

Abstract
Human DNA sequence variation data are useful for studying the origin, evolution, and demographic history of modern humans and the mechanisms of maintenance of genetic variability in human populations, and for detecting linkage association of disease. Here, we report worldwide variation data from a ≈10-kilobase noncoding autosomal region. We identified 75 variant sites in 64 humans (128 sequences) and 463 variant sites among the human, chimpanzee, and orangutan sequences. Statistical tests suggested that the region is selectively neutral. The average nucleotide diversity (π) across the region was 0.088% among all of the human sequences obtained, 0.085% among African sequences, and 0.082% among non-African sequences, supporting the view of a low nucleotide diversity (≈0.1%) in humans. The comparable π value in non-Africans to that in Africans indicates no severe bottleneck during the evolution of modern non-Africans; however, the possibility of a mild bottleneck cannot be excluded because non-Africans showed considerably fewer variants than Africans. The present and two previous large data sets all show a strong excess of low frequency variants in comparison to that expected from an equilibrium population, indicating a relatively recent population expansion. The mutation rate was estimated to be 1.15 × 10−9 per nucleotide per year. Estimates of the long-term effective population size Ne by various statistical methods were similar to those in other studies. The age of the most recent common ancestor was estimated to be ≈1.29 million years ago among all of the sequences obtained and ≈634,000 years ago among the non-African sequences, providing the first evidence from a noncoding autosomal region for ancient human histories, even among non-Africans.
Keywords: human origins, nucleotide diversity, rare variants, population expansion
As the Human Genome Project is entering its final sequencing phase, interest in DNA sequence variation in human populations is increasing rapidly. Recent studies of human DNA variation include regions containing, respectively, the genes for β-globin (1), lipoprotein lipase (2), pyruvate dehydrogenase E1 (PDHA1) (3), and angiotensin converting enzyme (4), an 8-kilobase (kb) segment of the dystrophin gene (5), and a noncoding region in Xq13.3 (6). To date, however, few large-scale worldwide surveys of DNA sequence variation in noncoding regions have been made, especially for autosomes. Such surveys are important because the level of sequence variation in noncoding regions, especially regions that are not closely linked to genes, should reflect to a large extent a balance between mutation and random drift and can serve as a neutrality standard for comparison with levels of variation in other regions. Such comparisons may provide much insight into the maintenance of genetic variability in human populations.
DNA sequence data from noncoding regions are also useful for studying human evolution. Indeed, as noncoding regions are not directly subject to natural selection, data from such regions may more accurately reflect human history than data from coding regions. The majority of past studies on human DNA variation, mainly from mitochondrial DNA, microsatellite DNA, and the Y chromosome, have revealed a relatively shallow human history. These observations have been taken as evidence for the out of African model for the origin of modern humans because this model postulates that a founder group emigrated from Africa about 100,000 years ago and completely replaced all of the indigenous populations outside of Africa (7, 8). However, the recent studies on the β-globin and the PDHA1 gene regions (1, 3) revealed an ancient genetic history of humans and suggested that human evolution has been more complex than depicted by the simple out of Africa model. It is therefore interesting to know whether long noncoding regions may yield further insight into human history.
In this study we selected a 10-kb noncoding region on chromosome 22 that appears to be an intergenic region and obtained sequence data from worldwide populations. A comparison with the data from Xq13.3 revealed many interesting features of sequence variation and a deep genetic history of humans.
Materials and Methods
DNA Samples.
Sixty-four individuals were collected worldwide from 16 populations in four major geographic areas, including 20 Africans (5 South African Bantu speakers, 1 !Kung, 2 Mbuti Pygmies, 2 Biaka Pygmies, 5 Nigerians, 5 Kenyans), 20 Asians (8 Chinese, 3 Japanese, 6 Indians, 3 Yakuts), 20 Europeans (6 Swedes, 2 Finns, 5 French, 5 Hungarians, 2 Italians), and 4 Oceanians (3 Papua New Guineans, 1 Melanesian). One common chimpanzee and one orangutan were used as the outgroup references.
Selection of a Noncoding Region.
An 11-kb region was selected from locus HSCN37F10, which is located between markers D22S280 and D22S86 on human chromosome 22q11.2 (GenBank accession no. Z69714). After excluding a region containing a partial Alu (449 bp total) and a region containing three copies of a 126-mer repeat (415 bp total), a total of ≈10,000 nucleotide sites were selected for sequencing. No known gene or potential coding region was found in this region in our search of GenBank or in the prediction by GRAIL-EXP or GenScan. The current GenBank data suggest that it is an intergenic region, about 9 kb downstream of a gene and about 9 kb upstream of another gene.
PCR and Sequencing.
Twenty-five pairs of primers were used to amplify the eight overlapping fragments that cover positions 1698 to 12424 in the 11-kb region and to sequence the PCR products (primer sequences are available upon request). Touch-down PCR (9) was used in a 25-μl reaction, which contained 0.4 μl starting dilution buffer (50 mM KCl/10 mM Tris⋅HCl, pH 7.0), 0.055 μg TaqStart antibody (CLONTECH), 0.5 U recombinant Taq polymerase (GIBCO/BRL), 1 × PCR buffer (50 mM KCl/20 mM Tris⋅HCl, pH 8.4), 240 μM dNTP, 1.5 mM MgCl2, 0.4 μM of each primer, and ≈100 ng of genomic DNA. The reactions were carried out by 14 cycles of denaturation (94°C, 45 s), annealing (62°C, 1 min, 0.5°C decrease per cycle), and extension (72°C, 2 min), followed by 30 cycles of denaturation (94°C, 45 s), annealing (55°C, 45 s), and extension (72°C, 1.5 min), and a final extension of 5 min at 72°C in the Perkin-Elmer GeneAmp PCR 9600 or 9700 system. The PCR products were purified by Wizard PCR Preps DNA purification resin kit (Promega) in 96-well column plates. Sequencing extension was performed according to the protocol of ABI Prism BigDye Terminator sequencing kits with AmpliTaq DNA polymerase, FS (Perkin–Elmer). The reaction mixture contained 2.0 μl of sequencing reagent premix, 0.2 μM sequencing primer, and 10–100 ng of purified PCR products in a 5-μl total volume. For high quality PCR products, only 1.6 μl of premix was used in a 4.0-μl reaction volume. After 20 s of hot start at 96°C, the reactions were incubated for 25 cycles of denaturation (96°C, 10 s), annealing (50°C, 5 s), and extension (60°C, 4 min). The extension products were purified by Sephadex G-50 (DNA grade, Amersham Pharmacia) in 96-well column plates and run on an ABI 377XL DNA sequencer using 4.25% gels (Sooner Scientific, Garvin, OK).
Sequence Analysis and Verification.
ABI DNA Sequence Analysis 3.0 was used for lane tracking and base calling. The data were then proofread and aligned in Sequence Navigator 1.01. The segment sequences were assembled manually or by using DNASTAR, and variant sites were identified in the aligned sequences. All of the nucleotides in the target region were sequenced by forward and reverse primers at least once in each direction; all non-high quality sequences were resequenced using either the original primers or new nested primers. Furthermore, ≈55% of the variant sites found in this region, mainly singletons and doubletons, were verified by restriction fragment length polymorphism (RFLP), by resequencing a region containing the variant site, or by sequencing PCR products amplified from clones. For RFLP analysis, DNASTAR was used to identify potential restriction enzyme recognition sites in a region containing the putative variant site and to determine a short region covering only one recognition site. The primers were specifically designed for each target site to cover that region. In some cases, the primers were designed to create a new RFLP site for a variant site which was not a restriction site. In subcloning, we first amplified a 10-kb long PCR product for each individual, then inserted it into the plasmid vector and subsequently sequenced the screened positive clones. The sequences of several clones for each insertion were compared to ascertain the haplotypes.
Statistical Analysis of Sequence Data.
For a DNA sequence subject to no natural selection, the mutation rate per sequence per generation (μ) is estimated by
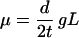 |
where d is the number of nucleotide substitutions per nucleotide site between two sequences, t is the divergence time between two species, L is the sequence length (bp), and g is the generation time (human g ≈ 20 years). Watterson's (10) and Tajima's (11) methods were used to estimate θ = 4 Neμ, where Ne is the effective population size.
Tajima's test (12) and Fu and Li's tests (13) were used to test the selective neutrality of the region studied (a program is available at http://hgc.sph.uth.tmc.edu/fu). Fu's (14) and Fu and Li's (15) methods were used to estimate the age of the most recent common ancestor (MRCA) of the DNA sequences in a sample. Given that the number of segregating sites in a sample of n sequences in a constant population is K, the conditional probability pn(t|K) for the age (t in 4Ne generations) of the MRCA was computed and used to compute the mode and mean of the age (T) of the MRCA in terms of years (14, 15); the mode and mean represent, respectively, the most likely value and the average value of T conditional on observing K segregating sites.
Results and Discussion
Distribution and Pattern of Sequence Variation.
We sequenced 9,901 nucleotide sites in the selected region in 64 humans, one chimpanzee, and one orangutan. The human consensus sequence was obtained from the alignment using the Sequence Navigator software. The human ancestral sequence was inferred by comparing the human sequences with the outgroup sequences. The GC contents of the human consensus, human ancestral, chimpanzee, and orangutan sequences are ≈46%, which is substantially higher than the genome average of 42%.
A total of 78 variant sites were originally found from the alignment of human sequences; 24 of them were mutations that were found in only one sequence in the sample (i.e., singletons), 22 were found in two sequences (i.e., doubletons), and 32 were found in more than two sequences (i.e., others). Forty-three sites (≈55%) were verified by RFLP, resequencing, or subcloning. We selected all 24 singletons, for two reasons. First, fluorescence trace reading is usually more difficult for a singleton because there is no other identical or similar trace pattern to compare with. Second, compared with errors for variants that have been observed more than once, an error at a singleton site has a stronger effect on statistical analyses of the data, e.g., the number of segregating sites. We found four errors. One of them was caused by missing a weak signal in the fluorescence trace in one individual, and the correction changed a putative singleton site to a doubleton site. For each of the three other errors, the putative singleton site was found to be actually a nonvariant site. We verified six doubleton sites and found no error. Among the 13 other variant sites verified, 7 errors were found. As these errors occurred at sites with high polymorphism, their effect on the data analyses should be minor. For example, the correction for the error at site 580 (10 As and 118 Ts among the 128 sequences) changed one T to A, so it has no effect on the number of segregating sites and has only a minor effect on heterozygosity.
After verification, the number of the variant sites among the 128 human sequences became 75, including 4 insertions/deletions (indels). On average, ≈8 variant sites per 1,000 bp were found in the region studied. The number and pattern of variant sites in Africans differed from those in non-Africans. The numbers of variant sites in African, Asian, European, and Oceanian sequences were 54, 32, 32, and 20, respectively. Furthermore, there were 31 unique variant sites (including 8 singletons) among the African sequences, whereas there were only 4 (4 singletons), 4 (3 singletons), and 6 (5 singletons) unique sites among the Asian, European, and Oceanian sequences, respectively. In total, the 40 African sequences showed 54 variant sites, whereas the 88 non-African sequences showed only 44 variant sites. Thus, the level of genetic diversity was higher in Africans than in non-Africans.
In Table 1, we compared the numbers of singletons, doubletons, and other types of variant sites in Africans and non-Africans in three data sets: our data, a 10-kb noncoding region in Xq13.3 (6), and a 24-kb gene region in 17q23 (4). The proportion of singletons in Africans and non-Africans was not significantly different in our data (χ2 =2.32, P = 0.13) and in the Xq13.3 region (χ2 = 0.09, P = 0.77) but was significantly different in the gene region in 17q23 (χ2 = 7.66, P = 0.006). We found fewer singletons in Africans than in non-Africans, whereas Rieder et al. (4) found the opposite in 17q23 (Table 1).
Table 1.
Comparison of the numbers of variant sites (including indels) in each DNA region among all human sequences studied, African or African American (Afr. Amer.) sequences, and non-African or European American (Eur. Amer.) sequences
| Type of variant | 10 kb
in 22q11.2
|
10 kb in Xq13.3 (ref. 6)
|
24 kb
in 17q23 (ref. 4)
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| All (128)* | Africans (40) | Non-Africans (88) | All (69) | Africans (23) | Non-Africans (46) | All (22) | Afr. Amer. (10) | Eur. Amer. (12) | |
| Singleton | 20 (2)† | 8 (1) | 12 (1) | 18 (0) | 11 (0) | 7 (0) | 26 | 22 | 4 |
| Doubleton | 23 (1) | 19 (0) | 4 (1) | 5 (0) | 5 (0) | 0 | 10 | 10‡ | 2‡ |
| Others | 32 (1) | 27 (1) | 28 (0) | 10 (0) | 8 (0) | 10 (0) | 42 | 38 | 38 |
| Total | 75 (4) | 54 (2) | 44 (2) | 33 (0) | 24 (0) | 17 (0) | 78 | 70 | 44 |
No. of sequences studied.
The number of the indels included is given in parentheses.
Two of them are shared by African Americans and European Americans.
Interestingly, in our sample doubletons outnumbered singletons, i.e., 23 to 20, whereas under the neutral Wright–Fisher model with a constant population size the expected number of doubletons should be half of that of singletons (16). This high number of doubletons was due to the presence of 19 doubletons in the African samples, in which 9 doubletons were shared by a South African Bantu-speaking black and a Kenyan, and four were shared by a South African Bantu-speaking black and the !Kung individual, although they did not have the same haplotypes. The data suggest substantial recent migration between these African populations.
When singletons and doubletons are considered jointly, there is a strong excess of low frequency variants in all three data sets in comparison with the number of the other types of variants; e.g., 43 (20 + 23) versus 32 in our data set (Table 1). These observations are in sharp contrast to the cases of the dystrophin and PDHA1 genes (3, 5). The excess might largely reflect relatively recent population expansions because such an excess is not expected from an equilibrium Wright–Fisher population. The stronger excess in Africans than in non-Africans might be because Africans have a weaker population subdivision than non-Africans, so that population expansion in Africa has a stronger effect on the number of low frequency alleles.
A comparison of all sequences, including the chimpanzee and orangutan sequences, revealed 463 variant sites, 47 of which were indels. The 463 variant sites were evenly distributed in this region, and the number of variant sites in human populations was also evenly distributed (data not shown).
Among the 47 indels in our data, 21 were single-nucleotide indels, and 5 were two-nucleotide indels. The size distribution of indels is similar to those obtained from 78 human pseudogenes (17) and from primate argininosuccinate synthetase-processed pseudogenes and their noncoding flanking sequences (18). The similar indel-size distribution in the noncoding region and pseudogenes indicates no selection in the region studied.
Mutation Pattern.
By comparing the human, chimpanzee, and orangutan sequences, the direction of 211 mutations could be inferred (data not shown); the proportion of transitional changes was 66%. For the 205 mutations for which the direction could not be inferred, the proportion of transitions was 73%. The overall proportion of transitions was 69%, which is substantially higher than the 59% estimated from pseudogenes (19). For those mutations whose direction could be inferred, the number of G/C to A/T mutations was 101, whereas that of A/T to G/C was 81. After normalization by the GC and AT contents, these numbers become 110 and 75, respectively, which are significantly different (χ2 =6.62, P = 0.01). This result indicates that G/C to A/T mutation occurs more frequently than A/T to G/C mutation, similar to the situation in 13 mammalian pseudogenes (G/C to A/T, 64.5%; A/T to G/C, 35.5%) (20). This is because G and C are in general more mutable than A and T, especially at CpG dinucleotide sites (19).
Neutrality Tests.
The assumption that the region under study is subject to no natural selection was tested using the sequence data. Using the critical points from the 5,000 samples we simulated, we found that Tajima's and Fu and Li's tests could not reject the neutral Wright–Fisher model, regardless of whether or not the indels were included in the tests (Table 2); Fu and Li's other tests (i.e., T, D*, T*) also gave the same conclusion. Note that both tests gave negative test values, indicating an excess of rare variants in the sample. In contrast, for the data from Xq13.3, the test values were statistically significant for rejecting the neutral Wright–Fisher model (Table 2). The rejection of neutrality may indicate linkage of this noncoding region to a functional gene subject to natural selection. In this light, it is interesting to note that the Xq13.3 region has a very low recombination rate (6).
Table 2.
Comparison of results of neutrality tests
| Test | Sample size | θ | Test value (probability) | Critical value (P = 0.05) |
|---|---|---|---|---|
| Present data | ||||
| Tajima's test | 128 | 13.1 | −1.03 (P > 0.1) | −1.36 |
| Fu and Li's D | 128 | 13.1 | −0.95 (P > 0.1) | −1.78 |
| Xq13.3 data | ||||
| Tajima's test | 69 | 6.9 | −1.57* (P < 0.03) | −1.40 |
| Fu and Li's D | 69 | 6.9 | −3.29* (P < 0.05) | −1.86 |
The critical points were obtained from 5,000 simulated samples. The indels were excluded in the tests. θ values were obtained from Watterson's method (10).
Significant at the 5% level.
Nucleotide Diversity.
Nucleotide diversity (π) is defined as the average number of nucleotide differences per site between two randomly chosen sequences from the population. The π value was calculated by the program DNASP 3.0 (21). It was 0.088% among all sequences, 0.085% among the African sequences, and 0.082% among the non-African sequences. Thus, there is virtually no difference in nucleotide diversity between Africans and non-Africans. This observation is in sharp contrast to that at the X-linked PDHA1 locus (3) and precludes a severe bottleneck during the evolution of modern non-Africans; however, a mild bottleneck could not be excluded because non-Africans showed considerably fewer variants than Africans (see above). The π values were 0.085% for Africans, 0.075% for Asians, 0.077% for Europeans, and 0.057% for Oceanians (Table 3). These are similar to the average value (0.11%) for silent sites in coding genes in white Americans (22), supporting the view of low nucleotide diversity in humans. The π value was largest between Africans and Europeans (0.108%) and smallest between Oceanians and Europeans (0.070%) (Table 3).
Table 3.
Nucleotide diversity (%) in different populations and between populations
| Population | African | Asian | European | Oceanian |
|---|---|---|---|---|
| African | 0.085 | |||
| Asian | 0.083 | 0.075 | ||
| European | 0.108 | 0.091 | 0.077 | |
| Oceanian | 0.093 | 0.079 | 0.070 | 0.057 |
The nucleotide diversity in the Xq13.3 noncoding region was low: only 0.033% among all of the sequences studied, 0.035% among the African sequences, and 0.030% among the non-African sequences. In comparison, the π values in the 24-kb region of the angiotensin-converting enzyme gene (17q23) and the 9.7-kb region of the lipoprotein lipase gene (8p22) were, respectively, 0.094% and 0.21% in the noncoding regions and 0.088% and 0.05% in the coding regions, being considerably higher than that in the Xq13.3 noncoding region. In an early study, the nucleotide diversities in the coding regions and their untranslated regions of 49 human genes were 0.049% and 0.036% among white Americans (22). The recent single nucleotide polymorphisms data from 106 and 75 human genes revealed about 15-fold variation of diversity among the genes studied (23, 24). These data suggest that nucleotide diversity varies greatly among genomic regions.
Mutation Rate, Parameter θ, and Effective Population Size Ne.
The average numbers of nucleotide substitutions were 133.8 between human and chimpanzee sequences and 272.8 between human and orangutan sequences. The mutation rates (μ) were estimated to be 1.13 × 10−9 and 1.17 × 10−9 per nucleotide per year by using a divergence time of 6 million years (Myr) between human and chimpanzee and a divergence time of 12 Myr between human and orangutan (Table 4). From these two values, the average mutation rate in this region was estimated to be 1.15 × 10−9 per nucleotide per year or 2.28 × 10−4 per sequence per generation. Different methods were applied to estimate the population parameter θ. It was estimated to be 9.12 by the average mutation rate and an effective population size of 10,000, 13.09 by Watterson's (10) method, and 8.70 by Tajima's (11) method. The higher θ value estimated by Watterson's method is due to an excess of singletons and doubletons.
Table 4.
Comparison of the estimated values of parameter θ, mutation rate μ (per sequence), and effective population size Ne
We used Watterson's and Tajima's θ values to estimate the effective population size Ne. Because the divergence time between human and their outgroups is not certain, we estimated Ne by the mutation rates based on different divergence times. The results were similar to the commonly accepted estimate of 10,000 (25) (Table 4). The lowest value (8,100) suggests that the long-term effective population size of humans is unlikely to be lower than 5,000.
The Age of the MRCA.
As the mutation rate estimated under the assumption of a divergence time of 6 Myr between chimpanzee and human was consistent with that under a divergence time of 12 Myr between orangutan and human, we chose the average mutation rate to compute the age of the MRCA of the human sequences sampled (Table 5). The estimated Ne from our data were close to the most commonly used value 10,000 (25). Based on these parameters, the mode of T (Tmode) was estimated to be 1,288,000 years ago; the mean estimate of T (Tmean) was 1,356,000 years ago; and the 95% confidence interval of T was between 712,000 and 2,112,000 years ago (Table 5, refs. 14 and 15). Tmode is preferred to Tmean because Tmode is the most likely value of T (26). At any rate, both Tmode and Tmean are significantly older than the previous estimates based on the Xq13.3 data (535,000 ± 119,000) and the β-globin gene data (750,000 ± 210,000), although they are younger than the estimate based on the PDHA1 gene data (1.86 Myr) (1, 3, 6). Because the effective population size is larger for an autosomal sequence than for an X chromosomal, mitochondrial, and Y chromosomal sequence, T is likely to be larger for an autosomal region.
Table 5.
The age (T, 103 years) of the MRCA of human sequences
| Sequences | Ne | Tmode | Tmean | 95% Interval |
|---|---|---|---|---|
| All samples | 10,000 | 1,288 | 1,356 | 712∼2,112 |
| 12,000 | 1,104 | 1,203 | 605∼1,949 | |
| 15,000 | 924 | 1,034 | 504∼1,728 | |
| Africans | 6,000 | 1,204 | 1,256 | 694∼1,882 |
| 8,000 | 1,158 | 1,203 | 646∼1,843 | |
| 10,000 | 1,032 | 1,105 | 576∼1,752 | |
| Non-Africans | 6,000 | 747 | 806 | 384∼1,330 |
| 7,000 | 678 | 756 | 353∼1,277 | |
| 8,000 | 634 | 713 | 333∼1,229 |
The average mutation rate (2.28 × 10−4 per sequence per generation) was used.
The genetic diversity patterns in Africans and non-Africans were different, so we estimated the TMRCA values for Africans and non-Africans separately. The TMRCA of Africans was about 0.4 Myr older than that of non-Africans (1,032,000 versus 634,000) (Table 5). However, the TMRCA of non-Africans from our data are older than the fossils for modern humans (see ref. 3) and is also older than those of non-Africans estimated from PDHA1, the β-globin gene, and mitochondrial DNA (1, 3, 8, 27). It indicates that even non-Africans have ancient genetic histories, at least at some regions. Of course, a substantial or large part of the genetic diversity in non-Africans at the region we studied might be due to migration from Africa. However, the genealogical depths at this region and the β-globin gene region (1) in non-Africans and the long separation time between African and non-African sequences at the PDHA1 region (3) suggest that the transformation from archaic to modern humans might have occurred in a subdivided population. This does not contradict the assumption of the African origin of modern humans and also does not imply independent evolution of modern characteristics in separate populations as implied by the multiregional model, because the transformation could have occurred through gene flow and natural selection (3). However, it does suggest that the origin and evolution of modern humans is more complex than depicted by the simple out of Africa model (1, 3), especially the assumption of a complete replacement of all indigenous populations outside of Africa by the African stock.
Acknowledgments
We thank Drs. J. B. Clegg, Marie Lin, Naymkhishing Sambuughin, and Maryellen Ruvolo for DNA samples and the two reviewers and N. Pearson for comments. This work was supported by National Institutes of Health Grants GM55759, GM30998, and GM59290.
Abbreviations
- kb
kilobase
- Myr
millions of years
- RFLP
restriction fragment length polymorphism
- MRCA
most recent common ancestor
- indel
insertions/deletions
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF291587–AF291652).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.200348197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.200348197
References
- 1.Harding R M, Fullerton S M, Griffiths R C, Bond J, Cox M J, Schneider J A, Moulin D S, Clegg J B. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- 2.Nickerson D A, Taylor S L, Weiss K M, Clark A G, Hutchinson R G, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E, Sing C F. Nat Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- 3.Harris E E, Hey J. Proc Natl Acad Sci USA. 1999;96:3320–3324. doi: 10.1073/pnas.96.6.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder M J, Taylor S L, Clark A G, Nickerson D A. Nat Genet. 1999;22:59–62. doi: 10.1038/8760. [DOI] [PubMed] [Google Scholar]
- 5.Zietkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd K K, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, et al. J Mol Evol. 1998;47:146–155. doi: 10.1007/pl00006371. [DOI] [PubMed] [Google Scholar]
- 6.Kaessmann H, Heiâig F, von Haeseler A, Pääbo S. Nat Genet. 1999;22:78–81. doi: 10.1038/8785. [DOI] [PubMed] [Google Scholar]
- 7.Stringer C B, Andrews P. Science. 1988;239:1263–1268. doi: 10.1126/science.3125610. [DOI] [PubMed] [Google Scholar]
- 8.Cann R L, Stoneking M, Wilson A C. Nature (London) 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 9.Don R H, Cox P T, Wainwright B J, Baker K, Mattick J S. Nucleic Acids Res. 1991;19:4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watterson G A. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 11.Tajima F. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y X, Li W H. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y X. Genetics. 1996;144:829–838. doi: 10.1093/genetics/144.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y X, Li W H. Science. 1996;272:1356–1357. doi: 10.1126/science.272.5266.1356. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y X. Theor Popul Biol. 1995;48:172–197. doi: 10.1006/tpbi.1995.1025. [DOI] [PubMed] [Google Scholar]
- 17.Gu X, Li W H. J Mol Evol. 1995;40:464–473. doi: 10.1007/BF00164032. [DOI] [PubMed] [Google Scholar]
- 18.Casane D, Boissinot S, Chang B H J, Shimmin L C, Li W H. J Mol Evol. 1997;45:216–226. doi: 10.1007/pl00006223. [DOI] [PubMed] [Google Scholar]
- 19.Li W H, Wu C-I, Luo C-C. J Mol Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 20.Li W H. Molecular Evolution. MA: Sinauer; 1997. pp. 31–33. , 237–267. [Google Scholar]
- 21.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 22.Li W H, Sadler L. Genetics. 1991;129:513–523. doi: 10.1093/genetics/129.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane C R, Lim E P, Kalyanaraman N, Nemesh J, et al. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 24.Halushka M K, Fan J B, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Nat Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- 25.Takahata N. Mol Biol Evol. 1993;10:2–22. doi: 10.1093/oxfordjournals.molbev.a039995. [DOI] [PubMed] [Google Scholar]
- 26.Fu Y X, Li W H. Mol Biol Evol. 1997;14:195–199. doi: 10.1093/oxfordjournals.molbev.a025753. [DOI] [PubMed] [Google Scholar]
- 27.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A C. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]