

Abstract
We examined 18 neuroblastoma cell lines and 32 primary single-copy MYCN tumor specimens to determine whether mutations of p73, a novel p53-related gene located in chromosome band 1p36.33, contribute to the genesis or progression of childhood neuroblastoma. By fluorescence in situ hybridization, 16 of the 18 cell lines, but only 3 of the 32 primary tumors, had evidence of a deleted p73 allele. Sequence analysis of the p73 coding region in the mRNAs expressed by these cell lines and tumors did not reveal inactivating mutations, suggesting that p73 is not homozygously inactivated in neuroblastoma. However, several novel splice forms of p73 mRNAs were identified, including one without exon 11 that predominated in multiple MYCN-amplified cell lines. Its encoded p73 protein differed from other splice forms in that the C-terminus was derived from an alternative reading frame. Further study of the functional properties of the protein encoded by this splice form of p73 will be needed to determine whether it contributes to the pathogenesis of childhood neuroblastoma with MYCN gene amplification.
Keywords: chromosome 1p deletion, neuroblastoma, p73, tumor suppressor gene
Introduction
Neuroblastoma, a childhood tumor arising in the adrenal medulla and sympathetic nervous system, is the most common extracranial solid tumor in infants and young children. The most frequent genetic abnormality identified in neuroblastoma is MYCN gene amplification [1], which has been shown by Brodeur, Seeger, and colleagues [2,3] to have clinical relevance, an observation since confirmed by many others [4–8]. MYCN gene amplification is found more often in cases with disseminated disease, is strongly associated with rapid tumor progression, and confers a poor prognosis, regardless of tumor stage.
Deletion of one allele of the distal short arm of chromosome 1 is a second clinically relevant genetic abnormality in neuroblastoma, suggesting that inactivation of one or more tumor suppressor genes in this region also contributes to oncogenesis during sympathetic nervous system development [9–13]. Studies by Caron et al. [14] have implicated at least two tumor suppressor loci on chromosomal bands 1p35-36 in this tumor. In their study, single-copy MYCN tumors with polymorphic markers from distal 1p36.2-3 showed preferential loss of the maternal allele (16 of 17 tumors analyzed). These findings suggest that a tumor suppressor gene (referred to here as NB1) is imprinted and inactivated when inherited from the father, so that deletion of the maternal allele alone results in complete loss of NB1 expression in the tumor. A second more proximal region of loss of heterozygosity (LOH) is associated with MYCN amplification and a random pattern of loss of parental alleles, suggesting that a separate gene (referred to as NB2) is located in the 1p35-36.1 region but is not affected by imprinting [14,15].
A new gene, designated p73, was recently identified and mapped to 1p36.3 [16], the region postulated to harbor NB1 in neuroblastoma. The predicted p73 protein shares significant amino acid sequence identity with the DNA-binding, transactivation, and oligomerization domains of p53, providing further support for its candidacy as a tumor suppressor gene. Like p53, p73 can activate transcription of the p21Waf1/Cip1 cell-cycle inhibitor and can also induce apoptosis, although expression of p73 is not induced by DNA-damaging agents, such as ultraviolet light or low-dose actinomycin D [16,17].
Initial studies have not revealed inactivating mutations in the coding regions of the p73 gene in a small number of both neuroblastoma and non-neuroblastoma cell lines or in human lung cancers [16,18]. However, preliminary indications that p73 is monoallelically expressed in neuroblastoma and normal human blood cells [16], together with its distal location on chromosome arm 1p, suggest that it may be the imprinted NB1 tumor suppressor gene of single-copy MYCN neuroblastomas [19,20]. These tumor cells generally do not grow well in vitro, and thus are under-represented among the neuroblastoma cell lines analyzed to date for inactivating mutations of p73. In addition, if one allele was inactivated by imprinting, only one allele would need to be inactivated in somatic neural precursors. Thus, it is important to look for inactivating mutations of p73 mRNA in primary neuroblastoma tumors lacking MYCN amplification or 1p deletion.
To test this hypothesis, we analyzed the p73 gene copy number by fluorescence in situ hybridization (FISH) and the mRNA sequence by reverse transcription-polymerase chain reaction (RT-PCR) in 32 primary neuroblastoma tumors lacking MYCN amplification. We also analyzed 18 neuroblastoma cell lines with and without MYCN amplification for both p73 deletions and inactivating mutations. Our analysis confirmed deletion of one allele of p73 in the context of 1p deletions but did not reveal homozygous deletion or inactivating mutations within expressed p73 sequences. However, alternative splice forms of p73 were detected and appeared to be preferentially expressed by MYCN-amplified neuroblastoma cell lines.
Materials and Methods
Cell Lines
Eighteen neuroblastoma cell lines (SJNB1 to SJNB20, excluding SJNB7 and SJNB19), all derived at St. Jude Children's Research Hospital, were used in this study [21]. Thirteen of the lines are characterized by MYCN amplification, whereas SJNB1, SJNB3, SJNB9, SJNB12 and SJNB16 have normal MYCN copy numbers [22] (Table 1). The cell lines have been passaged 60 to 70 times.
Table 1.
p73 Copy Number and mRNA Sequence Analysis in Neuroblastoma Cell Lines.
| Cell line | MYCN | p73 | Chromosome 1 | Nested Primer Sets | |||||
| Status | Copy No. | Copy No. | a | b | c | D | e | f | |
| SJNB1 | Sc | 2 | 4 | N | N | N | N | N; sf1 | N |
| SJNB3 | Sc | 2 | 2 | N | N | N | N | N; sf1 | N |
| SJNB9 | Sc | 2 | 4 | LDP | LDP | N | N | N; sf1 | N |
| SJNB12 | Sc | 3 | 4 | N | N | LDP | N | LDP | N |
| SJNB16 | Sc | 1 | 3 | LDP | LDP | LDP | N | LDP | LDP |
| SJNB2 | Am | 1 | 3 | N | N | N | N | N; sf2 | N |
| SJNB4 | Am | 1 | 2 | LDP | LDP | LDP | N | LDP | N |
| SJNB5 | Am | 1 | 2 | N | N | N | N | N; sf1, sf2, sf3, sf4 | N |
| SJNB6 | Am | 2 | 2 | N | N | N | N | N; sf1, sf2, sf3, sf6 | N |
| SJNB8 | Am | 2 | 3 | 629T-C | N | N | N | N; sf1, sf2, sf3, sf4 | N |
| SJNB10 | Am | 2 | 4 | N | N | N | N | N; sf1, sf2, sf3, sf6 | N |
| SJNB11 | Am | 2 | 3 | LDP | LDP | N | N | N; sf3 | N |
| SJNB13 | Am | 3 | 4 | LDP | LDP | LDP | N | LDP | N |
| SJNB14 | Am | 2 | 3 | N | N | N | N | N; sf1, sf2, sf3, sf4 | N |
| SJNB15 | Am | 1 | 2 | N | N | N | N | N; sf1, sf2, sf3, sf5 | N |
| SJNB17 | Am | 2 | 3 | N | N | N | N | N; sf2, sf3 | N |
| SJNB18 | Am | 1 | 2 | N | N | N | N | N; sf1, sf2, sf3, sf6 | N |
| SJNB20 | Am | 2 | 4 | N | N | N | N | N; sf1, sf2, sf3 | N |
Am, amplified; Sc, single copy; N, normal p73 sequence; LDP, lacking detectable product for sequence analysis; 629T-C, transition of T to C at nucleotide 629; sf, p73 transcript splice form containing all of the 14 exons (sf1) or lacking exon 13 (sf2), exon 11 (sf3), exons 11 + 13 (sf4), exons 11 + 12 (sf5), or 11 + 12 + 13 (sf6).
Primary Tumors
Thirty-two primary tumor specimens were obtained from the Pediatric Oncology Group (Table 2). All were classified as having single-copy MYCN genes. Nineteen were diploid and 12 were hyperdiploid. The samples were collected at diagnosis, before the administration of chemotherapy or radiation therapy. Specimens were immediately frozen in liquid nitrogen and stored at -80°C.
Table 2.
Clinical Features of Patients with Primary Neuroblastoma Lacking MYCN Amplification and Analysis of the p73 Gene in Tumor Specimens.
| Patient No. | Age (yr) | Sex | Tumor | DI | p73 | Chromosome 1 | Nested Primer Sets | |||||||
| Stage | Copy No | Copy No | a | b | C | D | e | e1 | e2 | f | ||||
| 1 | 1.9 | F | A | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 2 | 1.3 | M | C | 1.00 | 1 | 2 | N | N | N | N | N; sf1 | N | N | N |
| 3 | 4.7 | F | C | 1.00 | NT1 | NT | N | N | N | N | LDP | N | N | N |
| 4 | 1.4 | M | A | 1.00 | 2 | 3 | N | LDP | LDP | N | LDP | N | N | N |
| 5 | 12.2 | M | A | 1.00 | NT | NT | N | LDP | LDP | N | LDP | 135G-A | N | N |
| 6 | 1.2 | F | A | 1.00 | 2 | 2 | N | N | N | N | N; sf1 | N | N | N |
| 7 | 4.2 | M | D | 1.00 | NT | NT | N | N | LPD | N | LDP | N | N | N |
| 8 | 5.8 | M | B | 1.00 | NT | NT | N | N | N | N | LDP | N | N | N |
| 9 | 2.8 | M | A | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 10 | 15.3 | F | A | 1.00 | 2 | 2 | N | N | LDP | N | LDP | N | N | N |
| 11 | 2.0 | F | A | 1.00 | 2 | 2 | N | N | LDP | N | LDP | N | N | N |
| 12 | Newborn | M | DS | 1.00 | 1 | 2 | N | N | LDP | N | LDP | N | N | N |
| 13 | 1.2 | M | B | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 15 | 0.7 | M | C | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 16 | 1.4 | M | D | 1.18 | 2 | 2 | N | N | N | N | N; sf1 | N | N | N |
| 17 | Newborn | M | A | 1.00 | 2 | 2 | N | N | LDP | N | LDP | N | N | N |
| 18 | 6.2 | F | A | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 19 | 3.0 | F | B | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 20 | 0.9 | M | B | 1.00 | 2 | 2 | N | N | N | N | N; sf3 | N | N | N |
| 21 | 11.8 | F | A | 1.00 | 2 | 2 | N | N | N | N | LDP | N | N | N |
| 23 | 0.8 | M | A | 1.46 | NT | NT | N | N | N | N | LDP | N | N | N |
| 24 | 1.6 | M | A | NT | NT | NT | N | N | N | N | LDP | N | N | N |
| 25 | 1.5 | F | A | 1.83 | 3 | 3 | N | N | N | N | LDP | N | N | N |
| 26 | 0.7 | F | A | 1.31 | 3 | 3 | N | N | N | N | LDP | N | N | N |
| 27 | Newborn | M | A | 1.63 | 3 | 3 | N | N | LDP | N | LDP | N | N | N |
| 28 | 0.9 | M | A | 1.48 | 3 | 3 | N | N | N | N | N; sf1 | N | N | N |
| 29 | 0.1 | M | A | 1.18 | 3 | 3 | N | N | LDP | N | LDP | N | N | N |
| 30 | 1.4 | F | A | 1.25 | 4 | 4 | N | N | LDP | N | LDP | N | N | N |
| 31 | 0.7 | M | A | 1.69 | NT | NT | N | N | N | N | LDP | N | N | N |
| 32 | Newborn | M | A | 1.45 | NT | NT | N | N | N | N | LDP | N | N | N |
| 33 | 1.5 | F | A | 1.20 | 3 | 3 | N | N | LDP | N | LDP | N | N | N |
| 34 | Newborn | M | A | 1.30 | 2 | 2 | N | N | N | N | LDP | N | N | N |
NT, not tested; N, normal p73 sequence; LDP, lacking detectable product for sequence analysis; 1358G-A, transition of G to A at nucleotide 1358; sf, p73 transcript splice form containing all 14 exons (sf1) or lacking exon 11 (sf3).
Patient Data
The 32 patients ranged in age from 4 days to 15.3 years, with a median of 1.4 years. Twenty were older and 12 were younger than 1 year (20 boys and 12 girls). Twenty-two were classified as stage A, 4 as stage B, 3 as stage C, 2 as stage D, and 1 as stage DS (Table 2).
P1 Clone of the p73 Gene for FISH Analysis
In order to isolate a genomic clone corresponding to the p73 gene, we used PCR to amplify two p73 genomic fragments with oligonucleotide sets from both the 5′(ACCATGGACCAGATGAGCA; AGTGACCTCAAAGTGGTGGG) and 3′(AGCAGGGCCACGACTACAGC; TTGGGGATGGTGATGGTGTG) portions of the gene. These oligonucleotides were then used to screen a P1 library for the p73 gene (Genome Systems, Inc, St. Louis, MO, USA). One P1 clone was obtained and introduced by electroporation into Electro-Max DH10BTM cells (Gibcol BRL, Life Technologies). The transformed cells were then streaked onto kanamycin agar plates. Single colonies were picked and incubated at 35°C overnight. The bacterial cells were collected, and DNA was purified according to instructions from the company.
Fluorescence in Situ Hybridization
Colcemid (0.05 µg/mL) was added to cells growing in culture, and slides containing fixed metaphase cells were prepared as previously described [23]. The primary tumor cells were touched onto slides and then fixed with 75% methanol and 25% glacial acetic acid. The hybridization and fluorescence detection was performed under the reported conditions [24]. DNA from the p73 P1 clone was labeled with digoxigenin-11-dUTP or biotin-16-dUTP by nick translation. A control probe (pUC1.77), which is highly repeated in the chromosome 1 pericentromeric heterochromatin, was nick translated with biotin-16-dUTP and used in the hybridization mixture to verify the copy number of chromosome 1. Signals were detected by incubating the slides with fluorescein-conjugated sheep antidigoxigenin antibodies (Boehringer Mannheim) and Texas red-conjugated avidin (Vector Laboratories), followed by signal amplification with fluorescein-conjugated rabbit antisheep antibodies (Boehringer Mannheim) and biotinylated anti-avidin. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
RT-PCR
Total RNA was extracted from neuroblastoma cell lines with RNA STAT-60™ solution (Tel-Test, Inc, Friendswood, TX) according to a published procedure [25]. RT-PCR was performed as previously described ]26]. In brief, cDNAs were reverse transcribed from 5 µg of total RNAs in a 20 µL mixture by use of a cDNA synthesis kit (Boehringer Mannheim). Three microliters of the cDNA were amplified by PCR by using the p73-specific oligonucleotide primers (Table 3). Because p73 is expressed at low levels in normal human tissues [16], nested PCR was required for each of the amplifications. The positions and sequences of the oligonucleotide primers for the first RT-PCR and nested PCR are shown in Figure 1 and Table 3. β-Actin-specific oligonucleotides were used as a control for RNA integrity [27].
Table 3.
Primers Used in RT-PCR Analysis of the p73 Gene.
| Primer pairs | Sense Primers | Antisense Primers | Amplified Nucleotides |
| 1 | AGCGAGCTGCCCTCGGA | AAGTGACCTCAAAGTGGTGGGG | 78–501 |
| 2 | GATTCCAGCATGGACGTCTT | GATCTGGATGGGGCATGTCT | 246–599 |
| 3 | CCTCCAACACCGACTACCC | GCCGATAGGAGTCCACCAGT | 457–1281 |
| 4 | CCGAAAAGCTGATGAGGAC | GTGGCCATGGTTGTTGAG | 1013–1484 |
| 5 | CCGAAAAGCTGATGAGGAC | TGGGGATGGTGATGGTGT | 1013–1902 |
| 6 | GAGATGAGCAGCAGCCACAG | CTCCTGAGGCAGTTTTGGAC | 1503–2110 |
| A | GTGGGGAAGATGGCCCAGT | GACATGGTGTCGAAGGTGGAG | 102–439 |
| B | CCTGGAGGGCATGACTACAT | TGGCGATCTGGCAGTAGAG | 269–579 |
| C | CCCACCACTTTGAGGTCACT | TCAGCTCCAGGCTCTCTTTC | 481–1242 |
| D | CTTGGTGCCGGTGTGAAG | GCCGATAGGAGTCCACCAGT | 1128–1281 |
| E | GCCTGGAGCTGATGGAGTT | CTGTAGTCGTGGCCCTGCTT | 1231–1774 |
| F | TGAAGATCCCCGAGCAGTA | TTTGGACACACAGGAAGGA | 1702–2097 |
| e1 | AGCCACTGGTGGACTCCTATC | CTGTGGCTGCTGCTCATCT | 1258–1522 |
| e2 | GAGATGAGCAGCAGCCACAG | GTACTGCTCGGGGATCTTCA | 1503–1721 |
Pairs 1–6 were used for RT-PCR, a–f for the nested PCR, and e1 and e2 for nested PCR to amplify primary tumor samples.
Figure 1.
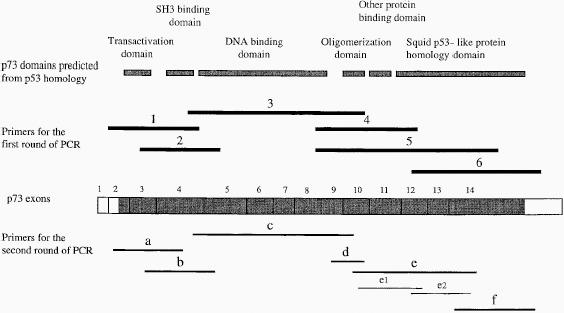
Structure of the p73 gene. The top row shows p73 domains predicted from amino acid sequence homology between this protein and p53 [16]. The bold lines in the second row indicate the location and the relative length of the cDNA sequences amplified with the 6 pairs of primers used in the first round of PCR. p73 Exon boundaries are shown in the third row [16]. The coding regions are indicated by solid boxes and the untranslated regions by open boxes. The fourth row shows the cDNA segments (a–f, e1, e2) used for direct sequence analysis, and after amplification with nested primer pairs in a second round of PCR, with products of the first round of RT-PCR serving as templates. PCR products a–f were analyzed by direct sequence techniques for mutations of the p73 coding sequence. Primers e1 and e2 were used for analysis of mutations in exons 11–13 only in primary tumors.
Sequencing
PCR products were sequenced directly as described previously with slight modifications [26]. The sequencing primers were those used for nested PCR. If there were several transcripts in one PCR product, then each band was gel-purified, excised, and sequenced directly.
Results
p73 Gene Copy Number in Neuroblastoma Cell Lines
To determine whether the p73 gene is included in 1p deletions in neuroblastoma cell lines, we performed FISH analysis with a P1 genomic clone that contained the entire p73 coding sequence. This probe was cohybridized with a second probe, pUC1.77, [28] that recognizes repeated sequences from proximal 1q to determine the number of chromosomes 1 in each cell line. Figure 2 shows examples of the results obtained by interphase and metaphase FISH analysis. Both p73 alleles were retained in cells from a neuroblastoma cell line (SJNB3) with 2 cytogenetically normal chromosomes 1 (Figure 2A and B). One allele of p73 is deleted in a neuroblastoma cell line (SJNB5) with a cytogenetically visible 1p36 deletion (Figure 2C and D). A tetraploid cell line (SJNB1) is characterized by retention of 2 p73 alleles and loss of p73 from the remaining chromosome 1 alleles with cytogenetically identical 1p deletions (Figure 2E and F).
Figure 2.
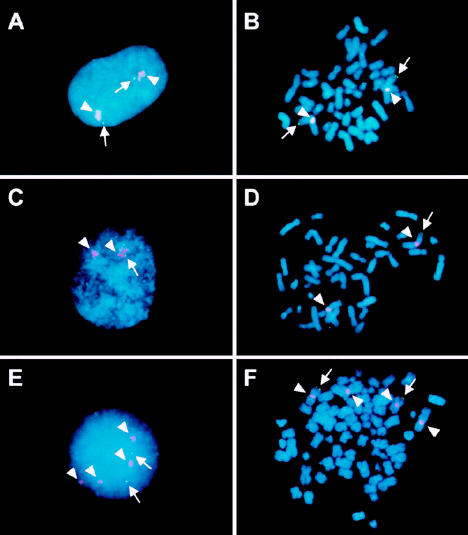
FISH analysis of the p73 gene copy number in SJNB3, SJNB5, and SJNB1 cell lines. Interphase (panels A, C, and E) and metaphase (panels B, D, and F) cells were hybridized with a p73 P1 clone (green) and the subcentromeric control (pUC1.77) probe (red). A,B. SJNB3 cells possess 2 chromosomes 1 (red) and 2 p73 (green) signals. C,D. SJNB5 cells have 2 chromosomes 1 but retain only a single p73 allele. E,F. SJNB1 cells have tetrasomy of chromosome 1 but only 2 copies of the p73 locus.
When applied to a series of 18 neuroblastoma cell lines, FISH analysis yielded distinct patterns of p73 loss (see Table 1). The high frequency of chromosome 1 trisomy or tetrasomy that accompanied 1p loss in these cell lines underscores the need to determine the number of chromosomes 1 per cell with the pUC1.77 probe, as well as the number of p73 gene copies. In 4 of the cell lines (SJNB4, SJNB5, SJNB15, and SJNB18), the analysis indicated classical 1p deletion with complete loss of 1 p73 gene locus in the context of 2 copies of chromosome 1 (see Table 1). Similarly, 2 cell lines (SJNB2 and SJNB16) had trisomy of chromosome 1, but only 1 of these chromosomes 1 retained the p73 gene on 1p see (Table 1). Thus, 6 of the 18 cell lines were monosomic for the p73 locus on chromosome 1. Four additional cell lines (SJNB1, SJNB9, SJNB10, and SJNB20) had tetrasomy of chromosome 1, but only 2 had copies of the p73 locus. By cytogenetic analysis, each of these lines had 2 apparently normal chromosomes 1, together with 2 chromosomes with morphologically identical 1p deletions, suggesting that the deletions occurred before reduplication, leading to tetraploidy. Six lines (SJNB8, SJNB11, SJNB12, SJNB13, SJNB14, and SJNB17) had trisomy or tetrasomy of chromosome 1 with deletion of 1 allele of the p73 gene. Only 2 of the cell lines (SJNB3, MYCN not amplified, and SJNB6, MYCN amplified) had disomy of chromosome 1 and lacked evidence of p73 gene deletion (see Table 1).
Mutational Analysis of p73 mRNA in Neuroblastoma Cell Lines
We used RT-PCR techniques to examine the sequence of p73 mRNA expressed by neuroblastoma cell lines. Relatively low levels of p73 mRNA expression by these cell lines precluded amplification of the entire coding region. Thus, for sequence analysis, we designed 6 pairs of primers for the first round of the analysis (see primer pairs 1 to 6, Figure 1), in which we amplified portions of the mRNA, and 6 pairs of internal primers to perform nested PCR (see primer pairs a to f, Figure 1).
Detectable RT-PCR products were obtained after the first round of PCR with RNA templates from subsets of the cell lines. For example, with primer set 3 (Figure 1), RT-PCR products were detected in RNAs of 7 of the 13 MYCN-amplified neuroblastoma cell lines and 1 of the 5 single-copy MYCN cell lines (Figure 3A). After nested PCR analysis with the internal primer set c, p73 cDNA fragments were detected in 11 of the 13 MYCN-amplified cell lines and 3 of the 5 single-copy MYCN cell lines (see Figure 3B and Table 1). Moreover, with a primer set that amplified mRNA sequences from exons 9 and 10 (see set d, Figure 1, and Table 1) and a control β-actin primer set for RNA integrity (see Figure 3D), products were detected in RNAs from each of the tumor cell lines, indicating that p73 mRNA was expressed. Presumably, the p73 mRNA levels were lower in tumors in which products could only be detected with the most effective primer set flanking exons 9 and 10.
Figure 3.
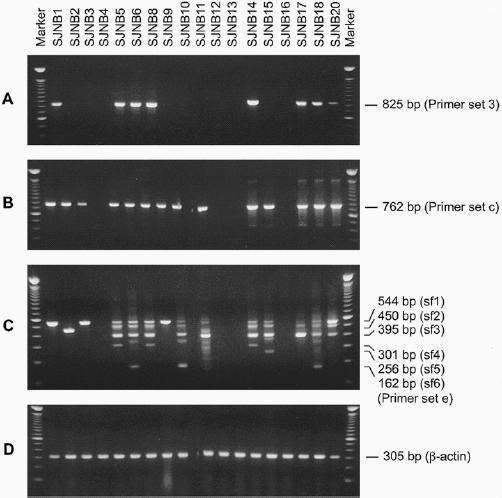
RT-PCR analysis of p73 mRNA in neuroblastoma cell lines. Agarose gel electrophoresis of p73 cDNA fragments amplified with different primer pairs. A. After the first round of RT-PCR amplification with primer set 3 (see Figure 1), PCR products were detected in 8 of 18 cell lines, including one single-copy MYCN cell line (SJNB1) and 7 MYCN-amplified lines (SJNB 5, 6, 8, 14, 17, 18, and 20). B. After nested PCR with primer set c, products were detected in 14 of the cell lines, but not in SJNB4, 12, 13, and 16. C. After a first round of RT-PCR with primer set 5 (see Figure 1) and nested PCR with primer set e, 6 different products were observed. The expected sizes of each of the bands are indicated on the right side of the figure. D. Primers specific for β-actin mRNA were used to amplify products from each cell line, demonstrating RNA integrity. The first and last lanes of each gel show the mobilities of markers consisting of a 100-bp ladder.
We then analyzed each of the amplifiable RT-PCR products from the neuroblastoma cell lines by direct DNA sequence analysis. The sequence of each product was normal, except for a T → C substitution in codon 173 of the RNA from SJNB8, which did not result in an alteration of the amino acid sequence (see Table 1). However, with the primer sets 5 and e, which amplify mRNA sequences between exons 10 and 14, 6 splice forms of p73 mRNA were detected in variable patterns among the 18 cell lines (see Figure 3C). The exons contained in each of these splice forms are illustrated in Figure 4, with a summary of the results presented in Table 1. The sf3 isoform predominated in most MYCN-amplified neuroblastoma cell lines. This finding appears important, because others have shown that the same isoform, referred to as γ, has little activity in growth suppression and efficiently forms heterocomplexes with other p73 isoforms [29]. In contrast to splice form sf1 (Figure 4A), the splice forms referred to as sf2, sf3, sf4, and sf6 alter the open reading frame, yielding shorter polypeptides of 499, 475, 555, and 404 amino acids, respectively (Figure 4A, B). The sf5 form results in an in-frame deletion of amino acids encoded by exons 11 and 12, producing a 540 amino acid polypeptide. Interestingly, each of the single-copy MYCN neuroblastoma cell lines either lacked detectable transcripts with primer sets 5 and e (SJNB12 and SJNB16) or had only transcripts containing each of these exons (SJNB1, SJNB3 and SJNB9), with no evidence of alternatively spliced mRNAs (see Figure 3C and Table 1).
Figure 4.
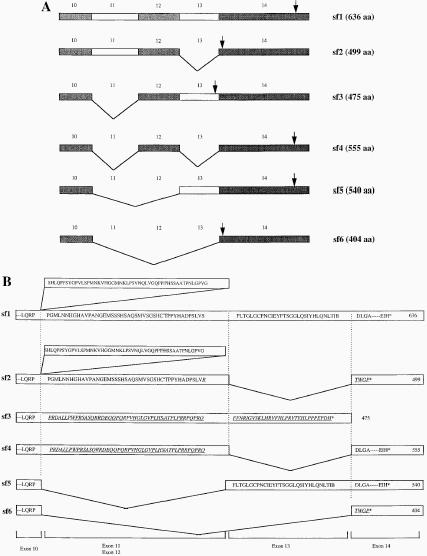
Schematic representation of alternative splice forms of p73 mRNAs. A. Sf1, splice form 1, contains an intact p73 C terminus from exons 10 to 14; sf2 lacks exon 13; sf3 lacks exon 11; sf4 lacks exons 11 and 13; sf5 lacks exons 11 and 12; and sf6 lacks exons from 11 to 13. The arrows indicate the location of the translational termination codon in each splice form. B. Predicted amino acid sequence encoded by p73 splice forms. Sf2, 3, 4, and 6 have out-frame deletions of exons 13, 11, 11 + 13, or 11 + 12 + 13, which interrupt the open reading frame and yield products of 499, 475, 555, and 404 amino acids. Sf5 has an in-frame deletion of amino acids encoded by exons 11 and 12, producing a 540 amino acid polypeptide. The underlined sequences indicate the shifted reading frame due to alternative splicing. The asterisk (*) indicates the stop codon of each transcript.
p73 Analysis in Primary Neuroblastomas With Single-Copy MYCN
Because of the relative paucity of cell lines with single-copy MYCN genes and initial reports suggesting that p73 is an imprinted gene [16], we extended our analysis to include 32 primary neuroblastoma tumors lacking MYCN amplification (Table 2). FISH analysis revealed that 2 diploid tumors (from cases 2 and 12) had monosomy of p73 and disomy of chromosome 1, indicating deletion of one p73 allele. An additional case (no. 4) had trisomy of chromosome 1 but only 2 copies of the p73 gene, also signifying 1p deletion, including the p73 locus. Thus, 3 of the 24 single-copy MYCN tumors evaluated by FISH showed deletion of 1p, in agreement with the expected frequency of 1p loss in this subgroup of neuroblastomas [12–15,30].
p73 mRNA levels were low in primary neuroblastoma samples, requiring nested PCR (see Figure 1) for sequence analysis of amplified segments. As shown in Table 2, we did not identify nonsense or missense mutations in the p73 mRNA expressed by these tumors. The frequency of novel C-terminal alternative splice forms identified in mRNAs from neuroblastoma cell lines could not be analyzed in most primary tumors, because the low levels of gene expression precluded analysis with the informative primer sets 5 and e (see Table 2). To analyze this portion of the p73 sequence in RNA from primary tumors, primer set e was used in place of set 5 to perform the first round of PCR reactions. Two new primer sets, e1 and e2, were used to generate nested PCR products from exons 10 through 14 in the primary tumors. The sequence of these products matched the normal p73 sequence, except for a 1 nucleotide difference identified in RNA from the primary tumor of case 5, which did not alter the predicted amino acid sequence (see Table 2).
Discussion
Despite the location of p73 within a region that is frequently deleted from 1 chromosomal allele in childhood neuroblastoma [16], we did not identify inactivating mutations in p73 mRNAs expressed by 32 single-copy MYCN primary neuroblastomas and 18 neuroblastoma cell lines, including 13 with MYCN gene amplification. Thus, p73 does not appear to be homozygously inactivated in this disease. By FISH analysis, many of the neuroblastoma cell lines in our study had MYCN gene amplification together with loss of one allele of p73. The lack of detectable mutations in transcripts expressed from the remaining allele is consistent with results of an earlier study suggesting that p73 does not function as a classical suppressor in tumors with MYCN gene amplification [16].
Our original hypothesis, that p73 might represent the 1p tumor suppressor thought to be preferentially inactivated in tumors with single copies of MYCN per haploid genome [31], was attractive for several reasons. Caron et al. [14] have documented preferential loss of the maternally derived chromosome in this form of neuroblastoma by LOH analysis, suggesting inactivation of the paternal allele of a suppressor gene through gametic imprinting. When we initiated this study, p73 was an attractive candidate for the putative suppressor gene; indeed, the data of Kaghad et al [16], had demonstrated monoallelic expression of p73 in a small number of normal persons heterozygous for an allelic polymorphism within the 5′noncoding exon of the gene. However, in a recently reported study, biallelic expression of p73 was detected in both primary neuroblastomas and a spectrum of normal tissues, indicating a lack of genomic imprinting [32]. Our study did not specifically address the issue of imprinting, but an RNA expression in each neuroblastoma shown to have deletion of one allele of p73 argues against imprinting as a mechanism of inactivation.
As reported here, only 3 of 26 MYCN single-copy primary tumors showed evidence of 1p loss by FISH analysis, in accord with 1p LOH in a relatively low percentage of tumors of this type [12–15,30]. The finding of a normal p73 mRNA sequence in the remaining tumors with sufficient levels of expression for RT-PCR analysis would appear to rule out biallelic inactivating lesions within the gene itself. The lack of mutations in our study is significant because single-point mutations in the region encoding the putative DNA binding domain of p73, analogous to those observed in p53 in human cancer cells, can block the antiproliferative effects of p73 in in vitro assays [16,17]. Thus, if p73 operates as a tumor suppressor in either MYCN-amplified or single-copy neuroblastoma, it likely operates through alternative mechanisms, such as haploinsufficiency or preferential expression of mRNA splice forms encoding dominant-interfering proteins. A recently reported analysis supports the conclusion that p73 is unlikely to be a tumor suppressor gene that is homozygously inactivated during neuroblastoma development [32].
Comparison of amino acid sequences between p73 and p53 revealed striking homology, particularly within the DNA-binding and oligomerization domains. Within the former, p73 shares 63% identity with p53, with conservation of residues known to be critical for sequence-specific interactions with DNA [16]. Functionally, p73 induces the expression of p53-responsive genes, such as p21Waf1/Cip1, and inhibits cell growth by inducing apoptosis [16,17]. The carboxyl-terminal region of p73, however, lacks sequence similarity to p53 but is homologous with recently identified invertebrate p53 orthologs [16], suggesting that these sequences have functional significance. In contrast to p53, p73 was shown to be expressed in two forms, alpha and beta, which have different coding sequences in this region. The beta splice form, corresponding to sf2 in our study (Figure 4A and B), lacks exon 13. As a result, exon 14 sequences are placed in an alternative reading frame, leading to truncation of the protein by a stop codon after only 5 amino acids have been contributed to those encoded by exon 12 [16].
We also identified several new splice forms by RT-PCR (Figure 4A and B), 1 of which lacked exon 11 (sf3) and was a common form of p73 expressed by multiple cell lines with MYCN gene amplification. Loss of exon 11 changed the p73 reading frame, so that sequences from exons 12 and 13 encode a protein with a C terminus that differs from either the alpha or beta form. This transcript was not identified in MYCN single-copy cell lines in the RT-PCR study, an observation that may be biologically significant, although studies of larger numbers of neuroblastoma tumors and cell lines will be required to confirm this association. Other splice forms of p73 were expressed at comparatively much lower levels than the alpha (sf1), beta (sf2), and sf3 forms (Figure 3C and 4).
Two of these splice forms were independently identified by De Laurenzi et al. [29], including sf3 and sf6, which they referred to as γ and δ, respectively. The sf3, or γ, isoform, which lacks exon 11 and encodes a unique C terminus was shown to form heterocomplexes with other p73 isoforms, but not with p53. Furthermore, this isoform was the least active of the 4 isoforms tested in terms of its ability to transactivate expression of p21WAF1/CIP1 or to inhibit the formation of cell colonies in clonogenic assays. The C-terminal domain of p53 performs an important regulatory role in that phosphorylation of amino acids in this region is required to activate sequence-specific DNA binding by the central region of the molecule [33–35]. The diminished growth suppressive activity of the sf3 form of p73 suggests that its C terminus also encodes a region with functional significance. Thus, the predominance of the sf3 isoform in most tumors with MYCN amplification suggests that it may sequester more active forms of p73 into inert complexes, leading to a reduced capacity of this protein to interfere with cell growth. Further analysis will be required to determine whether differential expression of the sf3 isoform contributes to oncogenesis through a dominant-negative mechanism.
Acknowledgements
We thank John Gilbert for editorial assistance, Doris Dodson for assistance with manuscript preparation, and Center for Biotechnology of St. Jude Children's Research Hospital for automated sequence analysis.
Abbreviations
- DAPI
4′,6-diamidino-2-phenylindole
- FISH
fluorescence in situ hybridization
- LOH
loss of heterozygosity
- RT-PCR
reverse transcription-polymerase chain reaction
Footnotes
This work was supported by the National Cancer Institute, NIH grants CA 39771, CA 71907, CA 31566 and CA 21765, and by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children's Research Hospital.
References
- 1.Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305:245–248. doi: 10.1038/305245a0. [DOI] [PubMed] [Google Scholar]
- 2.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 3.Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 4.Taylor SR, Locker J. A comparative analysis of nuclear DNA content and N-myc gene amplification in neuroblastoma. Cancer. 1990;65:1360–1366. doi: 10.1002/1097-0142(19900315)65:6<1360::aid-cncr2820650619>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 5.Cohn SL, Rademaker AW, Salwen HR, Franklin WA, Gonzales-Crussi F, Rosen ST, Bauer KD. Analysis of DNA ploidy and proliferative activity in relation to histology and N-myc amplification in neuroblastoma. Am J Pathol. 1990;136:1043–1052. [PMC free article] [PubMed] [Google Scholar]
- 6.Grady-Leopardi EF, Schwab M, Ablin AR, Rosenau W. Detection of N-myc oncogene expression in human neuroblastoma by in situ hybridization and blot analysis: Relationship to clinical outcome. Cancer Res. 1986;46:3196–3199. [PubMed] [Google Scholar]
- 7.Nakagawara A, Ikeda K, Tsuda T, Higashi K. N-myc oncogene amplification and prognostic factors of neuroblastoma in children. J Pediatr Surg. 1987;22:895–898. doi: 10.1016/s0022-3468(87)80583-3. [DOI] [PubMed] [Google Scholar]
- 8.Tsuda T, Obara M, Hirano H, Gotoh S, Kubomura S, Higashi K, Kuriowa A, Nakagawara A, Nagahara N, Shimizu K. Analysis of N-myc amplification in relation to disease stage and histologic types in human neuroblastomas. Cancer. 1987;60:820–826. doi: 10.1002/1097-0142(19870815)60:4<820::aid-cncr2820600418>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 9.Brodeur GM, Green AA, Hayes FA, Williams KJ, Williams DL, Tsiatis AA. Cytogenetic features of human neuroblastomas and cell lines. Cancer Res. 1981;41:4678–4686. [PubMed] [Google Scholar]
- 10.Brodeur GM, Tsiatis AA, Williams DL, Luthardt FW, Green AA. Statistical analysis of cytogenetic abnormalities in human cancer cells. Cancer Genet Cytogenet. 1982;7:137–152. doi: 10.1016/0165-4608(82)90010-3. [DOI] [PubMed] [Google Scholar]
- 11.Takeda O, Homma C, Maseki N, Sakurai M, Kanda N, Schwab M, Nakamura Y, Kaneko Y. There may be two tumor suppressor genes on chromosome arm 1p closely associated with biologically distinct subtypes of neuroblastoma. Genes Chromosomes Cancer. 1994;10:30–39. doi: 10.1002/gcc.2870100106. [DOI] [PubMed] [Google Scholar]
- 12.Schleiermacher G, Peter M, Michon J, Hugot J-P, Vielh P, Zucker J-M, Magdelenat H, Thomas G, Delattre O. Two distinct deleted regions on the short arm of chromosome 1 in neuroblastoma. Genes Chromosomes Cancer. 1994;10:275–281. doi: 10.1002/gcc.2870100409. [DOI] [PubMed] [Google Scholar]
- 13.White PS, Maris JM, Beltinger C, Sulman E, Marshall HN, Fujimori M, Kaufman BA, Biegel JA, Allen C, Hilliard C, Valentine MB, Look AT, Enomoto H, Sakiyama S, Brodeur GM. A region of consistent deletion in neuroblastoma maps within human chromosome 1p36.2-36.3. Proc Natl Acad Sci USA. 1995;92:5520–5524. doi: 10.1073/pnas.92.12.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caron H, Peter M, van Sluis P, Speleman F, de Kraker J, Laureys G, Michon J, Brugieres L, Voute PA, Westerveld A, Slater R, Delattre O, Versteeg R. Evidence for two tumour suppressor loci on chromosomal bands 1p35-36 involved in neuroblastoma: one probably imprinted, another associated with N-myc amplification. Human Mol Genet. 1995;4:535–539. doi: 10.1093/hmg/4.4.535. [DOI] [PubMed] [Google Scholar]
- 15.Cheng NC, Van Roy N, Chan A, Beitsma M, Westerveld A, Speleman F, Versteeg R. Deletion mapping in neuroblastoma cell lines suggests two distinct tumor suppressor genes in the 1p35-36 region, only one of which is associated with N-myc amplification. Oncogene. 1995;10:291–297. [PubMed] [Google Scholar]
- 16.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan J-C, Valent A, Minty A, Chalon P, Lelias J-M, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 17.Jost CA, Marin MC, Kaelin WG., Jr p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 18.Nomoto S, Haruki N, Kondo M, Takahashi T, Takahashi T, Takahashi T. Search for mutations and examination of allelic expression imbalance of the p73 gene at 1p36.33 in human lung cancers. Cancer Res. 1998;58:1380–1383. [PubMed] [Google Scholar]
- 19.Clurman B, Groudine M. Killer in search of a motive? Nature. 1997;389:122–123. doi: 10.1038/38116. [DOI] [PubMed] [Google Scholar]
- 20.Oren M. Lonely no more: p53 finds its kin in a tumor suppressor haven. Cell. 1997;90:829–832. doi: 10.1016/s0092-8674(00)80347-5. [DOI] [PubMed] [Google Scholar]
- 21.Johnson MR, Look AT, DeClue JE, Valentine MB, Lowy DR. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP-Ras. Proc Natl Acad Sci USA. 1993;90:5539–5543. doi: 10.1073/pnas.90.12.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro DN, Valentine MB, Rowe ST, Sinclair AE, Sublett JE, Roberts WM, Look AT. Detection of N-myc gene amplification by fluorescence in situ hybridization. Diagnostic utility for neuroblastoma. Am J Pathol. 1993;142:1339–1346. [PMC free article] [PubMed] [Google Scholar]
- 23.Morris SW, Valentine MB, Shapiro DN, Sublett JE, Deaven LL, Roberts WM, Foust JT, Cerretti DP, Look AT. Reassignment of the human CSF1 gene to chromosome 1p13-p21. Blood. 1991;78:2013–2020. [PubMed] [Google Scholar]
- 24.Lahti JM, Valentine M, Xiang J, Jones B, Amann J, Grenet J, Richmond G, Look AT, Kidd VJ. Alterations in the PITSLRE protein kinase gene complex on chromosome 1p36 in childhood neuroblastoma. Nat Genet. 1994;7:370–375. doi: 10.1038/ng0794-370. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Kong XT, Ida K, Ichikawa H, Shimizu K, Ohki M, Maseki N, Kaneko Y, Sako M, Kobayashi Y, Tojou A, Miura I, Kakuda H, Funabiki T, Horibe K, Hamaguchi H, Akiyama Y, Bessho F, Yanagisawa M, Hayashi Y. Consistent detection of TLS/FUS-ERG chimeric transcripts in acute myeloid leukemia with t(16;21) (p11;q22) and identification of a novel transcript. Blood. 1997;90:1192–1199. [PubMed] [Google Scholar]
- 27.Kong XT, Choi SH, Inoue A, Xu F, Chen T, Takita J, Yokota J, Bessho F, Yanagisawa M, Hanada R, Yamamoto K, Hayashi Y. Expression and mutational analysis of the DCC, DPC4, and MADR2/JV18-1 genes in neuroblastoma. Cancer Res. 1997;57:3772–3778. [PubMed] [Google Scholar]
- 28.Christiansen H, Schestag J, Bielke W, Schutz B, Rust G, Engel R, Beniers E, Christiansen NM, Lampert F. Chromosome 1 interphase-cytogenetics in 32 primary neuroblastoma of different clinical stages. Prog Clin Biol Res. 1991;366:99–105. [PubMed] [Google Scholar]
- 29.De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A, Falco M, Annicchiarico-Petruzzelli M, Levrero M, Melino G. Two new p73 splice variants, γ and δ, with different transcriptional activity. J Exp Med. 1998;188:1763–1768. doi: 10.1084/jem.188.9.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maris JM, White PS, Beltinger CP, Sulman EP, Castleberry RP, Shuster JJ, Look AT, Brodeur GM. Significance of chromosome 1p loss of heterozygosity in neuroblastoma. Cancer Res. 1995;55:4664–4669. [PubMed] [Google Scholar]
- 31.Versteeg R, Caron H, Cheng NC, van der Drift P, Slater R, Westerveld A, Voute PA, Delattre O, Laureys G, Van Roy N, Speleman F. 1p36: Every subband a suppressor? Eur J Cancer. 1995;31A:538–541. doi: 10.1016/0959-8049(95)00037-j. [DOI] [PubMed] [Google Scholar]
- 32.Kovalev S, Marchenko N, Swendeman S, LaQuaglia M, Moll UM. Expression level, allelic origin, and mutation analysis of the p73 gene in neuroblastoma tumors and cell lines. Cell Growth Differ. 1998;9:897–903. [PubMed] [Google Scholar]
- 33.Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 35.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]