

Abstract
Vascular endothelial growth factor (VEGF) plays a fundamental role in mediating tumor angiogenesis and tumor growth. Here we investigate the direct effect of a novel small molecule inhibitor of the Flk-1-mediated signal transduction pathway of VEGF, SU5416, on tumor angiogenesis and microhemodynamics of an experimental glioblastoma by using intravital multifluorescence videomicroscopy. SU5416 treatment significantly suppressed tumor growth. In parallel, SU5416 demonstrated a potent antiangiogenic activity, resulting in a significant reduction of both the total and functional vascular density of the tumor microvasculature, which indicates an impaired vascularization as well as significant perfusion failure in treated tumors. This malperfusion was not compensated for by changes in vessel diameter or recruitment of nonperfused vessels. Analyses of the tumor microcirculation revealed significant microhemodynamic changes after angiogenesis blockage such as a higher red blood cell velocity and blood flow in remnant tumor vessels when compared with controls. Our results demonstrate that the novel antiangiogenic concept of targeting the tyrosine kinase of Flk-1/KDR by means of a small molecule inhibitor represents an efficient strategy to control growth and progression of angiogenesis-dependent tumors. This study provides insight into microvascular consequences of Flk-1/KDR targeting in vivo and may have important implications for the future treatment of angiogenesis-dependent neoplasms.
Keywords: antiangiogenic therapy, protein tyrosine kinase, glioma, vascularization, vascular endothelial growth factor (VEGF)
Introduction
Angiogenesis is the continuous formation of new blood vessels from the existing vasculature. Without angiogenesis, most solid tumors do not progress to a clinically relevant size due to inadequate tissue oxygenation and nutritional supply [1]. The process of tumor angiogenesis is mediated by angiogenic growth factors and their cognate receptors via a paracrine mechanism [2]. Targeting of angiogenic signal transduction pathways represents a promising alternative in the treatment of neoplasms that are resistant to the therapeutic armamentarium of surgery, radiotherapy, and cytotoxic chemotherapy. Among these are high-grade gliomas (glioblastoma multiforme) that are the most malignant brain tumors, with a mean survival time of 9 to 12 months and a high resistance to conventional oncology therapy. Because they are among the most highly vascularized human tumors they should be ideal candidates for antiangiogenic therapy [3].
Vascular endothelial growth factor (VEGF), also known as vascular permeability factor (VPF), is an endothelial cell-specific mitogen in vitro, as well as a potent angiogenic growth factor and mediator of microvascular hyperpermeability in vivo [4,5]. This dimeric glycoprotein has a significant sequence homology with platelet-derived growth factor (PDGF) [6] and placenta growth factor (PlGF) [7] and is expressed in at least 4 different molecular isoforms of 206, 189, 165 and 121 amino acids as a result of alternative splicing of mRNA. Among these, VEGF165 is the predominantly expressed isoform in most human tissues, including the central nervous system [5]. The biological effects of VEGF are mediated by 2 high-affinity receptors, the class III protein tyrosine kinases (PTKs) VEGFR-1 (Flt-1) [8] and VEGFR-2 (Flk-1/KDR) [9], which are almost exclusively expressed on microvascular endothelial cells. These two receptors may serve distinct functions, including endothelial cell proliferation and chemotaxis, monocyte migration, and cell to cell or cell to matrix interaction [10–12].
A substantial body of evidence has emerged suggesting that the VEGF-Flk-1/KDR system is the dominant signal tranduction pathway regulating glioma-induced angiogenesis. First, temporal and spatial expression patterns of VEGF, Flk-1/KDR, and Flt-1 correlate significantly with the angiogenic activity in human gliomas [13,14]. Second, both administration of a neutralizing anti-VEGF antibody [15] and interference with the Flk-1/KDR-mediated signal transduction pathway by using a dominant-negative strategy [16] inhibited glioma growth in the athymic mouse. Third, Flt-1 is not associated with endothelial cell mitogenicity and endothelial cell chemotaxis, but rather with the regulation of endothelial cell to cell and cell to matrix interactions during vascular development [10,11]. Therefore, considerable interest has been developed in selective targeting of the VEGF-Flk-1/KDR signaling pathway for antiglioma therapy.
Small molecule inhibitors of tyrosine phosphorylation on Flk-1/KDR, which belong to the tyrphostins, quinoxalines and quinazolinones chemical classes, have been shown to possess antiangiogenic activities in vitro [17]. More recently, SU5416, a novel and selective inhibitor of the tyrosine kinase activity of Flk-1/KDR has been identified [18]. SU5416 and related compounds have been shown to function as adenine mimetics at the catalytic domain of the tyrosine kinase [19]. In in vitro studies, SU5416 exerts a potent, rapid, and long-lasting antiproliferative effect on en-dothelial cells without directly affecting growth of tumor cells in culture [18]. In vivo, parenteral administration of SU5416 resulted in an inhibition of growth of multiple tumor types of various tissue origins [18].
The objective of the present study was to assess the in vivo effect of SU5416 on tumor growth, tumor-induced angiogenesis, and tumor microcirculation and, thereby, to further define the mechanism of its in vivo action as well as the microhemodynamic consequences following Flk-1/KDR inhibition. The use of the dorsal skinfold chamber preparation in the athymic mouse [20] represents a unique tool to study the dynamic processes of angiogenesis and microvascular perfusion patterns of normal and neoplastic tissue (e.g., adenocarcinoma, high-grade glioma) by direct, continuous, and noninvasive means [21–24]. We report for the first time that tumor growth suppression by SU5416 is accompanied by 1) direct inhibitory effects on microvascular proliferation and thus a reduced total as well as functional vascular density, and 2) microhemodynamic changes with an increase in blood perfusion in individual remnant tumor vessels. These results are of considerable interest for the further characterization of the novel therapeutic concept of small-molecule Flk-1/KDR inhibitors and the understanding of the microcirculatory consequences of this antiangiogenic intervention, thus assisting in the design of future therapeutic strategies for Flk-1/KDR intervention in oncology.
Materials and Methods
Cells and Cell Culture
C6 rat glioma cells were cultured in HAM's F-10 culture medium in 12-well dishes at 37°C in humidified atmosphere with 5% carbon dioxide in air. A suspension of 5 x 105 cells was implanted into the skin chamber for tumor growth studies as previously described in detail [24].
Animals and Dorsal Skinfold Chamber Model
Athymic nude mice (nu/nu; male, 28–32 g) were bred and maintained within a specific pathogen germ-free environment. The technique for implantation of the dorsal skinfold chamber has been previously described [20,24]. Briefly, animals were anesthetized by subcutaneous injections of 7.5 mg ketamine hydrochloride and 2.5 mg xylazine per 100 mg body weight. Two symmetrical titanium frames were implanted into the dorsal skinfold of animals to sandwich the extended double layer of skin and create the dorsal skinfold chamber which consists of one layer of striated muscle, subcutaneous tissue, and epidermis. An observation window, covered with a glass cover slip, allowed for repeated intravital microscopic observations of the microvasculature of the tumor growing in the chamber. The backside of the chamber preparation remained open, allowing a 3-dimensional quantitation of the growth of the implanted glioma. Two days after chamber preparation, the coverslip of the dorsal skinfold chamber was temporarily removed and a suspension of tumor cells placed on the surface of the striated skin muscle. The animals tolerated the skinfold chambers well and showed no signs of discomfort or changes in sleeping and feeding behavior.
Experimental Protocol
Animals (n = 7) were treated daily with an intraperitoneal (IP) bolus SU5416 in DMSO (25 mg/kg in 50 µL DMSO), starting on the day of glioma cell implantation. The dose of SU5416 was selected by toxicity studies [18], identifying 25 mg/kg per day as the maximum tolerated dose that does not cause significant toxicity in the mouse. Furthermore, previous studies had shown that SU5416 at this dose has no effects on macrohemodynamics or rheologic parameters (TAT Fong, data not shown), thus ruling out the theory that potential microhemodynamic changes during treatment might be related to blood pressure or hematocrit alterations. Animals in the control groups received either the vehicle DMSO (n = 3, 50 µL) or saline (n = 3, 50 µL). Animals were weighed regularly and observed for behavioral changes. The macroscopic appearance of the skinfold chamber preparation and the implanted glioma was documented daily. Intravital multi-fluorescence microscopic studies of glioma growth, angiogenesis, and microcirculation were performed on days 6, 10, 14, 18, and 22 after glioma cell implantation. During the days of observation, the newly formed microvasculature within the fluorescently labeled glioma mass (intratumorally) and at the glioma periphery (peritumorally), i.e., outside the tumor and next to the tumor edge, was separately assessed. Measurements on vascular density and microhemodynamics included only newly formed tumor microvessels which can be clearly distinguished by their chaotic arrangement from the autochthonous host striated muscle microvessels displaying the typical parallel arrangement of the muscle capillaries [24,25]. Vascular densities were measured in 6 to 9 regions of interest per animal and per observation time point. Microvascular diameters as well as hemodynamic parameters were determined by analysing 5 to 10 microvessels per region of interest. At the end of the in vivo experiments (22 days after glioma cell implantation), the animals were sacrificed with an overdose of ketamine/xylazine, and the skinfold chamber preparations were processed for light microscopic analyses.
Intravital Multi-fluorescence Videomicroscopy
Intravital multi-fluorescence videomicroscopy (epi-illumination) was performed with a modified Axiotech vario microscope with a 100-W mercury lamp attached to a Ploema-Pak illuminator with an ultraviolet excitation wave length: 340–380 nm) and a blue (450–490 nm) filter block (Zeiss, Oberkochen, Germany). Observations were made with 3.2 x long distance, 10 x long distance, and 20 x water immersion working objectives (all Zeiss), resulting in magnifications of 71 x, 216 X, and 435 X, respectively. The glioma cells and microvasculature were visualized by means of a low-light level CCD (charge-coupled device) video camera (Cohu FK 6990, Pieper, Schwerte, Germany). Microscopic images were recorded with a S-VHS videosystem (Panasonic, Munich, Germany) for off-line analysis. Before tumor inoculation, C6 glioma cells were fluorescently labeled with the fluorescent vital dye Fast Blue (10 µg/mL; Sigma Chemical Co, St. Louis, MO) [26] for a 2-hour incubation period. With ultraviolet light the dye is characterized by a bright blue fluorescence with only little bleaching and persists through several cell generations. Because it demonstrates sufficient staining even after 3 weeks without affecting cell viability, it has been widely used in studies on glioma growth and glioma cell infiltration in vivo [26–28]. The specific fluorescence/background fluorescence ratio was still high enough at the end of the 22-day observation period to detect individual tumor cells and, thus, to precisely delineate the tumor mass from the surrounding unaffected host tissue. With this technique of tumor cell prelabeling, the C6 glioma exhibits a rapid tumor growth in the dorsal skinfold chamber model to a size of approximately 60 mm2 (400 mm3) after 22 days [24]. The vascular compartment, including angiogenic sprouts, newly formed microvessels, and the glioma microvasculature were visualized by contrast enhancement with 2% fluorescein isothiocyanate (FITC)-conjugated dextran (0.1 mL FITC-dextran150 intravenous; mol wt = 150,000; Sigma) under blue light epi-illumination. As previously described [24], extravasation of FITC-dextran150 was analyzed 15 minutes after administration, applying the following semiquantitative score: no extravasation (-); moderate extravasation (increased extravasal gray levels without negatively contrasted microvascular imaging) forming a patchy (+) or homogeneous (++) pattern; extravasation with negatively contrasted microvascular imaging +++).
Analyzed Parameters
All intravital microscopic measurements were performed by computer-assisted image analysis system (CAPIMAGE; Zeintl Software Engineering, Heidelberg, Germany). Tumor growth was assessed by measurement of the tissue area covered by the fluorescently labeled tumor mass within the chamber (mm2), and tumor volume (mm3) was calculated by using the equation V = 2/3 x A x h, where A is the area of the tumor mass and h is the thickness of the tumor bulging out at the chamber back side. The thickness of the tumor was measured after each intravital microscopic analysis with a caliper. Particular care was taken to avoid compression of the tissue and its microvasculature. Quantitative analysis of microcirculatory parameters included the angiogenic take rate (%), which was defined as the percentage of gliomas inducing the formation of red blood cell (RBC)-perfused microvessels; the tissue area covered by the newly formed microvascular network (mm2); the total vascular density (cm-1), which was defined as the length of all newly formed microvessels per area of interest and observation time point; the functional vascular density (cm-1), which was defined as length of RBC-perfused microvessels per area of interest and observation time point; microvessel diameters (µm), and microvascular RBC velocities (µm/s). A perfusion index PI was calculated as the percentage of the functional vascular density (FVD) and total vascular density (TVD) as follows: PI = FVD/TVD x 100. Blood flow rate Qv (in nanoliter/s) of individual microvessels was calculated according to Qv = p x (D/2)2 x RBCV/K, where RBCV represents the RBC velocity, D represents the microvessel diameter, and K is the Baker/Wayland factor (= 1.3) [29] considering the parabolic velocity profile of blood in microvessels. Newly formed glioma microvessels were not categorized into arterioles, capillaries, and venules because this classification is based on morphological and physiological criteria for normal tissue and might not be applicable to tumors [30].
Statistical Analyses
Quantitative data are given as mean values ± standard deviation. Mean values of microcirculatory data were calculated from the average values in each animal. For analysis of differences between the groups, post-hoc unpaired Bon-ferroni t test was used following one-way analysis of variance (ANOVA). Results with P values of < .05 were considered significant.
Histology
Upon completion of experiments, the glioma containing dorsal skinfold chamber preparations were excised, fixed over 48 hours with 10% paraformaldehyde, and embedded in paraffin. For histomorphological analyses, 5 µm sections were prepared and stained with hematoxylin/eosin.
Results
The effect of Flk-1/KDR tyrosine kinase inhibition on glioma growth, angiogenesis, and microcirculation was studied. Athymic nude mice were treated daily with SU5416 beginning on the day of glioma cell implantation. Control animals receiving the vehicle DMSO or saline (n = 3 each) showed no significant difference in any of the evaluated parameters and were thus grouped together (control group, n = 6) for graphic representation and statistical evaluation. In the current studies, no mortality was observed, and the sleeping and feeding behavior of the animals was not altered during the 22-day treatment and observation period. No significant differences were measured comparing the body weight of vehicle- and drug-treated animals (30.0 ± 2.4 g v 28.9 ± 4.3 g on the day of glioma cell implantation; 30.0 ± 1.4 g v 28.6 ± 4.4 g on day 6; 30.4 ± 2.6 g v 28.0 ± 5.2 g on day 14; and 29.9 ± 1 g v 27.2 ± 3.1 g on day 22 following glioma cell implantation).
The effect of SU5416 on early tumor growth is illustrated in Figure 1. The tumors remained dormant in both the control and treated groups between days zero and 6 after tumor cell implantation. Exponential tumor growth kinetics were observed in animals from the control group between days 6 to 22. Daily treatment with SU5416 resulted in a significantly lower tumor growth rate with tumor masses of up to 8% of that present in control animals by day 22 after implantation (Figure 1A). Inhibition of tumor growth was clearly preceded by a marked reduction of the tissue area covered by the newly formed glioma microvasculature in the SU5416-treated group, indicating a reduced initial tumor vascularization (Figure 1B).
Figure 1.
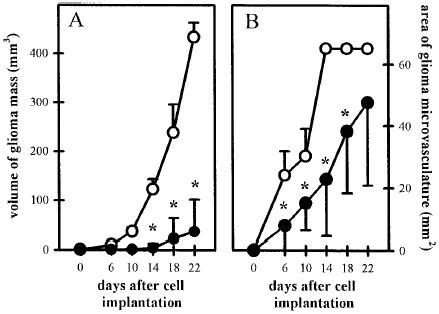
Influence of Flk-1 tyrosine kinase inhibition on C6 glioma growth as evaluated by monitoring the volume of glioma mass (A) and the tissue area covered by glioma microvasculature (B) from day zero to day 22 after implantation of C6 glioma cells. Animals were treated with saline or vehicle DMSO (50 µL/d IP; n = 6; open symbols) or SU5416 (25 mg/kg/d, IP; n = 7; closed symbols). The volume of the glioma mass was calculated from the area covered by the glioma mass (analyzed planimetrically by intravital microscopy) and the thickness of the glioma mass (see methodology section). The tissue area covered by the glioma microvasculature was analyzed planimetrically off-line with a computer-assisted image analysis system. The mean ± SD values are represented. Statistical analysis was performed by using ANOVA, followed by unpaired Student t test. (* P < .05 v control).
Figure 2 shows representative histological sections of C6 gliomas on day 22 after implantation into control or SU5416-treated animals. Specimens from control animals demonstrated a large tumor mass grown on the muscle and subcutaneous tissue of the chamber preparation. In contrast, tumors in SU5416-treated animals were markedly thinner. Analysis of tumors from both control and SU5416-treated animals revealed the presence of small anaplastic tumor cells characterized by cellular and nuclear polymorphism. Mitotic figures were seen in both groups. Areas of necroses, however, could not be observed in either control or treated tumors.
Figure 2.
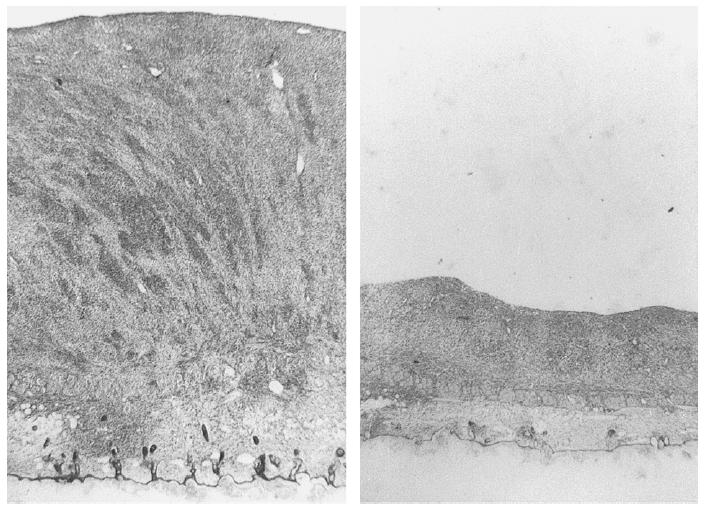
Histological examination of C6 gliomas on day 22 after glioma cell implantation into dorsal skinfold chamber preparation of nude mice. Animals were treated with the vehicle DMSO (50 µL/d, IP; left) or SU5416 (25 mg/kg/d, IP; right). (H and E staining of 5 µm sections).
To test the hypothesis that the inhibition of tumor growth by SU5416 was due to the antiangiogenic activity of SU5416, tumor-induced angiogenesis and tumor vascularization were assessed by using intravital multi-fluorescence videomicroscopy. In the control group, implantation of C6 gliomas into the skinfold chamber preparation induced local hyperemia, edema formation, and massive diapedesis of RBCs, leading to multiple intratumoral hemorrhages within the first days after tumor cell implantation (Figure 3). Initial signs of glioma-induced angiogenesis were observed between days 2 and 4 after glioma cell implantation. The process of angiogenesis was characterized by microvascular sprouts and sinusoidal vessel configurations originating from dilated and tortuously elongated host capillaries and postcapillary venules. By day 6, these newly formed microvessels first became branched and then subsequently interconnected to form a complex, RBC-perfused microvascular network in the peritumoral area (angiogenic take rateday6 = 6/6 = 100%) (Figure 4). By day 10, the area of the glioma mass was completely vascularized. The microvasculature was characterized by a chaotic angioarchitecture, heterogeneous blood perfusion, and increased permeability to macromolecules as indicated by gross extravasation (++) of the high molecular weight fluorescent marker FITC-dextran150. The glioma microvasculature was maintained during tumor growth between days 10 to 22. However, the microvascular network within the peritumoral and intratumoral areas revealed marked angioarchitectural differences. High angiogenic activity and vascular density were observed in peritumoral areas while the angiogenic activity within the glioma mass was decreasing centripetally in proportion to the distance toward the center (Figure 4). In contrast to the control group, chamber preparations from SU5416-treated animals did not show glioma-induced hyperemia, edema formation, or major intratumoral bleeding (see Figure 3). Glioma-induced angiogenesis with formation of a functional microvascular network was prevented in 5 of 7 treated animals (angiogenic take rateday6 = 29%) (Figure 4) until day 6 after glioma cell implantation. By day 10, however, a RBC-perfused microvascular network had been established in all treated animals (angiogenic take rateday10 = 100%). In comparison with control animals, the microvasculature of gliomas from SU5416-treated animals was characterized by a reduced angiogenic activity, heterogeneous vascularization, and loss of the peri-to-intratumoral vascular density gradient (Figure 4) between days 10 and 22. Extravasation of FITC-dextran150 was also markedly reduced and solely confined to angiogenically active microvascular areas (+).
Figure 3.
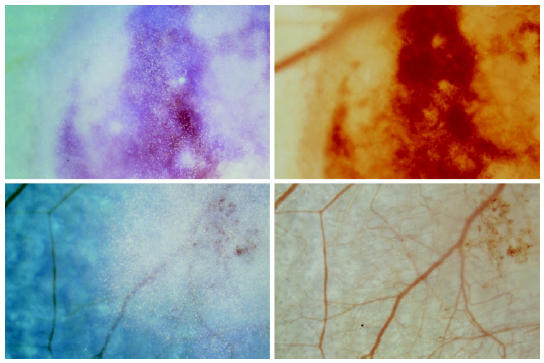
C6 gliomas on day 4 after glioma cell implantation and daily treatment with saline/DMSO (50 µL/d, IP, top panels) or SU5416 (25 mg/kg/d, IP, bottom panels) visualized by intravital microscopy applying transillumination (right row) and combined trans- and epi-illumination techniques (left row). Tumor cells are stained with Fast Blue (Sigma), allowing exact identification of tumor mass.
Figure 4.
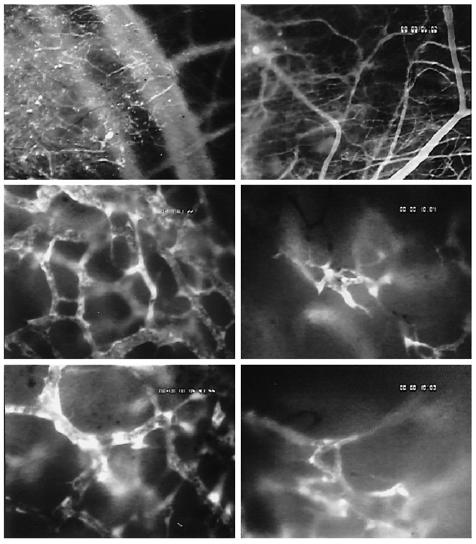
C6 glioma microvasculature in animals treated with DMSO (50 µL/d, IP; left row) or SU5416 (25 mg/kg/d, IP; right row) on day 6 (top panels) and 18 (middle and bottom panels) after glioma cell implantation. Intravital multi-fluorescence videomicroscopy, contrast enhancement with 2% FITC-dextran150 IV.
Figure 5 shows the quantitative analysis of total and functional vascular densities in control and treated animals. Throughout the entire observation period, the total vascular density of the glioma microvasculature was significantly reduced in tumors from SU5416-treated animals compared with those from control animals, supporting the existence of an inhibitory mechanism on microvascular proliferation of the drug based on blockage of angiogenesis (Figure 5A and B). A greater magnitude of reduction of the total vascular density was observed in areas with higher angiogenic activity (60%–70% in peritumoral v 30%–40% in intratumoral areas, respectively). In parallel, the functional vascular density was reduced, too, indicating significant perfusion failure and impaired vascularization in SU5416-treated tumors (Figure 5C and D). Again, these effects were more pronounced in the well-vascularized peritumoral than in the intratumoral areas.
Figure 5.
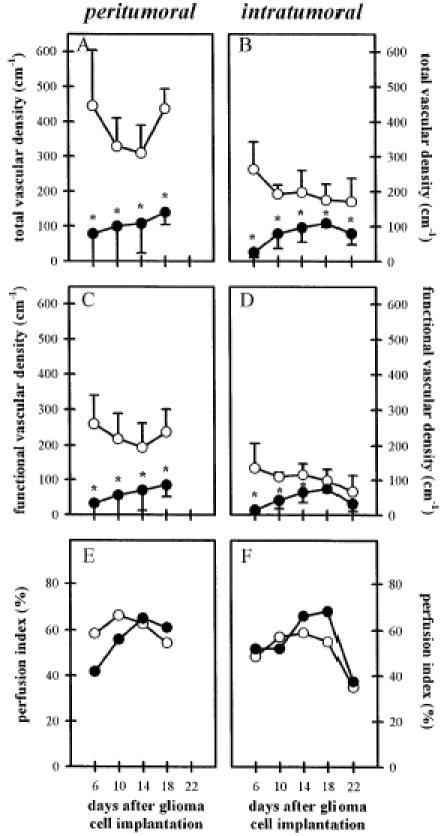
Influence of Flk-1 tyrosine kinase inhibition on C6 glioma angiogenesis as evaluated by total vascular density (A and B), functional vascular density (C and D), and perfusion index (E and F) within the peritumoral (left column) and intratumoral (right column) areas from days 6 to 22 after C6 glioma cell implantation. Animals were treated with saline/DMSO (50 µL/d IP; n = 6; open symbols) or SU5416 (25 mg/kg/d, IP; n = 7; closed symbols). Microcirculatory parameters were analyzed off-line by using a computer-assisted image analysis system. The mean ± SD values are represented. Statistical analysis was performed by using ANOVA followed by unpaired Student t test. (* P < .05 v control).
Significant antiangiogenic intervention may subsequently affect the microcirculation and microhemodynamics within the remaining glioma microvasculature. Antiangiogenic therapy leading to impaired tumor vascularization might be compensated by several microcirculatory mechanisms including recruitment of nonperfused microvessels, dilation of individual microvessels, and/or increase of microvascular RBC velocity [31]. Analysis of perfusion indices excluded a significant recruitment of nonperfused vessels as a compensatory mechanism because in both the control and SU5416-treated groups, 40% to 70% of newly formed microvessels were perfused with RBCs (Figure 5E and F). Similarly, no differences in microvascular diameters were observed (Figure 6A and B). In contrast, RBC velocity and blood flow within remnant intratumoral microvessels were higher in SU5416-treated tumors than in control tumors (Figure 6D and F). In peritumoral areas, this difference of microvascular RBC velocity and blood flow after inhibition of Flk-1/KDR activity was less pronounced (Figure 6C and E).
Figure 6.
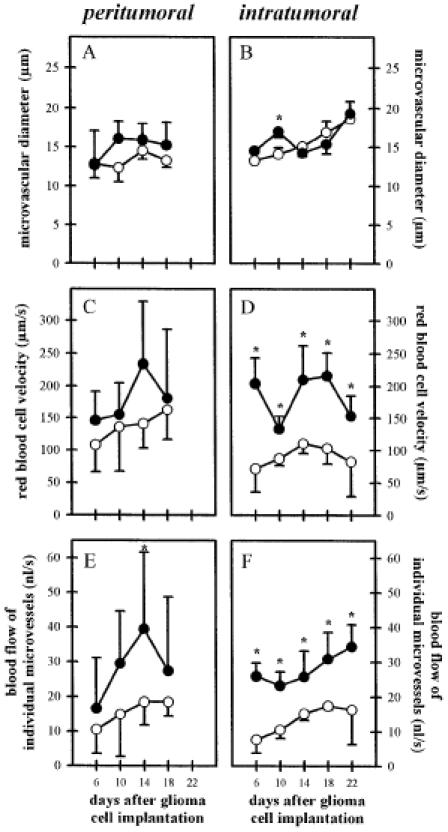
Influence of Flk-1 tyrosine kinase inhibition on C6 glioma microcirculation as evaluated by microvessel diameter (A and B), microvascular RBC velocity (C and D), and blood flow of individual microvessels (E and F) within the peritumoral (left column) and intratumoral (right column) areas for days 6 to 22 after C6 glioma cell implantation. Animals were treated with saline/DMSO (50 µl/d IP; n = 6; open symbols) or SU5416 (25 mg/kg/d, IP; n = 7; closed symbols). Microcirculatory parameters were analyzed off-line by using a computer-assisted image analysis system. The mean ± SD values are represented. Statistical analysis was performed by using ANOVA, followed by unpaired Student t test. (* P < .05 v control).
Discussion
We have demonstrated that targeting the Flk-1/KDR tyrosine kinase with a small molecule inhibitor represents an effective means to suppress tumor growth and progression in vivo. The principal novel finding of the present study is that the mechanism of tumor growth inhibition by the Flk-1/KDR tyrosine kinase inhibitor SU5416 in vivo is based on a direct inhibition of tumor-induced angiogenesis and vascularization and not mediated through alternative mechanisms independent of Flk-1-mediated microvascular proliferation. Moreover, a detailed in vivo analysis of the tumor microcirculation revealed that significant microhemodynamic changes accompanied the antiangiogenic intervention, including a higher RBC velocity and blood flow in individual remnant glioma vessels when compared with controls.
The use of C6 glioma cells implanted into a transparent dorsal skinfold chamber in athymic nude mice was based on several considerations, namely, 1) that C6 glioma xenografts have been shown to represent a prototype of angiogenesis-dependent experimental tumors [16]; 2) that the subcutaneous and muscle tissue are well established implantation sites to study C6 glioma growth [16,24,32,33] revealing growth behavior, vascularization process, and microvascular morphology similar to the intracerebral implantation site [24,32,34,35]; and 3) that subcutaneously implanted C6 gliomas are among the most extensively characterized experimental tumor models with respect to the functional expression of VEGF and Flk-1 in vivo [16,35]. Glioma growth, angiogenesis, and microcirculation were assessed by means of intravital multi-fluorescence videomicroscopy. The strength of this approach is 2-fold: First, in contrast to alternative procedures to assess tumor vascularization such as histological vessel counts [36]; 3-dimensional vascular corrosion casts [37]; microangiography by injection of dyes [38]; and blood flow measurements applying [14C]iodoantipyrine autoradiography [39], 133Xe-washout [40], laser-Doppler flowmetry [41], and perfusion weighted magnetic resonance microimaging [33], intravital multi-fluorescence videomicroscopy is currently the only experimental technique that allows for a direct, continuous, and noninvasive visualization of the glioma microvasculature at the level of individual microvessels [42]. Second, analyzing glioma angiogenesis by means of intravital multi-fluorescence videomicroscopy also allows for quantitative evaluation of various microcirculatory parameters, which appears to be mandatory because nutritive tissue perfusion not only depends on morphological criteria such as vascular density but also on microcirculatory and rheological parameters. The assessment of the microhemodynamic response may be relevant to gain more insight into mechanisms and consequences of antiangiogenic therapy. This is especially true when thinking about combinations of novel antiangiogenic compounds with conventional chemotherapy, an issue that has not yet been adequately appreciated in the field of tumor angiogenesis.
In accordance with previous reports [16–18,25,43], the present study confirmed the fundamental role of the VEGF-Flk-1/KDR signal transduction pathway in tumor angiogenesis. Recently, several molecular mechanisms regulating VEFG-Flk-1/KDR expression have been proposed. These include a genetic determination by oncogene activation and/or tumor suppressor gene inactivation [44,45], microenvironmental stress due to intratumoral hypoxia or nutritional deficiency [14,46–48], and induction by various growth factors and cytokines [49]. As a consequence, upon binding of VEGF to Flk-1/KDR, the intrinsic tyrosine kinase function is activated, which results in phosphorylation of the receptor, initiation of an intracellular multistep signaling pathway, and, finally, proliferation of the endothelial cells [50]. Potentially, inhibition of any step of this cellular signaling cascade will interfere with tumor-induced angiogenesis and tumor growth [51].
The selective inhibition of Flk-1/KDR activity with small molecule tyrosine kinase inhibitors such as SU5416 represents a novel approach to antiangiogenesis therapy. One advantage of this strategy is that using a small molecule inhibitor allows us to bypass the need for antibody and protein therapy. Furthermore, the lipophilic nature and low molecular weight of SU5416 guarantee a rapid endothelial uptake of the compound during its residence time within the vascular compartment and thus, a rapid binding to the intracellular domain of Flk-1/KDR [18]. Finally, SU5416 is efficacious at doses with no measurable toxicity in the mouse and is expected to be relatively safe compared to conventional cytotoxic therapy because Flk-1 is downregulated in angiogenically quiescent tissues [10].
We have chosen to analyze the newly formed glioma microvasculature separately in peritumoral and intratumoral areas. Previous studies using in situ hybridization have demonstrated that Flk-1 is expressed in both areas of C6 gliomas, suggesting similar mechanisms for intratumoral and peritumoral angiogenesis in gliomas [35]. In the present study, increased angiogenic activity and thus higher vascular density were consistently observed within peritumoral, in comparison with intratumoral, areas. This observation is in agreement with the hypothesis that high-grade gliomas, like most solid tumors, grow and expand at the tumor periphery, followed by a progressive failure in intratumoral vascularization and microvascular perfusion. In accordance with its inhibitory activity on endothelial cell proliferation, the most prominent antiangiogenic activity of SU5416 was observed within the peritumoral areas where most of the glioma-induced neovascularization occurs. Consequently, this observation points to regional differences in the susceptibility of the tumor microvasculature to the antiangiogenic intervention with SU5416. Apart from this antiangiogenic effect, inhibition of the Flk-1 tyrosine kinase with SU5416 also reduced the angiogenesis-associated microvascular hyperpermeability and diapedesis of RBCs.
To date, microhemodynamic changes following antiangiogenic therapy have received only minor attention. In the present study, we have demonstrated that suppression of glioma growth and angiogenesis by Flk-1 tyrosine kinase inhibition is accompanied by a higher microvascular RBC velocity and blood flow within individual remnant tumor microvessels than in control tumors. Based on previous reports demonstrating that small molecule Flk-1 inhibitors prepared in methylcellulose pellets reduce VEGF-related microvascular hyperpermeability, and based on our observation of a reduced extravasation of the high molecular marker FITC-dextran150 (mol wt = 150,000) following parenteral SU5416 treatment, we suggest that these microhemodynamic changes are due to a decreased interstitial pressure and thus, lower microvascular resistance as a consequence of reduced edema formation or ‘unpacking’ of tumor vessels as recently hypothesized by Folkman [52]. This is further strengthened by Boucher et al. [53], who were able to show that intratumoral interstitial fluid pressure is closely linked to tumor angiogenesis, thereby identifying interstitial hypertension in solid tumors as a function of their angiogenic activity. The authors' observation that the interstitial fluid pressure in angiogenically active solid tumors is high in the center and decreases centrifugally [51] well explains the reported regional differences of microhemodynamic changes (intratumoral v peritumoral) in our study. Nevertheless, the observed increase of RBC velocity and blood flow in individual remnant microvessels did not suffice to efficiently compensate for the reduced functional vascular density due to a significant decrease of the vascular surface exchange area (calculated from functional vascular density and microvascular diameters, data not shown) and increase of the intervascular spacing (i.e., diffusion distance).
In summary, inhibition of the Flk-1 tyrosine kinase by the small molecule inhibitor SU5416 has proven to be an effective and nontoxic strategy to control growth of angiogenesis-dependent tumors. For high-grade gliomas, which represent a prototype of angiogenesis-dependent tumors virtually resistant to currently available therapies, this novel compound represents a promising therapeutic alternative. The understanding of the microcirculatory and microhemodynamic consequences of antiangiogenic therapy in general and of Flk-1 tyrosine kinase inhibition in particular is mandatory because first compounds from this class of protein tyrosine kinase inhibitors have entered initial clinical trials.
Acknowledgements
This work was in part presented at the 89th Annual Meeting of the American Association of Cancer Research in New Orleans, L.O. and awarded with a young investigator award. We gratefully acknowledge the excellent technical assistance of Beate Wolf.
Abbreviations
- Flk-1
fetal liver kinase-1
- KDR
kinase insert domain-containing receptor
- flt-1
fms-like tyrosine kinase-1
- DMSO
dimethysulfoxide
- FITC
fluorescein isothiocyanate
- RBC
red blood cell.
Footnotes
This study was supported in part by the Forschungfond Mannheim 58/96 and by an EU-grant (BMH4-CT95-0878).
References
- 1.Folkman J. What is the evidence that tumors are angiogenesis-dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- 3.Brem S, Cotran R, Folkman J. Tumor angiogenesis: A quantitative method for histological grading. J Natl Cancer Inst. 1972;48:347–356. [PubMed] [Google Scholar]
- 4.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrara N, Houck K, Jakeman J, Leung DW. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev. 1992;13:18–32. doi: 10.1210/edrv-13-1-18. [DOI] [PubMed] [Google Scholar]
- 6.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 7.Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA. 1991;88:9267–9271. doi: 10.1073/pnas.88.20.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 9.Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NPH, Risau W, Ullrich A. High affinity VEGF binding and development expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 10.Waltenberger J, Claesso-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and FLT1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 11.Fong GH, Rossant J, Gertsenstein M, Breitman LM. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 12.Clauss M, Weich H, Breier G, Knies U, Rockl W, Waltenberger J, Risau W. The vascular endothelial growth factor receptor flt-1 mediates biological activities. J Biol Chem. 1996;30:17629–17634. doi: 10.1074/jbc.271.30.17629. [DOI] [PubMed] [Google Scholar]
- 13.Plate KH, Breier G, Weich HA, Mennell HD, Risau W. Vascular endothelial growth factor and glioma angiogenesis: Coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 14.Samoto K, Ikezaki K, Ono M, Shono T, Kohno K, Kuwano M, Fukui M. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res. 1995;55:1189–1193. [PubMed] [Google Scholar]
- 15.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 16.Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 17.Strawn LM, McMahon G, App H, Schreck R, Kuchler WR, Longhi MP, Hui TH, Tang C, Levitzki A, Gazit A, Chen I, Keri G, Orfi L, Risau W, Flamme I, Ullrich A, Hirth KP, Shawver LK. Flk-1 as a target for tumor growth inhibition. Cancer Res. 1996;56:3540–3545. [PubMed] [Google Scholar]
- 18.Fong TAT, Shawver LK, Sun L, Tang Ch, Kim YH, Schreck R, App H, Powell J, Vasile S, Wang XY, Risau W, Ullrich A, Hirth KP, McMahon GM. SU5416 is a potent and selective inhibitor of the VEGF receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 19.Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor on complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- 20.Vajkoczy P, Menger MD, Simpson E, Messmer K. Angiogenesis and vascularization of murine pancreatic islet iso-grafts. Transplantation. 1995;60:123–127. [PubMed] [Google Scholar]
- 21.Vajkoczy P, Olofsson MA, Lehr HA, Leiderer R, Hammersen F, Arfors K, Menger MD. Histogenesis and ultrastructure of pancreatic islet graft microvasculature. Evidence for graft revas-cularization by endothelial cells of host origin. Am J Pathol. 1995;146:1397–1405. [PMC free article] [PubMed] [Google Scholar]
- 22.Vajkoczy P, Lehr HA, Hubner C, Arfors K, Menger MD. Prevention of pancreatic islet xenograft rejection by dietary vitamin E. Am J Pathol. 1997;150:1487–1495. [PMC free article] [PubMed] [Google Scholar]
- 23.Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, Jain RK. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 24.Vajkoczy P, Schilling L, Ullrich A, Schmiedek P, Menger MD. Characterization of angiogenesis and microcirculation of high-grade glioma: An intravital multifluorescence microscopic approach in the athymic nude mouse. J Cereb Blood Flow Metab. 1998;18:510–520. doi: 10.1097/00004647-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Borgstrom P, Hillan KJ, Sriramarao P, Ferrara N. Complete inhibition of angiogenesis and growth of microtumors by anti-vascular endothelial growth factor neutralizing antibody: Novel concepts of angiostatic therapy from intravital videomicroscopy. Cancer Res. 1996;56:4032–4039. [PubMed] [Google Scholar]
- 26.Espinosa de los Monteros A, Bernard R, Tiller B, Rouget P, de Vellis J. Grafting of fast blue labeled glial cells into neonatal rat brain: differential survival and migration among cell types. Int J Dev Neurosci. 1993;11:625–639. doi: 10.1016/0736-5748(93)90051-e. [DOI] [PubMed] [Google Scholar]
- 27.Chicoine MR, Silbergeld DL. Assessment of brain tumor cell motility in vivo and in vitro. J Neurosurg. 1995;82:615–622. doi: 10.3171/jns.1995.82.4.0615. [DOI] [PubMed] [Google Scholar]
- 28.Chicoine MR, Silbergeld DL. Invading C6 glioma cells maintaining tumorigenicity. J Neurosurg. 1995;83:665–671. doi: 10.3171/jns.1995.83.4.0665. [DOI] [PubMed] [Google Scholar]
- 29.Baker M, Wayland H. On-line volume flow rate and velocity profile measurement for blood in microvessels. Microvasc Res. 1974;7:131–143. doi: 10.1016/0026-2862(74)90043-0. [DOI] [PubMed] [Google Scholar]
- 30.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48:7022–7032. [PubMed] [Google Scholar]
- 31.Intaglietta M, Zweifach BW. Microcirculatory basis of fluid exchange. Adv Biol Med Phys. 1974;15:111–159. doi: 10.1016/b978-0-12-005215-8.50011-5. [DOI] [PubMed] [Google Scholar]
- 32.Coomber BL, Stewart PA, Hayakawa EM, Farrell CL, Del Maestro RF. A quantitative assessment of microvessel ultrastructure in C6 astrocytoma spheroids transplanted to brain and to muscle. J Neuropathol Exp Neurol. 1988;47:29–40. doi: 10.1097/00005072-198801000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Abramovitch R, Meir G, Neeman M. Neovascularization induced growth of implanted C6 glioma multicellular spheroids: Magnetic Resonance Microimaging. Cancer Res. 1995;55:1956–1962. [PubMed] [Google Scholar]
- 34.Deane BR, Lantos PL. The vasculature of experimental brain tumours. Part 1. A sequential light and electron microscope study of angiogenesis. J Neurol Sci. 1981;49:55–66. doi: 10.1016/0022-510x(81)90188-x. [DOI] [PubMed] [Google Scholar]
- 35.Plate KH, Breier G, Millauer B, Ullrich A, Risau W. Upregulation of vascular endothelial growth factor and its cognate receptor in a rat glioma model of angiogenesis. Cancer Res. 1993;53:5822–5827. [PubMed] [Google Scholar]
- 36.Wesseling P, van der Laak J, de Leeuw H, Ruiter DJ, Burger PC. Quantitative immunohistochemical analysis of the microvasculature in untreated human glioblastoma multiforme. J Neurosurg. 1994;81:902–909. doi: 10.3171/jns.1994.81.6.0902. [DOI] [PubMed] [Google Scholar]
- 37.Orita T, Nishizaki T, Kamiryo T, Aoki H, Harada K, Okamura T. The microvascular architecture of human malignant glioma. Acta Neuropathol. 1988;76:270–274. doi: 10.1007/BF00687774. [DOI] [PubMed] [Google Scholar]
- 38.Bernsen HJJA, Rijken PFJW, Oostendorp T, van der Kogel AJ. Vascularity and perfusion of human gliomas xenografted in the athymic nude mouse. Br J Cancer. 1995;71:721–726. doi: 10.1038/bjc.1995.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whittle IR, Collins F, Kelly PAT, Ritchie I, Ironside JW. Nitric oxide synthase is expressed in experimental malignant glioma and influences tumour blood flow. Acta Neurochir (Wien) 1996;138:870–876. doi: 10.1007/BF01411266. [DOI] [PubMed] [Google Scholar]
- 40.Andrade SP, Hart IR, Piper PJ. Inhibitors of nitric oxide synthase selectively reduce flow in tumour-associated neovasculature. Br J Pharmacol. 1992;107:1092–1095. doi: 10.1111/j.1476-5381.1992.tb13412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foltz RM, McLendon RE, Friedman HS, Dodge RK, Bigner DD, Dewhirst MW. A pial window model for the intracranial study of human glioma microvascular function. Neurosurgery. 1995;36:976–984. doi: 10.1227/00006123-199505000-00014. [DOI] [PubMed] [Google Scholar]
- 42.Jain RK, Schlenger K, Hockel M, Yuan F. Quantitative angiogenesis assays: progress and problems. Nat Med. 1997;3:1203–1208. doi: 10.1038/nm1197-1203. [DOI] [PubMed] [Google Scholar]
- 43.Cheng SY, Huang HJ, Nagane M, Ji XD, Wang D, Shih CC, Arap W, Huang CM, Cavanee WK. Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA. 1996;93:8502–8507. doi: 10.1073/pnas.93.16.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grugel S, Finkenzeller G, Weindel K, Barleon B, Marmeé D. Both V-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. J Biol Chem. 1995;270:25915–25919. doi: 10.1074/jbc.270.43.25915. [DOI] [PubMed] [Google Scholar]
- 45.Kieser A, Weich HA, Brandner G, Marmé D, Kolch W. Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene. 1994;9:963–969. [PubMed] [Google Scholar]
- 46.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 47.Shweiki D, Neeman M, Itin A, Keshet E. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: Implications for tumor angiogenesis. Proc Natl Acad Sci USA. 1995;92:768–772. doi: 10.1073/pnas.92.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM. Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest. 1996;97:469–476. doi: 10.1172/JCI118437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai JC, Goldman CK, Gillespie GY. Vascular endothelial growth factor in human glioma cell lines: Induced secretion by EGF, PDGF-BB, and bFGF. J Neurosurg. 1995;82:864–873. doi: 10.3171/jns.1995.82.5.0864. [DOI] [PubMed] [Google Scholar]
- 50.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 51.Levitzki A, Gazit A. Tyrosine kinase inhibition: An approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 52.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 53.Boucher Y, Leunig M, Jain RK. Tumor angiogenesis and interstitial hypertension. Cancer Res. 1996;56:4264–4266. [PubMed] [Google Scholar]