm. Then, the vacuole membrane fuses with the lysosome to deliver the contents into the organelle lumen, where they are degraded and the resulting macromolecules recycled (Fig. 1).
Fig. 1.
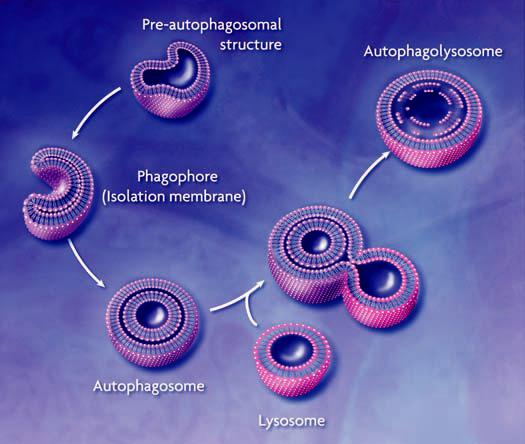
Conceptual model of macroautophagy. A sequestering membrane, termed a phagophore or isolation membrane, forms from the pre-autophagosomal structure. The source of the membrane is unknown but probably includes the endoplasmic reticulum and early secretory pathway. The isolation membrane enwraps cytosol and organelles; on completion, a double-membrane vesicle, the autophagosome or autophagic vacuole, is formed. The autophagosome acquires hydrolytic enzymes by fusing with the lysosome to generate an autophagolysosome, and the inner vesicle of the autophagosome is released into the lumen. The resulting autophagic body is broken down, allowing access to, and degradation and recycling of, the cargo.
Autophagy occurs at basal levels in most tissues and contributes to the routine turnover of cytoplasmic components. However, autophagy can be induced by a change of environmental conditions such as nutrient depletion. In addition to turnover of cellular components, autophagy is involved in development, differentiation, and tissue remodeling in various organisms (2). Autophagy is also implicated in certain human diseases. Paradoxically, autophagy can serve to protect cells but may also contribute to cell damage (Table 1). Here, we will summarize the current connections between autophagy and human disease and aging.
Table 1.
Possible roles of autophagy in health and disease.
| Disease state | Beneficial effects of autophagy | Negative effects of autophagy |
|---|---|---|
| Cancer | Acts as a tumor suppressor; may be involved in type II PCD in cancer cells, could limit cell size or may remove damaged organelles that could generate free radicals and increase mutations. | May allow survival of cancer cells within the nutrient-poor environment of a tumor, could prevent cell death, and may protect against some cancer treatments. |
| Liver disease | Allows removal of nonfunctional endoplasmic reticulum resulting from accumulation of aggregated α1-antitrypsin Z protein. | Increased mortality due to excessive mitochondrial autophagy. |
| Muscular disorder | Increased autophagy may compensate for defects in lysosome function. | Increased autophagy or defects in completing autophagy result in the accumulation of autophagosomes that may impair cell function. |
| Neurodegeneration | Allows the removal of protein aggregates before they become toxic. | May induce cell death in neurons that accumulate aggregated proteins. |
| Pathogen infection | Cellular defense against invasion by bacteria and viruses. | Subversion of the autophagic pathway allows pathogens to establish a replicative niche and supplies nutrients for growth. |
Programmed Cell Death
Autophagy is involved in programmed cell death (PCD). Type I PCD, apoptosis, is characterized by condensation of cytoplasm and chromatin, DNA fragmentation, and cell fragmentation into apoptotic bodies, followed by removal and degradation of the dying cells by phagocytosis. Type II PCD (autophagic) is characterized by the accumulation of autophagic vesicles (autophagosomes and autophagolysosomes) and is often observed when massive cell elimination is demanded or when phagocytes do not have easy access to the dying cells. One feature that distinguishes apoptosis from autophagic cell death is the source of the lysosomal enzymes used for most of the dying cells' degradation. Apoptotic cells use phagocytic cell lysosomes for this process, whereas cells with autophagic morphology use the dying cells' endogenous lysosomal machinery. It has been unclear whether autophagy directly executes cell death or is the secondary effect of apoptosis. A recent study, however, suggests that autophagy might cause cell death (3). Caspase inhibitor–induced autophagic cell death is severely affected by RNA interference (RNAi) with ATG7 and beclin 1 expression, two genes whose products are essential for autophagy (3).
Two key molecules that control PCD are members of the death-associated protein kinase (DAPk) family. Both DAPk and DAPk-related protein kinase-1 (DRP-1) promote death in a way that depends on their kinase activities. DAPk predominantly activates apoptosis through a caspase-dependent pathway (4). However, in mouse embryonic fibroblasts in which apoptosis cannot be activated, DAPk and DRP-1 instead induce autophagy (5). Another regulatory factor, tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL) is also implicated in the induction of caspase activity, autophagy, and, potentially, autophagic PCD, during lumen formation in an epithelial cell line (6). Inhibition of caspase activity alone does not block cell death during acinar cell morphogenesis, which suggests a role for caspase-independent autophagic PCD.
In PCD, the appearance of autophagic structures correlates with cell death; autophagy is not necessarily the cause of death. Also, the activation of autophagic cell death or its obstruction when autophagy genes are suppressed typically takes place in cells where apoptosis has been blocked through the use of inhibitors. Thus, the true physiological relevance of autophagic PCD is not clear. Autophagy may not only be a cause of cell death, it may also precede apoptosis as a defense mechanism. At low levels of stimulus, autophagy could represent a primary attempt to reestablish homeostasis; when the autophagic capacity is overwhelmed, apoptosis (and possibly type II PCD) is triggered (7). However, caspase activation precedes the appearance of autophagosomes during steroid-activated PCD of Drosophila salivary glands (8). In addition, caspase activity may inhibit autophagy through proteolysis of regulatory factors (9); in this case, inhibition of certain caspase activities might induce autophagy. Thus, the connections between type I and II PCD are complicated by the sharing of regulatory and mechanistic components.
Cancer
Autophagy probably factors into both the promotion and prevention of cancer, and its role may be altered during tumor progression. Inhibition of autophagy may allow the continuous growth of precancerous cells, and autophagy can act as a suppressor of cancer (10, 11). Later, as a tumor grows, cancer cells may need autophagy to survive nutrient-limiting and low-oxygen conditions, especially in the internal region of the tumor that is poorly vascularized (12). In addition, autophagy may protect some cancer cells against ionizing radiation (13), possibly by removing damaged macromolecules or organelles, such as mitochondria, which could protect against apoptosis and allow continued survival of transformed cells (14).
The class I phosphatidylinositol (PtdIns) 3-kinase/protein kinase B (Akt/PKB) signaling pathway promotes cell growth in response to mitogenic signals, and mutations in several proteins in this pathway cause a high percentage of common human malignancies (15). Class I PtdIns 3-kinase generates PtdIns(3,4)P2 and PtdIns(3,4,5)P3, which bind to the pleckstrin homology domain of Akt and its activator 3-phosphoinositide– dependent protein kinase-1 (PDK-1) (16) (Fig. 2). When the Akt signaling pathway is activated, autophagic degradation is reduced (17). Conversely, a dominant negative form of Akt induces a high rate of autophagy. Akt and PDK-1 activate other kinases, including mammalian target of rapamycin (mTOR), which negatively regulates autophagy. The tumor suppressor PTEN, which has a 3′-phosphoinositide phosphatase activity and antagonizes the PtdIns 3-kinase/Akt pathway, positively regulates autophagy (Fig. 2). Mutations in PTEN result in constitutive activation of the Akt signaling pathway and inactivation of autophagy and lead to tumor formation (17). Because Akt is centrally involved in regulating a range of substrates that participate in cell growth and survival (18), tumorigenesis resulting from activation of Akt may be due to a block in a pathway other than autophagy.
Fig. 2.
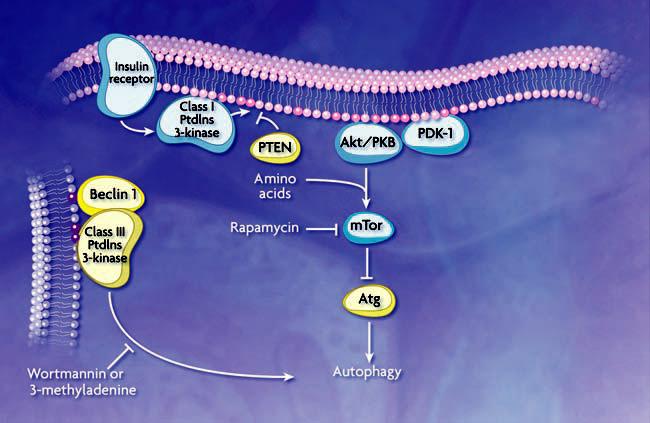
Schematic model of autophagic regulation. Stimulation of the class I PtdIns 3-kinase at the plasma membrane through the insulin receptor results in the generation of PtdIns(3,4)P2 and PtdIns(3,4,5)P3 (dark pink circles). These phosphoinositides allow binding and activation of Akt/PKB and its activator PDK-1. Along with amino acids, Akt/PKB activates mTor (additional components in this pathway are not depicted). Subsequent phosphorylation of a downstream effector, possibly analogous to Atg1 or other ATG gene products as demonstrated in yeast, inhibits autophagy. PTEN dephosphorylates 3′ phosphoinositides and antagonizes the action of class I PtdIns 3-kinase. A class III PtdIns 3-kinase complex, which includes Beclin 1/Atg6, generates PtdIns(3)P (purple circles) to control the membrane dynamics that are associated with autophagosome formation. Rapamycin inhibits mTor, while wortmannin and 3-methyladenine inhibit the class III PtdIns 3-kinase; the effect is to induce or inhibit autophagy, respectively. Autophagy is also regulated through heterotrimeric G proteins and other kinases and phosphatases that are not depicted.
Beclin 1 is a mammalian ortholog of yeast Atg6/Vps30, which is required for autophagosome formation and is monoallelically deleted in a high percentage of sporadic human breast, ovarian, and prostate carcinomas. In the established breast carcinoma cell line MCF7, the expression of Beclin 1 protein is decreased below a detectable level. Stable transfection of beclin 1 in MCF7 cells promotes autophagic activity and reduces tumorigenic capacity, which suggests that autophagic activity is associated with inhibition of cellular proliferation (19). In addition, beclin 1+/− heterozygous mice suffer from a high incidence of spontaneous tumors, and beclin 1−/− homozygous embryonic stem cells exhibit a decreased number of autophagic vesicles (20, 21). Autophagy may thus instigate tumor suppression, by causing cell death or by limiting cell growth.
Some pharmaceuticals used to treat cancer are likely to act through autophagy. For example, tamoxifen is used to treat certain types of breast cancer and may function by activating autophagy, possibly through the up-regulation of beclin 1 in a process mediated by ceramide (22). Other autophagy-inducing compounds, including the mTor inhibitor rapamycin and various analogs, are currently being tested in clinical trials in patients with malignant tumors, although the antitumor effects of inhibiting mTor could reflect its role in cell cycle regulation or translation rather than autophagy.
Muscular Disorder
Although many disorders associated with deregulated autophagy have been reported, most of them are observed in nonproliferative cells, such as muscle and neuronal cells, where the accumulation of damaged materials might be severe. The elevated accumulation of autophagic vesicles is a typical diagnosis in vacuolar myopathy. However, even though the responsible genes have been identified in many types of myopathy, their functions have not been directly linked to autophagy (23). Most analyses of myopathy that connect it to autophagy have been morphological. A malfunction of the lysosome is linked to a disorder that is accompanied by myopathy, Danon's disease (24). Genetic analysis of unrelated patients with Danon's disease identified mutations in the gene for lysosome-associated membrane protein 2 (Lamp2) (24). Furthermore, homozygous deletion of lamp2 in mice results in a phenotype typical of Danon's disease, massive accumulation of autophagic vesicles in many tissues (25). However, the mechanism of Lamp2 in autophagy is still unclear (26).
Also, it is not known whether the accumulation of autophagic vesicles in vacuolar myopathy results from the promotion of autophagosome formation or from the decrease in fusion of autophagosomes with lysosomes. For example, the lysosomotropic agent chloroquine induces a myopathy in cultured cells and rats similar to that of a human disorder termed distal myopathy with rimmed vacuoles, as well as inclusion body myositis (23, 27). Because chloroquine raises the lysosomal pH, which leads to inhibition of lysosome-autophagosome fusion and lysosomal protein degradation, the accumulation of autophagic vesicles in myopathy might be due either to a defect in autophagy, such as a malfunction in autophagosome maturation, or to a defect in lysosomal protein degradation. Finally, the transcription of ATG5 and ATG12 is elevated in inclusion body myositis (28). Thus, both increased formation and inhibited maturation of autophagosomes could lead to myopathy.
Neurodegeneration
The accumulation of autophagic vesicles has been observed in many neurodegenerative disorders such as Parkinson's, Huntington's, and Alzheimer's diseases and amyotrophic lateral sclerosis (Lou Gehrig's disease), or in transmissible spongiform encephalopathies (prion diseases), syndromes that are associated with proteins that misfold and aggregate. However, as with myopathies, it is not known whether this represents an increase in autophagy or a block in the consumption of autophagosomes. The effects of misfolded and aggregated proteins are not fully understood (29). Parkinson's disease (PD) is characterized by the accumulation of aggregates called Lewy bodies in neurons, as well as cell death of dopaminergic neurons in substantia nigra. Mutations in α-synuclein, a major protein in Lewy bodies, cause early-onset PD. For example, the expression of mutant [in which Ala53 is replaced by Thr (A53T)], but not wild-type, α-synuclein in a cultured cell line induces a massive accumulation of autophagic vesicles, as well as impairment of the ubiquitin-proteasome system (30). Because mutations in the ubiquitination system are also found in PD patients, the ubiquitin-proteasome system seems to function to remove misfolded proteins in the early stage of the disorder.
In contrast, defects in the ubiquitin-proteasome system do not seem to be causes of Huntington's disease (HD). Rather, eukaryotic proteasomes are unable to digest the abnormally expanded polyglutamine sequences that are found in the amino terminus of huntingtin, the accumulation of which causes HD (31). Additional work is required to clarify the effect of protein aggregates on proteasome function. For example, although transient expression of a mutant huntingtin fragment was reported to inhibit the ubiquitin-proteasome system (32), in vivo studies indicate that mutant huntingtin does not inhibit the activity of the 20S proteasome (33). This discrepancy could reflect differences in the levels of huntingtin expression. Finally, reduction in proteasome activity may result from caspase-dependent cleavage of proteasome subunits owing to aggregated protein-induced apoptosis (34).
Similar to the situation with mutant α-synuclein, the expression of mutant huntingtin also induces the accumulation of autophagic vesicles (35), and autophagy may protect against the toxic effects associated with protein aggregation. For example, mutant huntingtin and α-synuclein are targeted for autophagy in several experimental model cells (36, 37). Treatment of these cells with rapamycin, an inhibitor of mTor, not only promotes autophagic degradation of mutant huntingtin and α-synuclein but also prevents the accumulation of aggregates, which suggests a protective role for autophagy (36). A recent study indicates a mechanism for how aggregates may protect neuronal cells by inducing autophagy (38). mTOR appears to be packed into polyglutamine aggregates in cell models, transgenic mice, and samples from human brains of HD patients (38). Sequestration of mTOR inactivates kinase activity and induces autophagy. Why aggregated proteins such as α-synuclein, amyloid β-protein, and huntingtin cause neuronal cell death is not clear. Similarly, the interpretation of activated autophagy in neurodegenerative disorders is controversial (39).
In contrast to the protective function of autophagy, neuronal cell death may involve autophagy or lysosomal function. For example, constitutive activation of the δ2 glutamate receptor in lurcher mice results in the release of Beclin 1 and another protein, an isoform of the PDZ domain–containing protein (nPIST), from a sequestered protein complex; the resulting death of cerebellar Purkinje cells correlates with a redistribution of Beclin 1 that may indicate the induction of autophagy, the formation of autophagic structures, or both (40, 41). Which form of PCD, apoptotic or autophagic, executes neuronal cell death is still controversial. Both apoptosis and autophagic cell death have been observed in dopaminergic neurons of PD patients. The interaction of Beclin 1 with both nPIST and Bcl-2 suggests coordination between apoptotic and autophagic types of cell death. Because autophagy is often seen when apoptosis is suppressed (3, 5, 42), type II PCD may be a backup mechanism for apoptosis. Elevated levels of lysosomal proteinases such as cathepsins B and D have been observed in brain tissue from patients with Alzheimer's disease (43). Although it is possible that the increased lysosomal activity contributes to removal of neurotoxic materials, the loss of lysosomal integrity may cause cell death in neurodegenerative disorders, or there may be a combination of both effects. Thus, because autophagy is a catabolic mechanism that operates in stress conditions to recycle or remove cytoplasmic materials, misfolded proteins can be removed by autophagy as a protective measure (44). Alternatively, autophagy may induce cell death in neurons that accumulate aggregates in a way that results in a pathological condition, although at present there is no direct evidence that this occurs.
Pathogen Infection
One role of autophagy in cellular defense is to remove invading pathogens. Although bacterial pathogens that invade cells through endocytosis are usually delivered to lysosomes and degraded there (by a process termed phagocytosis), some of them escape the host defense mechanism by blocking or altering the maturation of the sequestering vesicle, the phagosome. For example, after entering into host cells through the endocytic pathway, the intracellular pathogen Listeria monocytogenes destroys the phagosome membrane using hemolysin, enters the cytoplasm, and multiplies there (Fig. 3). However, when infected cells are treated with chloramphenicol, an inhibitor of bacterial protein synthesis, after phagosome lysis, the bacteria are trapped by autophagosomes (45). Moreover, sequestration is enhanced by autophagic induction through serum withdrawal, whereas the autophagy inhibitors 3-methyladenine or wortmannin block uptake of this and other bacterial pathogens (46). Finally, a recent study shows that, when Streptococcus pyogenes escapes from the endosomes of nonphagocytic cells, it becomes engulfed within autophagosomes (47); subsequent autophagic degradation decreases the number of viable pathogens. Thus, autophagy can protect against bacteria that attempt to establish a replicative niche by entering the host cytoplasm.
Fig. 3.
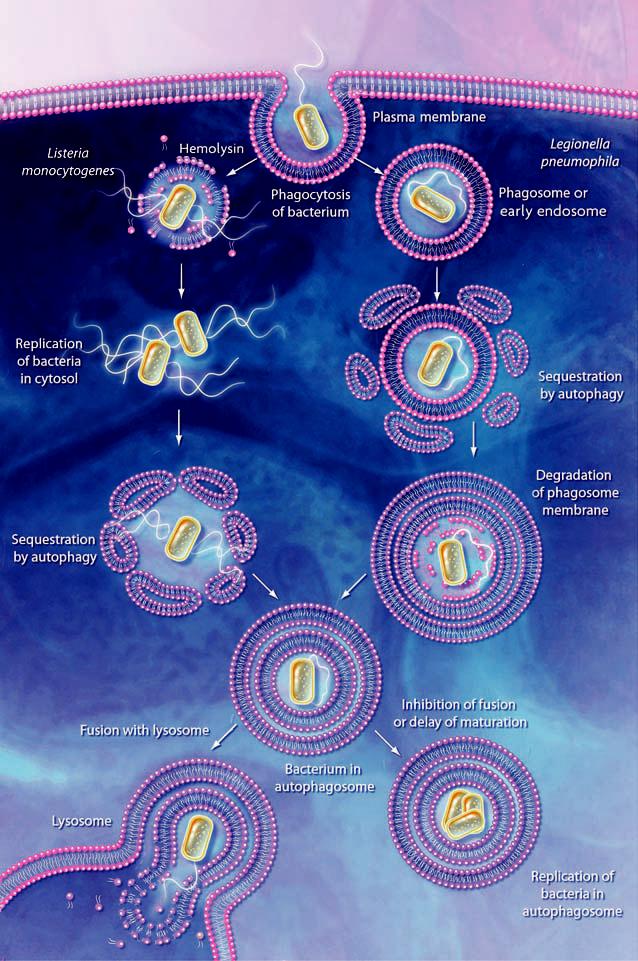
Action and subversion of autophagy during bacterial infection. Bacteria may be taken up by phagocytosis and the resulting phagosome can fuse with endosomes and then the lysosome; the bacteria are then degraded within the phagolysosome (not shown). Some pathogens such as L. monocytogenes, escape this pathway by lysing the phagosome membrane. The bacteria may subsequently become targets for autophagy. In the case of L. pneumophila, P. gingivalis, and B. abortus, the phagosome either fuses with, or becomes sequestered within, the autophagosome. Inhibition of autophagosome maturation or a delay in fusion with the lysosome, dependent on a type IV secretion system, allows the bacteria to replicate within the autophagosome and/or autophagolysosome (in the case of L. pneumophila) and possibly become resistant to lysosomal degradation. In addition, degradation of host cell proteins within the late autophagosome or autophagolysosome may supply the nutrients needed for growth of the pathogen.
Nevertheless, some types of bacterial pathogens such as Porphyromonas gingivalis, Brucella abortus, and Legionella pneumophila use the mechanism of autophagy to replicate by sheltering in autophagosome-like vesicles, thus subverting the autophagic pathway (48). After being taken up by phagosomes, these bacteria enter into double-membrane structures that exhibit features of autophagosomes, instead of completing the process of phagocytosis, although the mechanism by which bacteria are sequestered by autophagosomes is unclear. In many cases, the bacterial type IV secretion pathway is required for autophagic subversion. For example, Legionella pneumophila requires a type IV secretion system encoded by the icm/dot (intracellular multiplication/defect in organelle trafficking) genes to become established inside the autophagosome-like vesicles (49-51). These gene products may activate formation of the sequestering vesicles or may delay their maturation to allow the bacteria to remain within these normally transient compartments and to replicate. Accordingly, treatment of the infected cells with autophagy inhibitors directs the bacteria into a phagocytic pathway and increases the rate of delivery to the lysosome. In contrast, infection and intracellular replication of L. pneumophila in Dictyostelium discoideum appears to be independent of autophagy (52), which possibly reflects host-specific differences.
The appearance of autophagy-like structures also occurs after viral infection (53). Autophagy is induced through the activation of the double-stranded RNA-activated protein kinase R (PKR) following infection by herpes simplex virus (HSV) (54). PKR is a eukaryotic translation initiation factor–2α (eIF2α) kinase; phosphorylation of eIF2α by PKR results in translational arrest, which inhibits viral replication, and it also contributes to the induction of autophagy. HSV encodes a gene for a protein that antagonizes phosphorylation of eIF2α and thus acts to inhibit autophagy. Infection by HSV lacking this gene induces autophagy, whereas the wild-type virus does not. The induction of autophagy by the compromised HSV is not seen in either PKR-deficient cells or cells expressing only mutant eIF2α whose phosphorylation site is lost. Because viral replication is dependent on PKR inactivation (55), autophagy may be a mechanism of eliminating intracellular viruses. Another regulatory pathway that induces autophagy in response to viral infection may involve activation of receptor-interacting protein (RIP) (9). Caspase-8 inactivates RIP during apoptosis, but some viruses are able to inhibit caspase activity. The subsequent activation of RIP may trigger autophagy as an alternative means of killing the infected cell (3).
Host defense pathways exert strong evolutionary pressure on microbes to acquire the means to bypass the host's killing and clearance mechanisms, as noted above for certain bacteria. A similar situation likely exists for viral pathogens. For example, there are indications that the replication complex of certain RNA viruses is localized on the membrane of autophagosome-like structures that are induced by viral infection (46). It is not clear, however, whether the induction of autophagy contributes to the degradation of the replication complex or to viral replication. After infection, the replication complex of mouse hepatitis virus (MHV) localizes on a membrane structure that can be labeled with green fluorescent protein tagged LC3, GFP-LC3 (an ortholog of yeast Atg8), an autophagosome marker, in wild-type mouse embryonic stem cells (56). More important, recovery of extracellular virus decreases in Atg5−/− mouse embryonic stem cells by a factor of 1000, which suggests that components of the autophagic machinery are required for replication of MHV, and probably for other viruses, rather than for elimination of viral particles.
Aging
Recent genetic analyses in various organisms have resulted in the identification of genes involved in controlling longevity. The best-characterized pathway is the insulin/insulin-like growth factor 1 (IGF-1) pathway, which is highly conserved in eukaryotes from yeast to human (57). This signaling cascade includes a tyrosine kinase receptor, PtdIns 3-kinase and Akt/PKB, all of which are also involved in tumorigenesis as described above. In the case of the nematode Caenorhabditis elegans, inactivation of this cascade extends life-span up to 300% and increases heat and oxidative stress resistance, which may contribute to life-span extension. Because Akt/PKB controls the activity of Tor, the autophagy inhibitor, the down-regulation of the Akt/PKB pathway may also induce autophagy, so that autophagy may partly account for life-span extension. Along these lines, elimination or depletion of the TOR kinase/LET-363 also results in an increase in life-span (58). A loss-of-function mutation in the insulin-like tyrosine kinase receptor daf-2 extends life-span in C. elegans twofold; however, the inactivation of bec-1, the nematode ortholog of the yeast and mammalian VPS30/ATG6/beclin 1 gene that is essential for autophagy, by RNAi cancels this effect, which suggests that autophagy is required for the life-span extension resulting from down-regulation of the insulin/IGF-1 pathway (59). This agrees with the finding that the life-span extension resulting from RNAi-mediated inhibition of TOR is not additive with daf-2 (58).
The rate of autophagy decreases with age (60), which suggests a possible correlation between the two processes. Caloric restriction, which might induce autophagy, has positive effects on life-span extension (57, 60). The increase in longevity may be brought about by an increase in protection against oxidative damage, such as through the removal of damaged mitochondria (61), as well as by mechanisms involved in the repair and replacement of damaged DNA, proteins and lipids.
Conclusions
There are many connections between autophagy and human physiology; however, our understanding of autophagy, and particularly its role in human health and disease, is at a very early stage. Even the most fundamental question—whether autophagy plays a protective or a harmful role—is not clearly established for most diseases. Indeed, the consequences of autophagy may alter during the progression of a particular physiological condition. Furthermore, most studies have only shown that autophagy correlates with particular diseases or with cell death, not that it is causative. Accordingly, we need a better understanding of the effects of autophagy in vivo. The recent generation of animal models with tagged or deleted autophagy genes (20, 21, 59, 62, 63) has provided researchers with powerful tools to examine the induction of autophagy in a tissue- and time-dependent manner and has allowed insight into the effects of eliminating autophagic capacity. There remains a need for specific chemical or protein inhibitors and activators that can be used to regulate autophagy in vivo, possibly from pathogens that can alter the autophagic process.
A still unanswered question is whether the autophagic response can be precisely modulated to prevent or respond to disease; excessive autophagy could be just as deleterious as defective autophagy. A greater understanding of the regulatory pathways that control autophagy will be important in this regard. With respect to the molecular mechanisms of autophagy, many questions remain to be answered, including the source of the sequestering membrane, the mechanism of vesicle formation, and the means by which autophagy can become a selective process. For example, it is clear from studies in fungi that superfluous or damaged organelles can be targeted for specific elimination by autophagy. This targeting and recognition process may have special relevance to human health with regard to the removal of protein aggregates, as well as mitochondria and peroxisomes, organelles that generate and remove free radicals, respectively. In addition, mitochondria play a central role in the induction of apoptosis. Our understanding of autophagy at the molecular level is improving rapidly. Whether we can regulate autophagy to combat disease or promote health will be revealed.
References and Notes
- 1.Klionsky DJ, editor. Autophagy. Landes Bioscience; Georgetown, TX: 2004. pp. 1–303. [Google Scholar]
- 2.Levine B, Klionsky DJ. Dev. Cell. 2004;6:463. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Yu L, et al. Science. 2004;304:1500. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 4.Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A. Nature Cell Biol. 2001;3:1. doi: 10.1038/35050500. [DOI] [PubMed] [Google Scholar]
- 5.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. J. Cell Biol. 2002;157:455. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3438. doi: 10.1073/pnas.0400443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Enriquez S, He L, Lemasters JJ. Int. J. Biochem. Cell Biol. 2004;36:2463. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Lee C-Y, Baehrecke EH. Development. 2001;128:1443. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- 9.Chan FK-M, et al. J. Biol. Chem. 2003;278:51613. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 10.Gozuacik D, Kimchi A. Oncogene. 2004;23:2891. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 11.Ogier-Denis E, Codogno P. Biochim. Biophys. Acta. 2003;1603:113. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 12.Cuervo AM. Trends Cell Biol. 2004;14:70. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Paglin S, et al. Cancer Res. 2001;61:439. [PubMed] [Google Scholar]
- 14.Alva AS, Gultekin SH, Baehrecke EH. Cell Death Differ. 2004;11:1046. doi: 10.1038/sj.cdd.4401445. [DOI] [PubMed] [Google Scholar]
- 15.Blume-Jensen P, Hunter T. Nature. 2001;411:355. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 16.Meijer AJ, Codogno P. Int. J. Biochem. Cell Biol. 2004;36:2445. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Arico S, et al. J. Biol. Chem. 2001;276:35243. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 18.Hanada M, Feng J, Hemmings BA. Biochim. Biophys. Acta. 2004;1697:3. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Liang XH, et al. Nature. 1999;402:672. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 20.Qu X, et al. J. Clin. Invest. 2003;112:1809. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15077. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scarlatti F, et al. J. Biol. Chem. 2004;279:18384. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 23.Ueno T, Tanida I, Kominami E. In: Autophagy. Klionsky DJ, editor. Landes Bioscience; Geogetown, TX: 2004. pp. 264–286. [Google Scholar]
- 24.Nishino I, et al. Nature. 2000;406:906. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka Y, et al. Nature. 2000;406:902. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 26.Eskelinen E-L, et al. Mol. Biol. Cell. 2004;15:3132. doi: 10.1091/mbc.E04-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki T, et al. J. Biochem. (Tokyo) 2002;131:647. doi: 10.1093/oxfordjournals.jbchem.a003147. [DOI] [PubMed] [Google Scholar]
- 28.Kumamoto T, Ueyama H, Tsumura H, Toyoshima I, Tsuda T. Acta Neuropathol. (Berlin) 2004;107:59. doi: 10.1007/s00401-003-0774-2. [DOI] [PubMed] [Google Scholar]
- 29.Lipinski MM, Yuan J. Curr. Opin. Pharmacol. 2004;4:85. doi: 10.1016/j.coph.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. J. Neurosci. 2001;21:9549. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Mol. Cell. 2004;14:95. doi: 10.1016/s1097-2765(04)00151-0. [DOI] [PubMed] [Google Scholar]
- 32.Bence NF, Sampat RM, Kopito RR. Science. 2001;292:1552. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 33.Diaz-Hernandez M, et al. J. Neurosci. 2003;23:11653. doi: 10.1523/JNEUROSCI.23-37-11653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X-M, et al. Mol. Cell. 2004;14:81. doi: 10.1016/s1097-2765(04)00156-x. [DOI] [PubMed] [Google Scholar]
- 35.Kegel KB, et al. J. Neurosci. 2000;20:7268. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravikumar B, Duden R, Rubinsztein DC. Hum. Mol. Genet. 2002;11:1107. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 37.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. J. Biol. Chem. 2003;278:25009. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 38.Ravikumar B, et al. Nature Genet. 2004;36:585. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 39.Yuan J, Lipinski M, Degterev A. Neuron. 2003;40:401. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 40.Yue Z, et al. Neuron. 2002;35:921. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 41.Selimi F, Vogel MW, Mariani J. J. Neurosci. 2000;20:5339. doi: 10.1523/JNEUROSCI.20-14-05339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue L, Fletcher GC, Tolkovsky AM. Curr. Biol. 2001;11:361. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 43.Cataldo AM, Barnett JL, Pieroni C, Nixon RA. J. Neurosci. 1997;17:6142. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fortun J, Dunn WA, Jr., Joy S, Li J, Notterpek L. J. Neurosci. 2003;23:10672. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rich KA, Burkett C, Webster P. Cell. Microbiol. 2003;5:455. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 46.Kirkegaard K, Taylor MP, Jackson WT. Nature Rev. Microbiol. 2004;2:301. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakagawa I, et al. Science. 2004;306:1037. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 48.Dorn BR, Dunn WA, Jr., Progulske-Fox A. Cell. Microbiol. 2002;4:1. doi: 10.1046/j.1462-5822.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- 49.Vogel JP, Andrews HL, Wong SK, Isberg RR. Science. 1998;279:873. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- 50.Coers J, Monahan C, Roy CR. Nature Cell Biol. 1999;1:451. doi: 10.1038/15687. [DOI] [PubMed] [Google Scholar]
- 51.Swanson MS, Isberg RR. Infect. Immun. 1995;63:3609. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otto GP, et al. Mol. Microbiol. 2004;51:63. doi: 10.1046/j.1365-2958.2003.03826.x. [DOI] [PubMed] [Google Scholar]
- 53.Suhy DA, Giddings TH, Jr., Kirkegaard K. J. Virol. 2000;74:8953. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tallóczy Z, et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:190. [Google Scholar]
- 55.Leib DA, Machalek MA, Williams BRG, Silverman RH, Virgin HW. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6097. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. J. Biol. Chem. 2004;279:10136. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Longo VD, Finch CE. Science. 2003;299:1342. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- 58.Vellai T, et al. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 59.Melendez A, et al. Science. 2003;301:1387. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 60.Bergamini E, Cavallini G, Donati A, Gori Z. Int. J. Biochem. Cell Biol. 2004;36:2392. doi: 10.1016/j.biocel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Terman A, Brunk UT. Exp. Gerontol. 2004;39:701. doi: 10.1016/j.exger.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 62.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. Mol. Biol. Cell. 2004;15:1101. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Juhász G, Csikós G, Sinka R, Erdélyi M, Sass M. FEBS Lett. 2003;543:154. doi: 10.1016/s0014-5793(03)00431-9. [DOI] [PubMed] [Google Scholar]
- 64.Saftig P, Vogel M, Yuan J, Amer A, Baehrecke E, Codogno P, Cuervo AM, Kirkegaard K, Levine B, Neufeld T, Reggiori F, Rubinsztein D, Saftig P, Swanson M, Fairman JE. The authors would like to thank. for reading the manuscript; for helpful comments and/or providing information before publication; and. CMI, for the illustrations (copyrighted); however, none of the preceding people should be held responsible for incorrect information or interpretations of the published literature. D.J.K. is supported by Public Health Service grant GM53396 from NIH.