

Abstract
Clinical abnormalities in multiple sclerosis (MS) have classically been considered to be caused by demyelination and/or axonal degeneration; the possibility of molecular changes in neurons, such as the deployment of abnormal repertoires of ion channels that would alter neuronal electrogenic properties, has not been considered. Sensory Neuron-Specific sodium channel SNS displays a depolarized voltage dependence, slower activation and inactivation kinetics, and more rapid recovery from inactivation than classical “fast” sodium channels. SNS is selectively expressed in spinal sensory and trigeminal ganglion neurons within the peripheral nervous system and is not expressed within the normal brain. Here we show that sodium channel SNS mRNA and protein, which are not present within the cerebellum of control mice, are expressed within cerebellar Purkinje cells in a mouse model of MS, chronic relapsing experimental allergic encephalomyelitis. We also demonstrate SNS mRNA and protein expression within Purkinje cells from tissue obtained postmortem from patients with MS, but not in control subjects with no neurological disease. These results demonstrate a change in sodium channel expression in neurons within the brain in an animal model of MS and in humans with MS and suggest that abnormal patterns of neuronal ion channel expression may contribute to clinical abnormalities such as ataxia in these disorders.
Keywords: demyelinating diseases, gene transcription, ion channels
Multiple sclerosis (MS) has classically been considered to be a demyelinating disease in which clinical abnormalities are the result of damage to the insulating myelin sheath, which causes axonal conduction block (1, 2). Recently, increased attention has been focused on axonal degeneration in MS, and it has been suggested that axonal loss can produce persistent clinical deficits in MS (3, 4). Demyelination and axonal degeneration, however, do not necessarily underlie all of the clinical abnormalities that are observed in MS. The question of whether there are any changes in neurons that might affect their function in this disorder remains unanswered. In this regard, the possibility of molecular changes within neurons, such as deployment of abnormal repertoires of ion channels that would alter neuronal electrical signaling capability, has not received attention.
The generation and transmission of electrical impulse activity in neurons depend on ion fluxes through voltage-gated sodium channels, which consist of large (230–260 kDa) pore-forming α-subunits and smaller modulatory β-subunits. At least 10 different genes encode distinct sodium channel α-subunits with a common overall motif (5) but with different amino acid sequences that underlie different voltage dependences and kinetic properties. The expression of different sodium channel isotypes in different types of neurons endows them with distinct patterns of electroresponsiveness and impulse generation (6–11).
Sensory neuron-specific sodium channel SNS (alternatively termed PN3, SCN1OA, and Nav 1.8) is a tetrodotoxin-resistant “slow” sodium channel with a depolarized voltage dependence, slower activation and inactivation kinetics (12, 13), and more rapid recovery from inactivation (14, 15) than traditional “fast” channels. As a result of the distinct electrophysiological characteristics of SNS-type channels, the presence of these channels can alter the firing properties of neurons (16, 17). The expression of SNS sodium channels is highly specific within the uninjured nervous system. SNS channels are selectively expressed within primary sensory neurons of dorsal root ganglia (DRG) and the trigeminal ganglion and are not normally expressed within the brain (12, 13, 18, 19). Here we show that SNS sodium channels, which are not present in the cerebellums of control mice, are expressed within cerebellar Purkinje cells of mice with chronic relapsing experimental allergic encephalomyelitis (EAE). We also demonstrate SNS expression within cerebellar Purkinje cells in tissue obtained from patients with MS at autopsy, but not in control subjects with no neurological disease. The present findings demonstrate abnormal ion channel expression, a previously unrecognized molecular abnormality, in neurons within the brain in EAE and MS.
Materials and Methods
Chronic Relapsing EAE.
Chronic relapsing EAE was induced in 6- to 8-week-old Biozzi ABH mice after s.c. injection with 1 mg syngeneic spinal cord homogenate in Freund's complete adjuvant on days 0 and 7, as described previously (20). At the time of killing 60–80 days postinoculation, the injected mice had experienced three paralytic episodes and were in clinical remission. The mice with EAE used in this study demonstrated paresis of the hindlimbs and evidence of spasticity (21) and at this time consistently displayed lesions within the cerebellum, a feature that is not necessarily observed in monophasic EAE or in the early stages of chronic relapsing EAE in which spinal cord lesions are predominant (20). Immunostaining for myelin basic protein revealed patchy involvement of myelin in the cerebellums from mice with EAE examined in this study.
Mice were killed by CO2 intoxication and immediately perfusion-fixed through the heart, first with PBS solution and then with 4% paraformaldehyde in 0.14 M phosphate buffer, pH 7.4 at 4°C. After perfusion fixation, cerebellums were collected and placed in fresh fixative at 4°C. After 2–4 h, the tissue was transferred to a solution containing 4% paraformaldehyde and 30% sucrose in 0.14 M phosphate buffer and stored overnight at 4°C, before being snap-frozen in liquid N2. Fifteen-micrometer sections were cut and placed on poly-l-lysine-coated slides for examination with a Leitz Aristoplan microscope equipped with bright-field and Nomarski optics.
In Situ Hybridization.
Two RNA probes (SNS and SNS-3′-untranslated region) recognizing nucleotide sequences 1,476–2,129 and 6,078–6,443 (GenBank numbering) of rat sodium channel SNS were used to examine SNS mRNA expression in murine tissue (19). The first riboprobe hybridizes to a region within the intracellular loop joining domains I and II, and the second riboprobe hybridizes to a sequence within the 3′-untranslated region. The synthesis of the digoxigenin-substituted riboprobes has been previously described (22). Sections of control, EAE, and MS tissue were processed for in situ hybridization as previously described (17). Briefly, the sections were sequentially incubated in (1) 4% paraformaldehyde in 0.14 M phosphate buffer, 5 min; (2) PBS, three times, 2 min each; (3) proteinase K (10 μg/ml; Boehringer Mannheim, Indianapolis) in 100 mM Tris/50 mM EDTA, pH 8.0, 25 min at 37°C for mouse tissue and 10 min at 37°C for human MS tissue; (4) PBS, 3×, 2 min each; (5) 0.1 M triethanolamine, pH 8.0, 2 min; (6) 0.25% acetic anhydride in 0.1 M triethanolamine, 10 min; (7) 2× saline/sodium citrate buffer (SSC), three times, 2 min each; (8) prehybridization solution containing 50% formamide, 5× SSC, 1× Denhardt's solution, and 100 μg/ml salmon sperm DNA (Sigma), for 1–2 h; (9) hybridization solution containing 50% formamide, 1 mg/ml dextran sulfate, 5× SSC, 1× Denhardt's solution, 100 μg/ml salmon sperm DNA, and 0.5 ng/μl SNS sense or antisense riboprobe overnight at 58°C in a humified chamber; (10) 4× SSC, 5 min; (11) 2× SSC, two times, 10 min each; (12) RNase A (20 μg/ml in 10 mM Tris/500 mM NaCl, pH 8) at 37°C for 30–45 min; (13) 2× SSC, 10 min, two times; (14) 0.2× SSC, 20 min at 56°C, three times; (15) 100 mM Tris/150 mM NaCl, pH 7.5, 2 min; (16) 100 mM Tris/150 mM NaCl, pH 7.5, containing 2% normal sheep serum and 1% BSA, 15 min; (17) anti-digoxigenin antibody conjugated to alkaline phosphatase (Boehringer Mannheim; 1:500 in blocking solution) overnight at 4°C; (18) 100 mM Tris/150 mM NaCl, three times, 5 min each; (19) 100 mM Tris/100 mM NaCl/50 mM MgCl2, pH 9.5, three times, 5 min each; and (20) alkaline phosphatase color substrate (nitroblue tetrazolium/X-phos). The reaction was stopped with 10 mM Tris/1 mM EDTA, pH 8, and the glass slides were cover-slipped with Aqua-Poly/Mount (Polysciences, Warrington, PA).
Immunocytochemistry.
Immunocytochemical methods for detection of SNS protein were performed as previously described (19). Briefly, cryosections ofcontrol and EAE cerebellums and DRG were incubated sequentially in (1) PBS containing 5% normal goat serum, 2% BSA, and 0.1% Triton X-100, 30 min; (2) primary SNS antibody (antibody 10169#1, 1:500), overnight at 4°C; (3) PBS, six times, 5 min each; (4) goat anti-rabbit IgG/biotin, 1:250; (5) PBS, six times, 5 min each; (6) avidin/horseradish peroxidase, 1:250; (7) PBS, six times, 5 min each; (8) 0.4% diaminobenzidine and 0.003% hydrogen peroxide in PBS, 7 min; and (9) PBS containing 0.02% sodium azide. Affinity-purified antibody 10169#1 was generated against the polypeptide sequence CENHQAASPASMMSSEDLAPY corresponding to amino acid residues 1,041–1,062 of rat SNS (12). The antibody yielded no visible staining of control rat brain and spinal cord tissue sections. In addition, antibody 10169#1 stained subpopulations of DRG neurons, and this staining was eliminated by preadsorption with 100–500 molar excess of immunizing peptide.
MS Tissue.
Postmortem tissue was obtained from the NeuroResource Tissue Bank at the Institute of Neurology, London (23); 1 cm3 tissue blocks were placed in OCT mounting medium (R. A. Lamb, London, UK) on cork slides, then gently stirred for 9 s in isopentane precooled in liquid nitrogen before storage in air-tight containers at −70°C, and subsequently studied by using in situ hybridization.
The RNA probe used to examine MS tissue recognizes nucleotides 310–765 (GenBank accession no. NM 006514) of human sodium channel SNS. The synthesis of digoxigenin-substituted riboprobes has been previously described (22). Cryosections of control and MS cerebellums were processed for SNS protein detection in the same manner as EAE cerebellums, with antibody K107 (24), raised against a synthetic peptide corresponding to the SNS C terminus, which reacts with the human antigen.
Results
Cerebellums from 6- to 8-week-old clinically normal control mice and mice with EAE were hybridized with two riboprobes complementary to two distinct sequences of rat sodium channel SNS mRNA. Both probes yielded similar results. Cerebellums from control mice did not display SNS hybridization signal (Fig. 1c), consistent with previous observations in the rat (12, 13, 19). In contrast, there was substantial hybridization signal with both probes for SNS mRNA in Purkinje cells from all (n = 3) mice with EAE that were examined. In most regions, >95% of the Purkinje cells displayed at least moderate-to-heavy SNS signal (Fig. 1a and b). This high level of SNS hybridization signal was not detectable in sections from the sensorimotor cortex or red nucleus of mice with EAE. Only background levels of signal were observed in EAE cerebellums hybridized with sense riboprobes (Fig. 1d).
Figure 1.

Sensory neuron-specific sodium channel SNS is abnormally expressed in cerebellar Purkinje cells of mice with chronic relapsing EAE (a and b; two different mice). SNS mRNA signal was observed in all mice with EAE after hybridization with each of two SNS-specific antisense riboprobes [SNS-3′-untranslated region riboprobe (a); SNS riboprobe (b); n = 3 for each probe). SNS mRNA signal was not detectable in cerebellums from control mice (n = 3) (c) or after hybridization with sense riboprobes (d). (a, ×120; b–d, ×200.)
To show that the up-regulation of SNS was not part of a global increase in the expression of all sodium channels, cerebellums of these mice were also hybridized with a riboprobe specific for sodium channel NaN, a distinct tetrodotoxin-resistant sodium channel that is expressed, like SNS, in DRG and trigeminal ganglion neurons and not in brain (25). NaN hybridization signal was not present in either control or EAE cerebellums (data not shown).
To determine whether transcription of SNS mRNA is paralleled by production of SNS protein, we examined control and EAE cerebellums by using immunocytochemical methods with an antibody directed against SNS (19). Control cerebellums did not display SNS immunostaining (Fig. 2b). In contrast, Purkinje cells in EAE cerebellums exhibited robust immunostaining with the SNS antibody (Fig. 2a). Staining was prominent in most Purkinje cell bodies and extended into the proximal parts of apical dendrites.
Figure 2.
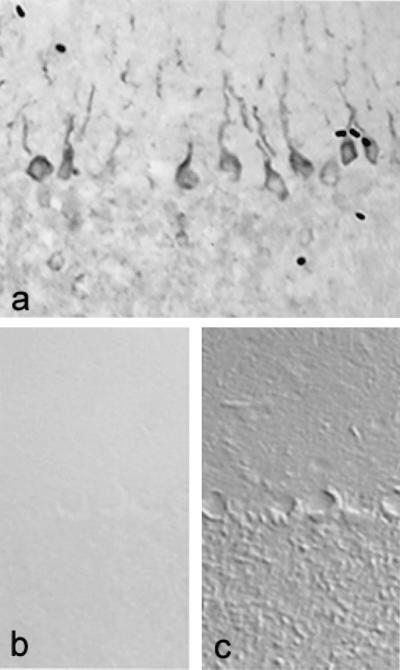
SNS sodium channel protein is abnormally expressed in Purkinje cells of mice with chronic relapsing EAE. SNS immunostaining in Purkinje cells from mice with EAE extends into proximal regions of the apical dendrites (a). SNS is not present in Purkinje cells from control mice (b and c). (b) Bright field image. (c) Nomarski image of the same field as b; note that SNS staining is not present in Purkinje cells from control brains. (×220.)
Postmortem cerebellar samples (death to freezing time, 14–26 h) were studied from three patients (aged 29–55 years) with disabling secondary progressive MS. Disease duration ranged from 7 to 20 years. All of the subjects had displayed cerebellar deficits on examination, and microscopic examination revealed MS plaques in the white matter adjacent to the cerebellar granule cell layer in each case. All of the patients had been treated for spasticity with baclofen, and none had received interferon β. Cerebellums from two control subjects with no neurological disease who died from coronary disease with pulmonary embolism and cardiac arrest (death to freezing time, 7–10 h) were hybridized in parallel with cerebellums from MS patients with a riboprobe specific for human SNS mRNA. Tissue from other parts of the central nervous system was not studied.
SNS mRNA hybridization signal was not present in Purkinje cells of control cerebellums (e.g., Fig. 3c). In contrast, Purkinje cells from two of three subjects with MS exhibited a distinct SNS hybridization signal. In some Purkinje cells, the SNS hybridization signal was intense (Fig. 3a and b). The pattern of Purkinje cells with SNS signal in the MS cerebellum sections suggests that not all Purkinje cells express SNS; clusters of SNS-positive Purkinje cells were separated from other Purkinje cells expressing SNS by distances of 50–500 μm or more. The lack of SNS signal in some MS Purkinje cells may indicate that SNS was not up-regulated in these cells or may reflect lability of SNS transcripts. SNS hybridization signal was not detectable after hybridization with sense riboprobes (Fig. 3d).
Figure 3.

SNS mRNA is abnormally expressed in cerebellar Purkinje cells from brains obtained postmortem from patients with MS. A distinct SNS hybridization signal is present in Purkinje cells from two subjects with MS (a and b) after hybridization with a riboprobe specific for human SNS mRNA, but it was not detectable in tissue obtained from control subjects without neurological disease (c). No SNS hybridization signal is detectable in MS cerebellum after hybridization with sense riboprobe (d). (a, ×95; Inset, ×230; b–d, ×130.)
Having demonstrated the abnormal expression of SNS mRNA in the cerebellum in MS, we next asked whether SNS protein is present. Immunocytochemistry with an antibody directed against SNS (24) demonstrated immunolabeling of Purkinje cells in MS cerebellums (Fig. 4a and b), but not in control cerebellums (Fig. 4c). SNS immunostaining was present in both of the two MS cerebellums studied, including one in which SNS mRNA had not been detected. The pattern of SNS immunostaining in Purkinje cells in cerebellums from MS subjects was similar to that observed for SNS mRNA, with clusters of Purkinje cells displaying distinct SNS immunolabeling.
Figure 4.
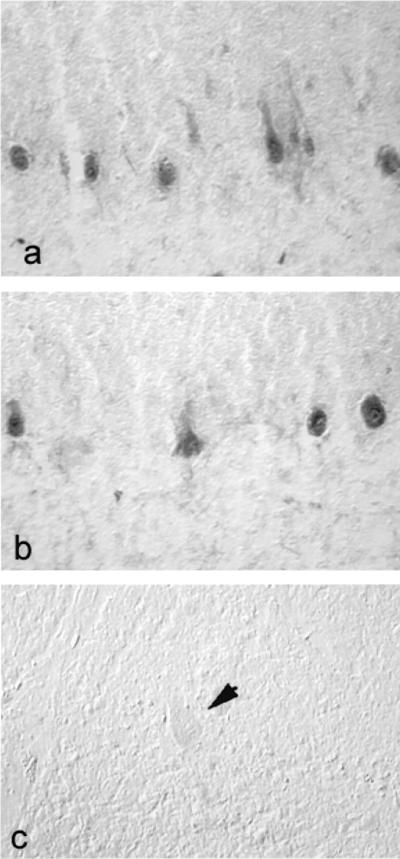
SNS protein is abnormally expressed in Purkinje cells from patients with MS, but not from control subjects without neurological disease. (a and b) SNS immunostaining is present in cerebellar Purkinje cells of subjects with MS. (c) SNS immunolabeling is not present in Purkinje cells from control subjects with no neurological disease (arrow indicates a Purkinje cell). (×220.)
Discussion
Our results demonstrate expression of sodium channel SNS in Purkinje cells within the cerebellum in mice with chronic relapsing EAE and in humans with MS. In the present study, as in earlier studies (12, 13, 18, 19), expression of SNS was not detected within the cerebellum of controls without neurological disease. We observed increased SNS mRNA expression in Purkinje cells in mice with EAE by using two different ribroprobes directed at two nonoverlapping domains within the SNS transcript: one sequence in the coding region and the other in the 3′-untranslated region. These domains were chosen because their nucleotide sequences are distinct from those of other sodium channels. Neither probe yielded detectable hybridization signal in control tissue, and both provided the same result, with antisense (but not sense) probes yielding a distinct SNS signal in Purkinje cells but not in other cell types of the cerebellum in EAE. SNS protein was present in a similar distribution in cerebellums from mice with EAE, but not in control mice. Our observations in MS parallel those in EAE, with Purkinje cells, but no other cell types within the cerebellum, showing aberrant expression of SNS mRNA and protein.
A number of factors are known to influence sodium channel expression and might contribute to up-regulation of SNS expression in EAE and MS. Axonal degeneration is known to occur in EAE (21) and in MS (3, 4), and axotomy has been shown to up-regulate expression of some sodium channels, although down-regulating others, in DRG neurons (26–28) and facial nucleus neurons (29). There is evidence that electrical activity (30, 31) and exposure to neurotrophic factors, such as nerve growth factor and glial-derived nerve growth factor (32–34), may regulate expression of sodium channels including SNS, and these might be altered in response to inflammation of the central nervous system. Changes in SNS gene expression, which result in the insertion of increased numbers of SNS sodium channels within the membrane of DRG neurons, have been observed in association with inflammation within the projection fields of their axons (35). Finally, it is possible that injury to myelin-forming cells and/or demyelination per se may trigger changes in ion channel expression. There is evidence that oligodendrocytes produce membrane-associated (36) and/or soluble protein signal molecules (37) that modulate sodium channel expression in axons. Elevated expression of type II sodium channels has been observed along hypomyelinated axons of shiverer mouse brain, and it has been suggested that the density and location of these channels are regulated by myelination (38). Interestingly, there is abnormal expression of SNS sodium channels in Purkinje cells of the taiep rat, which have lost their myelin as a result of a genetic abnormality that produces degeneration of oligodendrocytes after myelin has formed (19).
The abnormal expression of SNS in Purkinje cells in EAE and MS may have important functional implications. SNS channels produce a slowly inactivating tetrodotoxin-resistant sodium current with relatively depolarized voltage dependence (12, 13). SNS sodium currents in DRG neurons exhibit rapid recovery from inactivation (14) that appears to be due, at least in part, to the presence of a specific tripeptide insertion within the D4S3-S4 linker of the SNS channel (15). Patch–clamp studies have demonstrated that altered SNS mRNA expression in DRG neurons is paralleled by changes in the sodium currents that they produce (16, 39, 40). Introduction of SNS-like sodium channels, together with more conventional rapidly inactivating tetrodotoxin-sensitive sodium channels, changes the input–output properties of neurons, causing them to alter the pattern of impulses that they produce in response to stimuli (17, 41). Purkinje cells without pathological changes produce multiple sodium currents that interact to determine the firing properties of these cells (42, 43). Mutations of sodium channels that are expressed in Purkinje cells result in altered firing behavior in these cells in jolting mice, and it has been suggested that this may produce cerebellar ataxia (44, 45).
In summary, the present results demonstrate abnormal expression of sodium channel SNS, which is not normally detectable within the brain, within Purkinje cells in mice with chronic relapsing EAE and in humans with MS who had exhibited cerebellar deficits. These results demonstrate a previously unrecognized molecular change, i.e., abnormal ion channel expression in neurons within the brain in MS and EAE. It is well established that normal cerebellar functioning depends on precise firing of electrical impulses in Purkinje cells (46). These findings suggest that mistuning of neurons, because of aberrant ion channel expression, may contribute to clinical abnormalities in demyelinating diseases such as multiple sclerosis.
Acknowledgments
We thank G. Pryce and W. Hormuzdiar for technical assistance, S. Tate for antibody K107, and R. Herzog for assistance in the production of the human SNS probes. This work was supported in part by Grant RG-1912 from the National Multiple Sclerosis Society and by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs (to S.G.W.) and from the Multiple Sclerosis Society of Great Britain and Northern Ireland (to D.B.). We also thank the EPVA and the PVA for support, including a gift that supports the Yale–London Collaboration.
Abbreviations
- MS
multiple sclerosis
- DRG
dorsal root ganglia
- EAE
experimental allergic encephalomyelitis
References
- 1.McDonald W I. Brain. 1974;97:179–196. doi: 10.1093/brain/97.1.179. [DOI] [PubMed] [Google Scholar]
- 2.Matthews W B. McAlpine's Multiple Sclerosis. 2nd Ed. Edinburgh: Churchill Livingstone; 1991. pp. 341–370. [Google Scholar]
- 3.McDonald W I, Miller D H, Barnes D. Neuropathol Appl Neurobiol. 1992;18:319–334. doi: 10.1111/j.1365-2990.1992.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 4.Trapp B D, Peterson J, Ransohoff R M, Rudick R, Mork S, Bo L. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 5.Catterall W A. Physiol Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 6.Llinas R. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 7.Honmou O, Utzschneider D A, Rizzo M A, Bowe C M, Waxman S G, Kocsis J D. J Neurophysiol. 1994;71:1627–1638. doi: 10.1152/jn.1994.71.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crill W E. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 9.Stuart G, Sakmann B. Neuron. 1995;5:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 10.Parri H R, Crunelli V. J Neurosci. 1998;18:854–867. doi: 10.1523/JNEUROSCI.18-03-00854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka M, Cummins T R, Ishikawa K, Black J A, Ibata Y, Waxman S G. Proc Natl Acad Sci USA. 1999;96:1088–1093. doi: 10.1073/pnas.96.3.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akopian A N, Sivilotti L, Wood J N. Nature (London) 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 13.Sangameswaran L, Delgado S G, Fish L M, Koch R D, Jakeman L B, Stewart G R, Sze P, Hunter J C, Eglen R M, Herman R C. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 14.Elliott A A, Elliott J R. J Physiol (London) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dib-Hajj S D, Ishikawa I, Cummins T R, Waxman S G. FEBS Lett. 1997;416:11–15. doi: 10.1016/s0014-5793(97)01154-x. [DOI] [PubMed] [Google Scholar]
- 16.Akopian A N, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr B J, McMahon S B, Boyce S, et al. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 17.Schild J H, Kunze D L. J Neurophysiol. 1997;78:3198–3209. doi: 10.1152/jn.1997.78.6.3198. [DOI] [PubMed] [Google Scholar]
- 18.Novakovic S D, Tsoumaka E, McGivern J G, Haraguchi M, Sangameswaran L, Gogas K R, Eglen R M, Hunter J D. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Black J A, Fjell J, Dib-Hajj S, Duncan I D, O'Connor L T, Fried K, Gladwell Z, Tate S, Waxman S G. NeuroReport. 1999;10:913–918. doi: 10.1097/00001756-199904060-00004. [DOI] [PubMed] [Google Scholar]
- 20.Baker D, O'Neill J K, Wilcox C, Gschmeissner S, Butter C, Turk J L. J Neuroimmunol. 1990;28:261–270. doi: 10.1016/0165-5728(90)90019-j. [DOI] [PubMed] [Google Scholar]
- 21.Baker D, Pryce G, Craxford J L, Brown P, Huffman J W, Pertwee R G. Nature (London) 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- 22.Black J A, Dib-Hajj S, McNabola K, Jeste S, Rizzo M A, Kocsis J D, Waxman S G. Mol Brain Res. 1996;43:117–132. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- 23.Newcombe J, Cuzner M L. J Neural Trans. 1993;39:155–163. [PubMed] [Google Scholar]
- 24.Coward K, Plumpton C, Pacer P, Birch R, Carlstedt T, Tate S, Bountra C, Anand P. Pain. 2000;85:41–50. doi: 10.1016/s0304-3959(99)00251-1. [DOI] [PubMed] [Google Scholar]
- 25.Dib-Hajj S D, Tyrrell L, Black J A, Waxman S G. Proc Natl Acad Sci USA. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waxman S G, Kocsis J D, Black J A. J Neurophysiol. 1994;72:466–470. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dib-Hajj S D, Black J A, Felts P A, Waxman S G. Proc Natl Acad Sci USA. 1996;93:14950–14954. doi: 10.1073/pnas.93.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Black J A, Cummins T R, Plumpton C, Chen Y H, Hormuzdiar W, Clare J J, Waxman S G. J Neurophysiol. 1999;82:2776–2785. doi: 10.1152/jn.1999.82.5.2776. [DOI] [PubMed] [Google Scholar]
- 29.Iwahashi Y, Furuyama T, Inagaki S, Morita Y, Takagri H. Mol Brain Res. 1994;22:341–345. doi: 10.1016/0169-328x(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 30.Offord J, Catterall W A. Neuron. 1989;2:1447–1452. doi: 10.1016/0896-6273(89)90190-6. [DOI] [PubMed] [Google Scholar]
- 31.Sashihara S, Waxman S G, Greer C A. NeuroReport. 1997;8:1289–1293. doi: 10.1097/00001756-199703240-00046. [DOI] [PubMed] [Google Scholar]
- 32.Toledo-Aral J J, Brehm P, Halegoua S, Mandel G. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- 33.Dib-Hajj S D, Black J A, Cummins T R, Kenney A M, Waxman S G. J Neurophysiol. 1998;79:2668–2676. doi: 10.1152/jn.1998.79.5.2668. [DOI] [PubMed] [Google Scholar]
- 34.Fjell J, Cummins T R, Dib-Hajj S D, Fried K, Black J A, Waxman S G. Mol Brain Res. 1999;67:267–282. doi: 10.1016/s0169-328x(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka M, Cummins T R, Ishikawa K, Dib-Hajj S D, Black J A, Waxman S G. NeuroReport. 1998;9:967–972. doi: 10.1097/00001756-199804200-00003. [DOI] [PubMed] [Google Scholar]
- 36.Black J A, Waxman S G, Hildebrand C. J Neurocytol. 1985;14:887–907. doi: 10.1007/BF01224803. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan M R, Meyer-Franke A, Lambert S, Bennett V, Duncan I D, Levinson S R, Barres B A. Nature (London) 1997;386:724–728. doi: 10.1038/386724a0. [DOI] [PubMed] [Google Scholar]
- 38.Westenbroek R E, Noebels J L, Catterall W A. J Neurosci. 1992;12:2259–2267. doi: 10.1523/JNEUROSCI.12-06-02259.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cummins T R, Waxman S G. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cummins T R, Dib-Hajj S D, Black J A, Akopian A N, Wood J N, Waxman S G. J Neurosci. 1999;19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elliott J R. Brain Res. 1997;754:221–226. doi: 10.1016/s0006-8993(97)00072-3. [DOI] [PubMed] [Google Scholar]
- 42.Llinás R, Sugimori M. J Physiol. 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raman I M, Bean B P. J Neurosci. 1997;17:4157–4166. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohrman D C, Smith M R, Goldin A L, Harris J, Meisler J M H. J Neurosci. 1996;16:5993–5999. doi: 10.1523/JNEUROSCI.16-19-05993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raman I M, Sprunger L K, Meisler M H, Bean B P. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 46.Ito M. The Cerebellum and Motor Control. New York: Raven; 1982. [Google Scholar]