

Abstract
To explore how distal mutations affect binding sites and how binding sites in proteins communicate, an ensemble-based model of the native state was used to define the energetic connectivities between the different structural elements of Escherichia coli dihydrofolate reductase. Analysis of this model protein has allowed us to identify two important aspects of intramolecular communication. First, within a protein, pair-wise couplings exist that define the magnitude and extent to which mutational effects propagate from the point of origin. These pair-wise couplings can be identified from a quantity we define as the residue-specific connectivity. Second, in addition to the pair-wise energetic coupling between residues, there exists functional connectivity, which identifies energetic coupling between entire functional elements (i.e., binding sites) and the rest of the protein. Analysis of the energetic couplings provides access to the thermodynamic domain structure in dihydrofolate reductase as well as the susceptibility of the different regions of the protein to both small-scale (e.g., point mutations) and large-scale perturbations (e.g., binding ligand). The results point toward a view of allosterism and signal transduction wherein perturbations do not necessarily propagate through structure via a series of conformational distortions that extend from one active site to another. Instead, the observed behavior is a manifestation of the distribution of states in the ensemble and how the distribution is affected by the perturbation.
Most cellular processes, which are facilitated by proteins, are modulated by effectors. The basic features of such a mechanism of regulation are the presence of multiple binding sites for various ligands and communication between these binding sites, which often are situated many angstroms apart. An understanding of the ground rules of this regulatory mechanism requires a quantitative definition of the functional linkages between these binding sites. In 1964, Wyman introduced the thermodynamic concept of linked functions to establish a quantitative formulation for describing the mutual influence of binding sites on each other (1). Linkage theory is based on thermodynamic principles, is applicable to all biological systems, and exhibits quantitative predictive power. Although Wyman's theory provides the mathematical relationships, it does not address the mechanism through which different binding sites communicate. Thus, besides functional linkage, there are underlying structural–thermodynamic linkages that define the mechanism of site–site communication.
Despite a significant body of literature showing that information is transmitted through biological systems via a series of inter- and intramolecular communication events (refs. 2–5 and references therein), a quantitative predictive theory of structural linkage analogous to the Wyman Linkage Theory is not available. Recently, however, a theoretical approach was established to treat the native state of a protein as an ensemble of conformational states (6–9). A consequence of this approach is the ability to identify a number of fundamental aspects of cooperativity in proteins (9). We report the application of this approach to Escherichia coli dihydrofolate reductase (DHFR) (10–22) to investigate the mechanism of communication between binding sites and the susceptibility of these binding sites to mutations at other regions in the protein. The ensemble-based approach successfully captures the observations reported in the literature on site–site interaction and long-range mutational perturbations.
Results and Discussion
The Native State of DHFR Is an Ensemble.
Proteins under native conditions undergo both small- and large-scale conformational fluctuations that define the native-state ensemble (6). In studying these fluctuations, we developed the corex algorithm to model the native state (6–8). By using the high-resolution structure as a template, the corex algorithm generates a large number of different conformational states through the combinatorial unfolding of a set of predefined folding units (6). By means of an incremental shift in the boundaries of the folding units, an exhaustive enumeration of partially unfolded states is achieved for a given folding unit size. The Boltzmann weight of each state [Ki = exp(−ΔGi/RT)] is determined from the calculated Gibbs energy (23–29), and the probability of each state (Pi) is determined by:
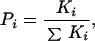 |
1 |
where the summation is over all states in the ensemble.
From the probabilities calculated in Eq. 1, an important statistical descriptor of the equilibrium is determined for each residue in the protein (6–8). Defined as the residue stability constant, κf,j, this descriptor is the ratio of the summed probability of all states in the ensemble in which a particular residue j is in a folded conformation (ΣPf,j) to the summed probability of all states in which that residue is in an unfolded conformation (ΣPnf,j):
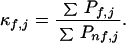 |
2 |
The important feature of the residue stability constants is that they provide a measure of the local stability around each residue that can be experimentally verified by comparison to hydrogen exchange protection factors (6–9).
The energetic connectivities in DHFR were investigated by performing a corex analysis (6–8) on the crystal structure of the DHFR⋅folate⋅NADP+ ternary complex [Protein Data Bank accession no. 7dfr (19)] with the folate and NADP+ molecules removed. To achieve a higher-resolution analysis than previously described (6), the corex algorithm was modified to use a Monte Carlo sampling strategy (30). In separate analyses, the Monte Carlo sampling was shown to provide equivalent results to those obtained from the original corex sampling (data not shown). Within the context of this Monte Carlo sampling, the residue stability constant (Eq. 2) becomes:
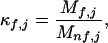 |
3 |
where Mf,j and Mu,j are the weighted number of states that residue j is folded or unfolded.
The residue stability constants for DHFR (Eq. 3) are shown in Fig. 1A. High stability constants signify residues that are folded in the majority of highly probable states under native conditions, whereas lower stability constants signify residues that are unfolded in many of those states (6–8). DHFR is characterized by regional variations in stability (Fig. 1A), even under native conditions. In general, residues with higher stability constants lie in β strands 1, 6, and 8 and α helices 1, 2, and 4, whereas lower stability constants are found in several of the loop regions that separate the elements of regular secondary structure. Against this background, special note is made of the region spanning residues 60–90, which is the least stable portion of DHFR even though it contains α-helix 3 and β-strand 4. An interesting feature of DHFR (Fig. 1A) is that a subset of residues located in unstable regions are involved with either NADPH (e.g., 62–65, 76–78) or folate (e.g., 94) binding.
Figure 1.

The ensemble of DHFR conformations. (A) Calculated residue stability constants for DHFR. The locations of secondary structural elements are indicated. Residues involved in folate (triangles) and NADP (squares) are also indicated. (B) Time-averaged (or single-molecule) representation of residue stability constants color coded on the high-resolution structure (19). Residues shown in red are stable and fluctuate the least, whereas residues shown in yellow fluctuate the most. Shades intermediate between red and yellow represent moderately stable regions. (C) Ensemble-averaged (or instantaneous) representation showing the most probable conformational states in the DHFR ensemble. Residues shown in red are folded in each state, whereas residues shown in yellow are unfolded. This figure was prepared by using the program molmol (38).
The stability constants (Fig. 1A) are depicted in two ways. In the time-averaged representation (Fig. 1B), some regions are seen as being more flexible or less stable than other regions. If a single molecule were to be viewed over a time course, regions colored yellow in Fig. 1B would be observed to fluctuate to a greater extent than those colored red. The ensemble-averaged representation (Fig. 1C) provides the equilibrium distribution of states in the ensemble that would be obtained at any instant; the regions colored red in each state are folded, whereas residues colored yellow are unfolded. Fig. 1 highlights the notion that proteins are dynamic molecules that undergo both small- and large-scale conformational fluctuations. Even though the probability of these fluctuations is minute, such that they can be detected only by hydrogen exchange experiments, they are nonetheless captured by our analysis as demonstrated previously (6–9).
An Ensemble View of Cooperativity.
Cooperativity in proteins is the result of energetic coupling between different regions. Within the context of an ensemble-based description of the equilibrium, cooperativity is manifested in the relative probabilities of the different states in the ensemble. For regions that are highly coupled energetically, the probability of states in which both regions are folded or unfolded is greater than the probability of states in which only one is folded. Thus, as the corex algorithm provides reasonable estimates for the probabilities of the different states in the ensemble (6–9), it captures, albeit implicitly, the network of cooperative interactions in the protein. This fact implies that the cooperativity between different structural and functional elements of a protein can be ascertained through an analysis of those regions of the protein that are folded in the states with the highest probabilities. If two residues, j and k, are both folded or unfolded in the majority of highly probable states, the residue stability constants (Eq. 3) will be identical for both residues. As such, a perturbation that affects residue j by a specific amount will necessarily affect residue k by that same amount. This reasoning can be extended to investigate the energetic coupling between groups of residues as well. Here we define the coupling between two residues or groups of residues as the connectivity, and we identify two distinct types of connectivity. The first describes how changes at individual residues are propagated (i.e., point mutations), whereas the other describes how changes to groups of residues are propagated (i.e., site-specific binding).
Residue-Specific Connectivities.
In describing the coupling between two residues, we introduce the residue-specific connectivity (RSC). In the context of the Monte Carlo sampling method used here (30), we use the correlation function to define the RSC as:
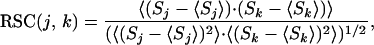 |
4 |
where Sj and Sk denote the folding state of residue j and k (if a residue is folded in a particular state, S = 1; if the residue is unfolded, S = −1), and < Sj > and < Sk > denote the average folding state of residues j and k over the ensemble. A positive value of the RSC indicates that a stabilization of residue j or k results in a stabilization of residue k or j (i.e., they display positive cooperativity), whereas a negative value indicates a stabilization of residue j or k leads to a destabilization of residue k or j (i.e., they display negative cooperativity). A value of zero means there is no correlation, and the residues are not energetically coupled.
Because Eq. 4 provides the mutual susceptibility of each residue to perturbations at every other residue, it can be used to probe the thermodynamic domain structure in proteins (9). For DHFR, the RSCs shown in Fig. 2 reveal two energetic domains. The major domain consists of the N- and C-terminal portions of DHFR (labeled A and C in Fig. 2). These regions are distal in sequence but are proximal in the three-dimensional structure and are energetically coupled, as indicated by the positive RSCs in the region labeled D. The second or minor domain consists of residues ≈60–90 (labeled B in Fig. 2). The energetic domain assignments described here agree well with those identified from structural analyses (19–22).
Figure 2.

Calculated RSC for each pair of residues in DHFR. Red corresponds to a large positive energetic connectivity, blue to the smallest connectivity, and purple to a negative energetic connectivity, as indicated in the color bar. Residues 1–34 (labeled A), residues 60–90 (labeled B), and residues 110–159 (labeled C) each behave cooperatively. Additionally, residues 1–34 and 110–159, although distal in sequence, are also positively cooperative (labeled D).
In addition to providing thermodynamic domain assignments, the RSCs can also be used to explore how mutational effects propagate from their point of origin. This is highlighted in Fig. 3, which shows the correlation of three different residues in DHFR (22, 67, and 145) to the rest of the structure. As seen in the figure, there are differences in both the magnitude and the extent to which mutational effects propagate. Residue 22 (Fig. 3A), for example, is energetically coupled to a large number of residues in the major domain, whereas residues 67 (Fig. 3B) and 145 (Fig. 3C) are coupled to a much smaller number of residues in the minor and major domain, respectively.
Figure 3.
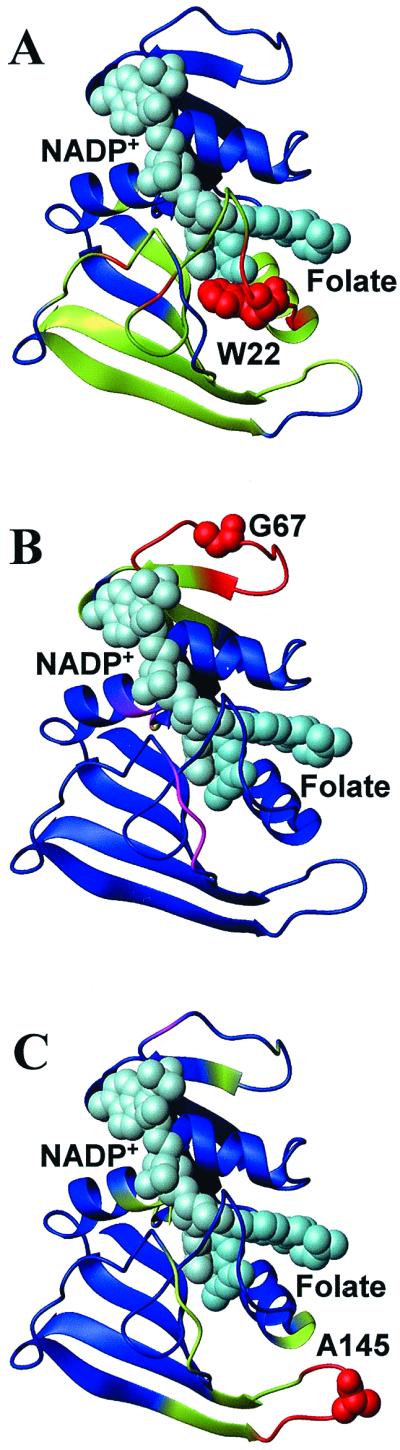
Ribbon representation of the crystal structure of DHFR with NADP+ (light blue) and folate (dark blue). Mutated residues are labeled and are shown as van der Waals' surfaces. DHFR is color coded according to the magnitude of the RSCs to residues (A) 22, (B) 67, and (C) 145; red is the greatest effect, green is intermediate, blue is the least, and purple/fuchsia indicates the negative effect. This figure was prepared by using the program molmol (38).
One of the hallmark features of DHFR is the experimental observation that mutations at distal regions of the protein often propagate many angstroms through the structure and preferentially affect the affinity of DHFR for either NADPH or folate (14–18). We are interested in whether a correlation exists between how each binding site is affected by a mutation (Fig. 3) and the experimentally observed effect on binding. In general, it is not expected that the effect of a mutation on the stability of a binding site should necessarily scale with the change in binding affinity. Nonetheless, as shown in Fig. 3 and Table 1, mutational effects in DHFR do propagate over many angstroms, and the degree to which a particular binding site is energetically coupled to the mutated residue correlates with the effect of that mutation on the affinity for each ligand. Although this correlation clearly provides no mechanistic details about how binding affinity is affected by the mutation, the correlation in behavior supports the accuracy of the connectivity information provided by Eq. 4 and demonstrates the ability of the algorithm to map effects over long distances.
Table 1.
Comparison of experimentally measured effects of mutations on ligand binding with the computed effect on residue-specific connectivity between the mutated residues and each binding site
| Mutants | Distance, Å*
|
Relative effect on Km† | Relative effect on κf of binding-site residues‡ | |
|---|---|---|---|---|
| Folate | NADPH | |||
| 22 | 14 | 18 | Folate > NADPH | Folate > NADPH |
| 67 | 23 | 17 | NADPH > folate | NADPH > folate |
| 113 | 12 | 18 | Folate > NADPH | Folate > NADPH |
| 121 | 19 | 15 | NADPH > folate | NADPH > folate |
| 145 | 21 | 28 | No effect | No effect |
The distance between a mutated residue and ligand-binding sites is the average distance between the mutated residue and all the residues in folate or NADPH-binding site.
Measured effects of mutation on Km for folate and NADPH (10–18), e.g., folate > NADPH means the effect of mutation on Km, for folate is larger than the effect on Km for NADPH.
The predicted effects of mutations on ligand-binding sites are obtained by averaging RSC values between the mutation site and each residue in the folate or NADPH-binding site (effectx = (∑i=1nx RSC(j, k))/nx), where k is the mutated residue, and the summation is over all nx residues in the binding site for ligand x.
Functional Connectivities.
Experimentally, it has been established that binding in either the NADPH- or folate-binding sites affects the affinity of DHFR for the other ligand (22). The nature of this effect can be cast in terms of the ensemble. As noted, the ensemble of DHFR conformations that exist under native conditions in the absence of both folate or NADPH is characterized by states in which many of the loop regions are unfolded, a subset of which are involved in binding to either ligand. In the presence of either ligand, however, those states whose binding-site residues are folded will bind ligand and will be preferentially stabilized over those states in which all or part of the active site is unfolded (31). Thus, in addition to the pairwise correlations described by the RSCs, it is necessary to identify those joint correlations between the binding site as a whole and the rest of the protein. Accordingly, we define the functional connectivity (FC) as the connectivity between the entire binding site for a given ligand, x, and every residue in the protein:
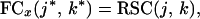 |
5 |
where j* and k* are defined in one of two ways. For all residues, j and k, not involved in the binding pocket for ligand x, Sj* = Sj and Sk* = Sk in Eq. 4. For residues in the binding site for ligand x, however, Sj* and/or Sk* are the average folding states over all of the nx residues in the binding site for ligand x:
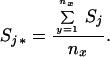 |
6 |
The difference between RSCs and FCs is noteworthy. RSCs are determined by correlating the probabilities of states in which two particular residues are folded, independent of the folded state of other residues. FCs, on the other hand, correlate the probability of a residue being folded with the probability of a group of residues being folded. As such, FCs effectively amplify connectivity information that is often not seen in an analysis of the RSCs. This result highlights the difficulties in experimentally deriving the functional connectivities in proteins from the effects of single site mutants.
Figs. 4 and 5 show the FCs for the NADPH- and folate-binding sites, respectively. In general, the two binding sites are positively coupled, as the binding site of either ligand shows positive FCs to many of the residues of the other binding site. Consistent with experimental results (22), this observation is indicative of positive cooperativity between the sites. It is also noted that, although the connectivity is greatest near the affected site, some of the affected residues are distal in structure. For example, in the folate FCs (Fig. 5) there is a significant effect in the loop containing residues 63–68. The negative values imply that stabilization of the folate-binding site is concomitant with a decrease in the stability of that loop. The predicted destabilization of the loop on binding ligand at the folate site is especially intriguing, as it has been observed experimentally in the same region. Specifically, in x-ray crystallographic analyses (16, 20–21), the binding of methotrexate to DHFR (at the folate site) was shown to induce a decrease in B factors throughout the protein, while largely increasing the mobility of the loop consisting of residues 63–72.
Figure 4.

FCs of the NADPH site. (A) FCs of the NADPH site showing the residues involved in NADP+ and folate binding as orange and green triangles. (B) High-resolution structure of DHFR (19) with van der Waals' surface of NADP+ in its respective binding site. The structure is color coded according to the magnitude of the FCs in A. Red corresponds to a positive FC, blue to no FC, and purple/fuchsia to a negative FC, as indicated in the color bar in Fig. 2. This figure was prepared by using the program molmol (38).
Figure 5.

FCs of the folate site. (A) FCs for the folate-binding site represented as described in Fig. 4 for the NADPH site. (B) High-resolution structure of DHFR (19) with van der Waals' surface of folate in its respective binding site. The negative FCs between the folate site and residues 64–68 (labeled A) are highlighted in B. Note the absence of a propagation pathway from the folate site. This figure was prepared by using the program molmol (38).
The importance of this result is 3-fold. First, the affected loop is more than 15 Å from the folate-binding site, indicating that the ensemble approach adequately models the propagation of binding energy through DHFR. Second, the residues that surround the affected loop (Fig. 5B) show no connectivity to the folate site, indicating that residues can be energetically coupled in the absence of a visible connectivity pathway. This result undermines the mechanical view of signal propagation wherein binding or mutational effects are propagated to distal parts of the protein through a series of conformational distortions. Third, and equally important, these results contradict the classic view that binding “freezes out” protein conformations, resulting in a decrease in motion (or dynamics) (32). The experimental observation that binding can increase dynamics in some systems (20–21, 33–37) emphasizes the importance of entropic contributions. The success of our approach in capturing this effect implies that the entropic contributions are adequately represented in the ensemble view as implemented by the corex algorithm (6–9).
Although the approach presented here contains many simplifying assumptions, which includes the fact that only the structure of the ternary complex (7dfr) was analyzed, the algorithm is nonetheless successful in capturing significant details regarding the energetic connectivities in DHFR. The reason for this success is rooted in the origins of energy propagation. As Eqs. 4 and 5 reveal, it is the number and relative probability of the low-energy states that determine the magnitude and extent to which binding and mutational effects are propagated throughout the structure. For DHFR, the differences in Ln κf between residues 60–90 and the rest of the protein (Fig. 1A) indicate that states with this region unfolded are greater than 7.0 kcal/mol more stable than states with other regions unfolded. Consequently, most mutational effects, which rarely account for more than 2 kcal/mol, are unlikely to significantly affect the energetic hierarchy of states and thus the connectivity pattern. It is this relationship between the distribution of states in the ensemble and the connectivities between the different functional elements that allows proteins to tolerate significant sequence diversity while maintaining complex biological behavior.
Conclusions
The propagation of energy through three-dimensional structure represents the physical basis for allosterism and signal transduction in biological systems. The success of our ensemble-based approach in modeling this behavior has two significant implications. First and most importantly, whereas Wyman's linkage theory provides the mathematical relationships governing site–site interactions, the ensemble-based theory described here (6–9, 31) provides the mechanism through which the structure is linked with the observed phenomena.
Second, as noted above, the effects seen in DHFR are the result of changes in states that are only minutely populated. Thus, our analysis suggests that a complete understanding of energy propagation in biological macromolecules cannot be reconciled only in terms of the bonds that are made or broken within the context of one or even a few conformational states. Instead, this phenomenon is more accurately viewed as resulting from a redistribution of the conformational ensemble in response to binding. In thermodynamic terms, this behavior results not only from enthalpic contributions, which are easily identified through inspection of the high-resolution structure (i.e., changes in the numbers and types of interactions), but also from significant entropic (possibly dynamic) contributions, which are difficult, if not impossible, to deduce from structure alone.
Finally, signal propagation in biological systems occurs through a series of inter- and intramolecular events; a molecule binds ligand, and the energy of binding propagates through the structure and affects a second site. In this study, we have investigated how two binding sites on the same protein are energetically connected by effectively decoupling binding from propagation. Our approach correlates the behavior of each residue to the respective binding sites as if each binding site behaved as a whole. In reality this need not be the case: different parts of the binding site may behave independently. In ensemble terms, states with only parts of the active site folded may retain significant affinity, and these states may have functional relevance. Indeed, the large stability difference between many active site residues in DHFR suggests that the energy of folding certain parts of the active site may not be compensated to the same extent by the binding energy. The significance of these states awaits further study.
Acknowledgments
This work was supported by National Science Foundation Grant no. 9875689 (V.J.H.), by National Institutes of Health Grant GM-45579, and by Robert A. Welch Foundation Grants H-0013 and H-1238 (J.C.L. and H.P.). We thank Drs. Ernesto Freire and James O. Wrabl for helpful discussions, Drs. Xiaodun Jing, Zhaoqing Zhang, and Ping Sheng for stimulating discussions on Monte Carlo sampling methods and the development of Eq. 4. Dr. John Wooll, Dr. Mark White, Emily Platzer, and Lisa Pipper for technical assistance, and Dr. Robert O. Fox for critical evaluation of the manuscript.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Abbbreviations: DHFR, dihydrofolate reductase; RSC, residue-specific connectivity; FC, functional connectivity.
See commentary on page 11680.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.220240297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.220240297
References
- 1.Wyman J. Adv Protein Chem. 1964;19:223–286. doi: 10.1016/s0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 2.Di Cera E. Adv Protein Chem. 1998;51:59–119. doi: 10.1016/s0065-3233(08)60651-8. [DOI] [PubMed] [Google Scholar]
- 3.Ackers G K. Adv Protein Chem. 1998;51:185–253. doi: 10.1016/s0065-3233(08)60653-1. [DOI] [PubMed] [Google Scholar]
- 4.Di Cera E. Thermodynamic Theory of Site-Specific Binding Processes in Biological Macromolecules. Cambridge CB2 1RP: Syndicate of the University of Cambridge; 1995. [Google Scholar]
- 5.Wyman J S, Gill J. Binding and Linkage: The Functional Chemistry of Biological Macromolecules. Mill Valley, CA: Univ. Sci. Books; 1990. [Google Scholar]
- 6.Hilser V J, Freire E. J Mol Biol. 1996;262:756–772. doi: 10.1006/jmbi.1996.0550. [DOI] [PubMed] [Google Scholar]
- 7.Hilser V J, Freire E. Proteins Struct Funct Genet. 1997;27:171–183. doi: 10.1002/(sici)1097-0134(199702)27:2<171::aid-prot3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 8.Hilser V J, Townsend B D, Freire E. Biophys Chem. 1997;64:69–79. doi: 10.1016/s0301-4622(96)02220-x. [DOI] [PubMed] [Google Scholar]
- 9.Hilser V J, Dowdy D, Oas T G, Freire E. Proc Natl Acad Sci USA. 1998;95:9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Falzone C J, Wright P E, Benkovic S J. Biochemistry. 1992;32:7826–7833. doi: 10.1021/bi00149a012. [DOI] [PubMed] [Google Scholar]
- 11.Falzone C J, Wright P E, Benkovic S J. Biochemistry. 1994;33:439–442. doi: 10.1021/bi00168a007. [DOI] [PubMed] [Google Scholar]
- 12.Epstein D M, Benkovic S J, Wright P E. Biochemistry. 1995;34:11037–11048. doi: 10.1021/bi00035a009. [DOI] [PubMed] [Google Scholar]
- 13.Sawaya M R, Kraut J. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 14.Fierke C A, Benkovic S J. Biochemistry. 1989;28:478–486. doi: 10.1021/bi00428a011. [DOI] [PubMed] [Google Scholar]
- 15.Warren M S, Brown K A, Farnum M F, Howell E E, Kraut J. Biochemistry. 1991;30:11092–11103. doi: 10.1021/bi00110a011. [DOI] [PubMed] [Google Scholar]
- 16.Ohmae E, Iriyama K, Ichihara S, Gekko K. J Biochem. 1996;119:703–710. doi: 10.1093/oxfordjournals.jbchem.a021299. [DOI] [PubMed] [Google Scholar]
- 17.Cameron, C. E. & Benkovic, S. J. Biochemistry36, 15792–15800. [DOI] [PubMed]
- 18.Ohmae E, Ishimura K, Iwakura M, Gekko K. J Biochem. 1998;123:839–846. doi: 10.1093/oxfordjournals.jbchem.a022013. [DOI] [PubMed] [Google Scholar]
- 19.Bystroff C, Oatley S J, Kraut J. Biochemistry. 1990;29:3263–3277. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- 20.Bolin J T, Filman D J, Matthews D A, Hamlin R C, Kraut J. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 21.Bystroff C, Kraut J. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- 22.Fierke C A, Johnson K A, Benkovic S J. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 23.D'Aquino J A, Gómez J, Hilser V J, Lee K H, Amzel L M, Freire E. Proteins Struct Funct Genet. 1996;25:143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 24.Gómez J, Hilser V J, Xie D, Freire E. Proteins Struct Funct Genet. 1995;22:404–412. doi: 10.1002/prot.340220410. [DOI] [PubMed] [Google Scholar]
- 25.Xie D, Freire E. J Mol Biol. 1994;242:62–80. doi: 10.1006/jmbi.1994.1557. [DOI] [PubMed] [Google Scholar]
- 26.Murphy K P, Freire E. Adv Protein Chem. 1992;43:313–361. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- 27.Baldwin R L. Proc Natl Acad Sci USA. 1986;83:8069–8072. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K H, Xie D, Freire E, Amzel L M. Proteins Struct Funct Genet. 1994;20:68–84. doi: 10.1002/prot.340200108. [DOI] [PubMed] [Google Scholar]
- 29.Habermann S M, Murphy K P. Protein Sci. 1996;5:1229–1239. doi: 10.1002/pro.5560050702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 31.Freire E. Proc Natl Acad Sci USA. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Froloff N, Windemuth A, Honig B. Protein Sci. 1977;6:1293–1301. doi: 10.1002/pro.5560060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akke M, Skelton N J, Kordel J, Palmer A G, Chazin W J. Biochemistry. 1993;32:9832–9844. doi: 10.1021/bi00088a039. [DOI] [PubMed] [Google Scholar]
- 34.Olejniczak E T, Zhou M M, Fesik S W. Biochemistry. 1997;36:4118–4124. doi: 10.1021/bi963050i. [DOI] [PubMed] [Google Scholar]
- 35.Yu L, Zhu C X, Tse-Dinh Y C, Fesik S W. Biochemistry. 1996;35:9661–9666. doi: 10.1021/bi960507f. [DOI] [PubMed] [Google Scholar]
- 36.Stivers, J. T., Abeygunawardana, C. & Mildvan, A. S. Biochemistry35, 16036–16047. [DOI] [PubMed]
- 37.Zidek L, Novotny M V, Stone M J. Nat Struct Biol. 1999;6:1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]
- 38.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]