

Abstract
Fibulin is a broadly conserved component of the extracellular matrix (ECM). Previous studies have shown that Caenorhabditis elegans FIBULIN-1 (FBL-1) controls the width of the gonad (Hesselson, D., C. Newman, K.W. Kim, and J. Kimble. 2004. Curr. Biol. 14:2005–2010; Kubota, Y., R. Kuroki, and K. Nishiwaki. 2004. Curr. Biol. 14:2011–2018; Muriel, J.M., C. Dong, H. Hutter, and B.E. Vogel. 2005. Development. 132: 4223–4234). In this study, we report that FBL-1 also controls developmental growth and that one isoform of fibulin-1, called FBL-1C, controls both functions by distinct mechanisms. A large FBL-1C fragment, including both epidermal growth factor (EGF) and fibulin-type C domains, is responsible for constraining gonadal width, but a much smaller fragment containing only two complete EGF repeats (EGF1-2C+) is critical for developmental growth. We suggest that the larger fragment serves a scaffolding function to stabilize the basement membrane and that the smaller fragment provides a regulatory function at the cell surface or within the ECM to control growth.
Introduction
The ECM is an important source of developmental cues. ECM proteins can stimulate cell migration (Giannelli et al., 1997), regulate the availability of signaling molecules (Kirkpatrick et al., 2004; Kreuger et al., 2004), and promote proliferation and differentiation (Nikolova et al., 2006). Fibulin-1 is a broadly conserved ECM protein (Timpl et al., 2003). However, functional analyses of this conserved class of ECM proteins remain in their infancy.
The Caenorhabditis elegans FIBULIN-1 (FBL-1) protein is homologous to mammalian Fibulin-1 (Barth et al., 1998). Both proteins share a common domain architecture with an N-terminal signal sequence, three anaphylatoxin (AT) repeats, nine EGF-like repeats, and an alternatively spliced fibulin-type (FIB) C-terminal domain (Fig. 1 A). The AT domain is related to inflammatory peptides that are released during activation of the complement system (Kohl, 2001). EGF-like repeats often mediate protein–protein interactions and can function as signaling ligands (Prigent and Lemoine, 1992). The mammalian Fibulin-1 EGF domain is required for binding to other basement membrane proteins, including nidogen-1 and fibronectin (Sasaki et al., 1995; Tran et al., 1997). In C. elegans, the second EGF repeat is alternatively spliced, generating a choice between a canonical EGF repeat (EGF2C) and a divergent EGF repeat (EGF2D) with a 39-aa insertion. The FIB domain defines the fibulin family of proteins, which includes five proteins in mammals but only FBL-1 in C. elegans. Mammalian and nematode Fibulin-1 homologues share the C and D alternatively spliced FIB domains (Barth et al., 1998). The FIB domain may control the specificity of physical interactions because mammalian Fibulin-1C has a 30-fold higher affinity than Fibulin-1D for nidogen-1 (Sasaki et al., 1995).
Figure 1.

A fbl-1 null allele. (A, top) fbl-1 gene modified from Muriel et al. (2005). Alternative splicing of exons C and D generates FBL-1C and -1D. (bottom) FBL-1C and -1D proteins. (B) Duration of larval development, measured in hours from L1 to mid-L4. n = 25. (C) Adult body size measured in micrometers squared. n = 10. (B and C) Data are given as the mean ± SEM (error bars). (D–F) Mid-L4 larvae; genotypes are given in the figure. Lines indicate the gonadal width. Arrowheads point to the distal ends of gonads. Bars, 20 μm.
Targeted deletion of mouse fibulin-1 has revealed a key role in development. fibulin-1 knockout mice have capillary defects, resulting in extensive hemorrhaging and perinatal death (Kostka et al., 2001). FBL-1 deregulation may lead to human disease. A balanced translocation that alters the ratio of the C and D forms of FBL-1 is linked to polydactyly, implicating FBL-1 in digit development (Debeer et al., 2002). FBL-1 may also play a role in cancer metastasis. The overexpression of FBL-1D but not -1C reduces the invasiveness of human fibrosarcoma cells in immunodeficient mice (Qing et al., 1997). Thus, it is likely that alternative splicing generates FBL-1 isoforms with specialized biological activities.
fbl-1 has multiple roles in C. elegans development. fbl-1 mutants are sterile with expanded somatic gonads (Hesselson et al., 2004; Kubota et al., 2004; Muriel et al., 2005), which is reminiscent of the enlarged capillaries found in fibulin-1 knockout mice. fbl-1 mutants are smaller than wild-type animals in overall body size and progress through larval stages at a slower rate than wild-type animals. These defects may reflect structural abnormalities in the feeding and digestive organs, the pharynx, and intestine, respectively (Muriel et al., 2005). The fbl-1 gene is expressed in the intestine (Hesselson et al., 2004; Kubota et al., 2004; Muriel et al., 2005), whereas gon-1 is expressed in both body wall muscle and distal tip cells (Blelloch and Kimble, 1999). However, both proteins are secreted: FBL-1C localizes to the gonad, pharynx, and intestine, whereas FBL-1D is associated with neuronal cells and flexible fibers (Hesselson et al., 2004; Kubota et al., 2004; Muriel et al., 2005). These different patterns of localization in C. elegans are consistent with the distinct binding properties of the two mammalian isoforms in vitro (Sasaki et al., 1995).
We previously described an antagonistic interaction between fbl-1 and gon-1/ADAMTS (a secreted a disintegrin and metalloprotease with thrombospondin repeats protein; Hesselson et al., 2004). GON-1 is required for the migration of gonadal distal tip cells (Blelloch and Kimble, 1999). Fibulin also affects distal tip cell migration; fbl-1 mutations can suppress the gon-1 migration defect (Hesselson et al., 2004). Furthermore, gon-1 mutations suppress the distended gonad of fbl-1 mutants, revealing an antagonistic relationship between these two regulators.
In this study, we report an unexpected synthetic lethality between gon-1 and fbl-1 null mutants. Using FBL-1 transgenes, we have defined the alternatively spliced EGF2C repeat as being critical for both larval progression in fbl-1 gon-1 double mutants and normal growth in fbl-1 single mutants. Indeed, a small fragment including EGF2C provides these essential FBL-1 functions. In contrast, most of the FBL-1C protein is required for gonadogenesis, revealing context-specific requirements for different portions of FBL-1C in larval development.
Results and discussion
Isolation and phenotypic analysis of a fbl-1 null allele
The fbl-1(q750) deletion allele removes genomic DNA encoding the C-terminal half of the FBL-1 protein (Hesselson et al., 2004). Therefore, this deletion encodes the N-terminal half of FBL-1 and may not be a null allele. To isolate a null allele, we used primers flanking the 5′ end of the fbl-1 gene to isolate fbl-1(q771), a 682-bp deletion that removes the N terminus of the fbl-1 locus, including 141 bp of the 5′ flanking sequence and the predicted transcriptional start site (Fig. 1 A). By RT-PCR using primers located 3′ of the fbl-1(q771) deletion and 5′ of the fbl-1(q750) deletion, products of the expected sizes were detected in wild type and fbl-1(q750) but not in fbl-1(q771) (see Materials and methods section fbl-1 transcript analysis). We conclude that the fbl-1(q771) allele does not produce an abundant mRNA product and that it is likely a null allele.
The phenotypes of fbl-1(q771) and fbl-1(q750) homozygotes were compared. fbl-1(q771) mutants have a reduced growth rate (Fig. 1 B), small size (Fig. 1 C), and uncoordinated kinking movement, which are defects previously observed in fbl-1(q750) mutants (Hesselson et al., 2004). Similarly, both fbl-1(q771) and fbl-1(q750) animals are sterile and have an expanded gonad compared with wild type (Fig. 1, D–F). Therefore, the fbl-1(q771) and fbl-1(q750) single mutants have similar phenotypes.
Synthetic larval arrest in fbl-1(q771) gon-1(0) double mutants
Because fbl-1(q771) and fbl-1(q750) single mutants have similar phenotypes, we predicted that these alleles would also have similar genetic interactions. The fbl-1(q750) mutation suppresses migration defects of gon-1(0), and gon-1(0) suppresses fbl-1(q750) gonadal expansion defects, revealing an antagonistic relationship between these two regulators (Hesselson et al., 2004). Surprisingly, fbl-1(q771) gon-1(0) double mutants arrested as young larvae (Fig. 2, compare A with D), whereas the fbl-1(q750) gon-1(0) double mutants grew to adulthood, as do the respective single mutants (Fig. 2, B and C). The fbl-1(q771) gon-1(0) larval arrest is not caused by extraneous linked mutations because an independently derived deletion allele, fbl-1(tk45), had a similar synthetic larval arrest with gon-1(0) (Fig. 2 E). The synthetic interaction is specific because the depletion of nidogen or the ADAM protease MIG-17 did not cause larval arrest in fbl-1(q771) nid-1(0) and fbl-1(q771) mig-17(0) double mutants (unpublished data). Therefore, both fbl-1 and gon-1 affect progression through larval development.
Figure 2.

Synthetic lethality of fbl-1(0) gon-1(0) double mutants. (A–J) Anterior to left; genotypes are given in the figure. (A–F) Body size comparisons of young adults (A–C) and double mutants (D and E) 72 h after L1. (F) Young adult rescued by EGF1-2C+ fragment. (G and H) Pharynx comparison of wild-type L4 (G) and double mutants at 48 h after L1 (H). Lines indicate anterior and posterior bulbs. (I and J) Intestine comparison of wild-type L4 (I) and double mutants (J) 48 h after L1. Arrowheads indicate gut lumen width; arrows point to bacteria in the lumen. Gut granules are depleted in the double mutant; compare boxed regions.
Next, we examined the stage and possible causes of larval arrest in fbl-1(q771) gon-1(0) double mutants. Double mutants completed embryogenesis and proceeded to the second larval stage. Details of this characterization are described in Materials and methods (see Phenotype characterization). No defect in pharyngeal function was found, and pharyngeal size was comparable in fbl-1(q771) gon-1(0) double mutants and wild type (Fig. 2, G and H). Intestinal morphology was altered in the double mutant, but its lumen was not obstructed (Fig. 2, I and J; and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200608061/DC1). Therefore, the cause of larval arrest remains unknown. Regardless, we conclude that fbl-1 and gon-1 are synthetically lethal, which suggests a redundancy of these two regulators.
FBL-1 domains required for larval progression
The synthetic lethality of fbl-1(q771) gon-1(0) double mutants provided a simple assay for analyzing the function of individual FBL-1 domains. Specifically, we generated a series of transgenes in which the fbl-1 5′ flanking sequence was used to drive the expression of FBL-1 fragments tagged with VENUS. For each transgene, we scored double mutants for progression to adulthood (Fig. 3). The expression of all transgenes was verified by the visualization of VENUS using confocal microscopy, and most were also verified by the detection of proteins in Western blots (see Figs. 5 and S1).
Figure 3.

EGF2C is critical for the rescue of fbl-1(q771) gon-1(0) larval arrest. (A–E, left) Protein diagrams show FBL-1 domains in each transgene. Each construct was expressed using 4.5 kb of the fbl-1 5′ flanking sequence. Domains depicted as in Fig. 1. VENUS, green oval. (right) n = 25 for each transgene. Gonads were variable in rescued animals.
Figure 5.

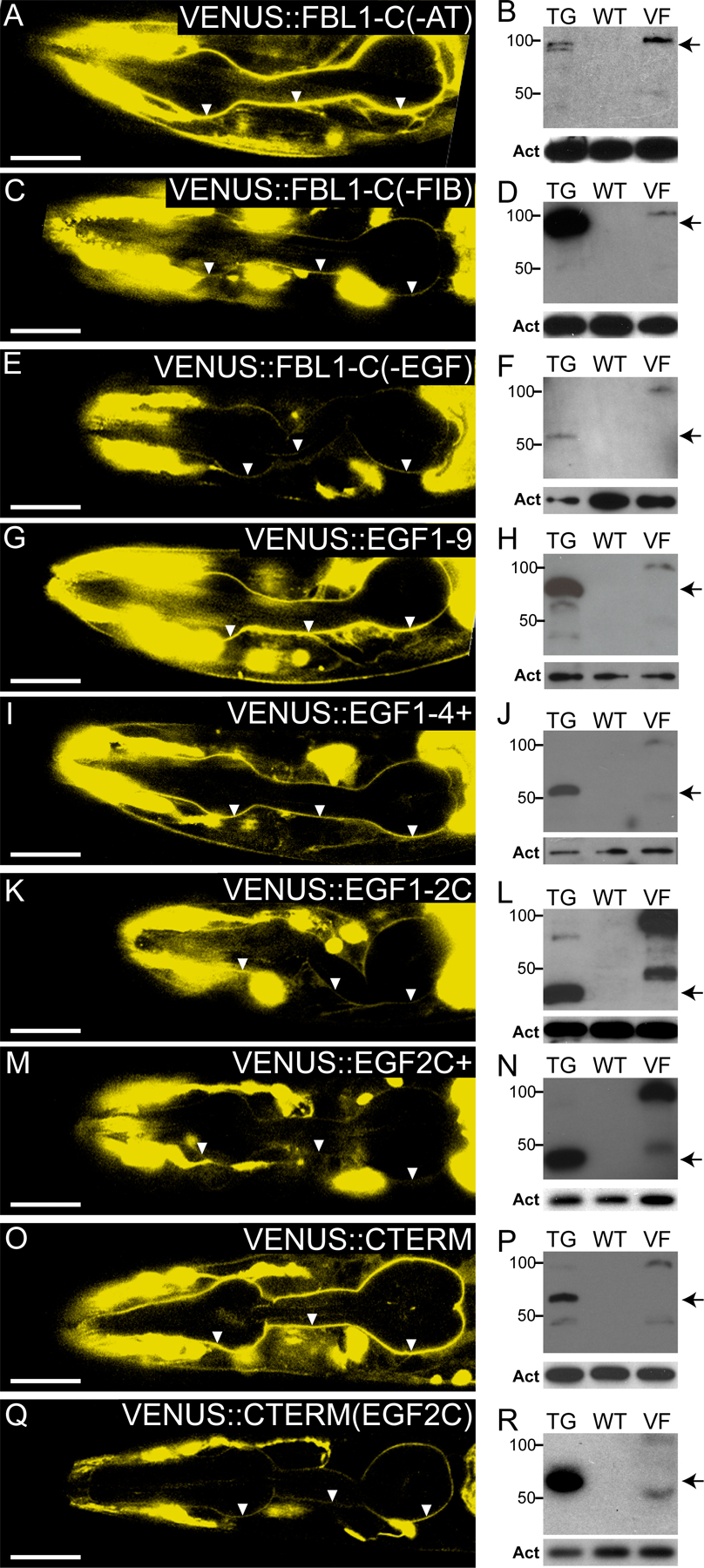
EGF2C and FIBC domains are redundant signals for localization to pharyngeal basal lamina. (left and middle) Heads of fbl-1(q771) L4 larvae; transgenes are noted in the figure. Arrowheads indicate pharyngeal basal lamina; anterior to the left. Arrows indicate fluorescence from the coinjection marker. Bars, 20 μm. (left) 0.8-μm confocal slices. (middle) Differential interference contrast image of the same animal. (A, F, L, O, and R) Constructs with either EGF2C or FIBC localize to pharyngeal basal lamina. (I) VENUS∷FBL-1D not in pharyngeal basal lamina. (right) Anti-VENUS Western blots. Both full-length FBL-1 isoforms are predicted to migrate at ∼100 kD, but FBL-1D only accumulates an ∼50-kD degradation product. TG, extract from transgenic strain noted in the left column; WT, extract from wild-type animals not carrying transgene; VF, extract from animals carrying VENUS∷FBL-1; Act, actin loading control. Arrows mark the predicted sizes of transgenic proteins. Arrowheads mark abundant degradation products.
Normally, the fbl-1 gene produces alternatively spliced mRNAs encoding two distinct proteins, FBL-1C and -1D (Fig. 1 A). The full fbl-1 gene with VENUS inserted at the N terminus rescued larval arrest of fbl-1(q771) gon-1(0) double mutants (Fig. 3 A). The animals grew to adulthood and had a gon-1(0) phenotype. To learn which alternatively spliced transcript was responsible for rescue, we expressed VENUS∷FBL-1C and -1D transgenes in fbl-1(q771) gon-1(0) double mutants. VENUS∷FBL-1C rescued larval arrest, whereas VENUS∷FBL-1D did not (Fig. 3 A).
To define the essential domains within FBL-1C, we generated a set of transgenes that were deleted for individual domains (Fig. 3 B). Rescue was observed with transgenes deleted for either AT or FIB domains, but deletion of the EGF domain abolished rescue (Fig. 3 B). Furthermore, the EGF domain was sufficient for rescue: VENUS∷EGF1-9, which encodes all nine EGF repeats but no other FBL-1 domain, was sufficient for rescue (Fig. 3 B). Therefore, the EGF repeats are both necessary and sufficient for rescue.
The EGF domains from the rescuing VENUS∷FBL-1C transgene and the nonrescuing VENUS∷FBL-1D transgene differ in the second EGF repeat as a result of the alternative splicing of exons 5C and 5D (Fig. 1 A). To ask whether EGF2C is necessary for rescue, we swapped EGF2 between the two isoforms to generate VENUS∷EGF2D/FIBC and VENUS∷EGF2C/FIBD (Fig. 3 C). Only VENUS∷EGF2C/FIBD was capable of rescue, demonstrating that alternatively spliced EGF2 repeats have different functions (Fig. 3 C).
Because EGF2C is required for rescue, we tested fragments of the EGF domain to define a minimal rescuing fragment, VENUS∷EGF1-2C+, which contained EGF repeats 1, 2C, and part of EGF3 (Figs. 2 F and 3 D). For simplicity, we dub this fragment EGF1-2C+. Removal of either EGF1 or the remainder of EGF3 from EGF1-2C+ eliminated rescue (Fig. 3 D). Either the single EGF2C requires the flanking sequence for correct folding/secretion or additional features within EGF1 and EGF3 contribute to rescue. In an attempt to distinguish between these possibilities, we placed EGF2C into the C-terminal half of the EGF domain, but this transgene had no rescuing activity (Fig. 3 E). The simplest explanation is that some additional features within the minimal EGF1-2C+ fragment are required for FBL-1C function. Indeed, the EGF1-2C+ fragment contains residues that affect FBL-1 function and localization in other developmental contexts (Kubota et al., 2004). Alternatively, the C-terminal half of FBL-1 may poison the biological activity of EGF2C.
Rescue of fbl-1 single mutant defects by deletion transgenes
To learn whether the EGF domain of FBL-1C also functions in the presence of wild-type GON-1, we scored VENUS-tagged FBL-1 transgenes for rescue of both the slow growth defect and small body size of fbl-1(q771) single mutants. All constructs that rescued the larval arrest of fbl-1(q771) gon-1(0) double mutants also rescued the fbl-1(q771) single mutant phenotypes (Fig. 4, A and B). Indeed, the same minimal EGF1-2C+ fragment rescued both the development rate and body size (Fig. 4, A and B).
Figure 4.

Rescue of fbl-1(q771) defects. (A–C) All tests performed in wild-type (N2) or fbl-1(q771) single mutants carrying indicated transgene. (A) Duration of larval development measured in hours from synchronized L1 to mid-L4. n = 25 animals for each strain. (B) Adult body size 24 h after mid-L4. n = 10 animals for each strain. (A and B) Mean ± SEM (error bars). *, P < 0.001 by t test. (C) Gonadal width and fertility. n = 20 animals for each strain.
Because fbl-1(q771) single mutants are sterile and have a distended gonad, we also measured gonadal width and fertility in the transgenic animals. In contrast to the rescue of growth and body size, both the EGF domain of FBL-1C and the FIBC domain were required for the rescue of gonadal width (Fig. 4 C). Indeed, most EGF repeats of FBL-1C are likely required for the control of gonadal width because deletion from the larger fragment of either the four N-terminal repeats or four C-terminal repeats abolished rescue (unpublished data). The AT domain was dispensable (Fig. 4 C). We conclude that FBL-1 has different molecular functions in its controls of larval growth and organ width.
VENUS∷FBL-1 localization
Full-length VENUS∷FBL-1C localizes to pharyngeal and gonadal basal laminas (Fig. 5 A, gonad not depicted; Hesselson et al., 2004; Kubota et al., 2004; Muriel et al., 2005). However, the localization of individual domains had not been assayed. We found that transgenes containing either EGF2C or FIBC localized to both pharyngeal and gonadal basal laminae (Fig. 5, A, F, L, O, and R; Fig. S1; and not depicted). In contrast, VENUS∷FBL-1D did not localize to either basal lamina (Fig. 5 I; Kubota et al., 2004; Muriel et al., 2005). Importantly, the rescue of gonadal width required both the EGF domain of FIB-1C and the FIBC domain (Fig. 4 C), but each of these domains was individually sufficient for localization. Furthermore, EGF1-2C+ failed to rescue gonadal width but was localized to the gonadal basal lamina. We conclude that the localization of FBL-1 fragments to the basal lamina is not sufficient for function.
Conclusions
This study focuses on the C. elegans FBL-1 ECM protein and its control of two biological processes: organ width and developmental growth. In fbl-1 null mutants, the normally slender gonad becomes distended and misshapen, an effect first seen in L4 larvae (Hesselson et al., 2004; Kubota et al., 2004; Muriel et al., 2005). The developmental defect in growth was mild in fbl-1 single mutants but severe in fbl-1(0) gon-1(0) double mutants, which arrest as L2 larvae. Using transgenic assays, we found that most of the FBL-1C protein, including both EGF and FIBC domains, is required for the control of organ width, but only a few EGF repeats are responsible for growth. Therefore, FBL-1C is likely to affect organ width and larval growth by separate molecular mechanisms.
Structural abnormalities in the basal lamina are likely to cause organ distension in fbl-1 null mutants. Overexpression of the C. elegans homologue of collagen XVIII, which is called CLE-1, rescues the gonadal defect of fbl-1 mutants (Muriel et al., 2006). Therefore, extra CLE-1 compensates for the loss of FBL-1, suggesting a similar role for these two conserved ECM proteins in the integrity of the basal lamina. In mammals, the EGF and FIBC domains of Fibulin-1 contain multiple binding sites for a variety of ECM molecules, including nidogen (Timpl et al., 2003). Both EGF and FIBC domains are needed for FBL-1C control of gonadal width. Therefore, FBL-1C is likely to control organ width by providing a scaffold that stabilizes ECM assembly within the gonadal basal lamina.
Major structural abnormalities in the basal lamina do not appear to cause the developmental growth defects of fbl-1 single mutants or fbl-1 gon-1 double mutants. Instead, a fragment containing only EGF1, EGF2C, and part of EGF3 controls the normal rate of larval growth and correct body size. We suggest that EGF2C or the EGF1-2C+ fragment may control growth as a signaling ligand. One precedent for this idea is TGF-α, which contains a single EGF repeat that binds to and signals through EGF receptors (Massague, 1983). An alternative possibility is that the EGF1-2C+ fragment antagonizes interactions within the ECM and, thereby, permits remodeling of the basal lamina that is critical for growth.
The fbl-1(0) gon-1(0) synthetic lethality suggests that FBL-1 and the GON-1 secreted metalloprotease act redundantly to promote larval progression, which is an idea that contrasts with the antagonistic activities of FBL-1 and GON-1 during gonadogenesis (Hesselson et al., 2004). Perhaps these distinct relationships can be explained by the two different functions of FBL-1C. For example, in the control of gonadal width, FBL-1C might stabilize the ECM, whereas the GON-1 protease promotes ECM breakdown and remodeling. In contrast, in the control of developmental growth, EGF1-2C+ and GON-1 might produce redundant signals.
The use of an alternatively spliced EGF repeat may be relevant to mammalian fibulin. Mammals contain five genes encoding fibulin-like proteins, and mammalian Fibulin-1 is most similar to C. elegans FBL-1 (Timpl et al., 2003). For example, Fibulin-1 and FBL-1 have the same types of motifs in the same number, and both have alternatively spliced FIBC and FIBD domains. However, unlike C. elegans FBL-1, the EGF domain of mammalian Fibulin-1 is not alternatively spliced; it contains the same EGF repeats in both C and D isoforms (Argraves et al., 1990). In contrast, mammalian Fibulin-2 and -3 are subject to alternative splicing within their EGF domains (Lecka-Czernik et al., 1995; Grassel et al., 1999). The functions of the alternatively spliced EGF repeats in these other fibulins have not been explored, but our findings suggest the possibility that they may have signaling activity. Perhaps the single C. elegans fbl-1 gene encompasses features that have been distributed to multiple fibulin genes in mammals.
Materials and methods
Nematode analysis
All strains were derivatives of wild-type Bristol strain N2 and were maintained at 20°C. fbl-1(q771) was made by standard methods (Kraemer et al., 1999); fbl-1(q771) removes F56H11∷15,541 through F56H11∷16,223. The fbl-1(tk45) deletion introduces a stop codon after the first EGF repeat (Kubota et al., 2004). Other strains include fbl-1(q750) (Hesselson et al., 2004), gon-1(q518) (Blelloch and Kimble, 1999), mig-17(k174) (Nishiwaki et al., 2000), and nid-1(cg119) (Kang and Kramer, 2000).
fbl-1 transcript analysis
Total RNA was extracted from 50 L4s of each genotype. We performed 37 cycles of RT-PCR at 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s using forward 5′-GATTCGACTTGGCACCAG-3′ and reverse 5′-GAATGATCCAGGGGTGTTC-3′ primers. eft-3 was amplified from each sample as a positive control.
Transgenes
Fragments of genomic fbl-1 were fused to VENUS under the control of the fbl-1 promoter and injected at 5 ng/μl with either neuronal coinjection marker ttx-3∷dsRED or rol-6(gf) at 20 ng/μl. See supplemental methods for construct details (available at http://www.jcb.org/cgi/content/full/jcb.200608061/DC1).
FBL-1 localization
VENUS∷FBL-1 localization was assayed in fbl-1(q771) mutants, which lack endogenous FBL-1 protein. Images were obtained on a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) with a 63× NA 1.4 objective lens. The brightness of images was uniformly modified in Photoshop (Adobe).
Western blots
Extract prepared from 20 transgenic L4s was loaded per lane. Proteins were separated on 4–20% gradient gels (Cambrex). Blots were probed with JL-8 anti-GFP antibody at 1:1,000 (Stratagene) and detected with an HRP-conjugated secondary antibody at 1:40,000 (Jackson ImmunoResearch Laboratories). Each blot was reprobed with an anti-actin antibody as a loading control.
Phenotype characterization
Embryonic lethality.
Embryonic lethality was assessed by scoring dead embryos among an entire brood from a single fbl-1(q771) gon-1(0) hermaphrodite.
Staging larval arrest.
20 synchronized fbl-1(q771) gon-1(0) L1s were grown for 24 h and scored for L1-specific alae; all had exited the L1 stage. Mixed stage fbl-1(q771) gon-1(0) double mutants were fixed and stained with MH27, a monoclonal antibody that highlights junctions and permits visualization of vulval precursor morphology. Most had undivided vulval precursors showing a failure to progress through the L3 stage.
Pharyngeal function.
Pharyngeal contraction rates were counted in both wild-type and fbl-1(q771) gon-1(0) double mutants at hatching and at 48 h after hatching.
Gut function.
Larvae were fed a mixture of OP50 bacteria and fluorescently labeled beads (Bangs Beads). After 1 h, they were moved to a plate with only OP50, and, after 5 min, they were mounted. Beads were visualized by fluorescence microscopy.
Rescue assays
Larval arrest.
Animals were synchronized by hatching in M9 and incubated for at least 144 h. Adulthood was determined by vulval morphology. Wild-type animals reach adulthood within 52 h. If transgenic double mutants did not reach adulthood within 144 h, they were scored as not rescued. For each strain, 25 animals were scored.
Length of larval development.
Animals were placed singly on plates, and time to mid-L4 was scored by vulval morphology. For each strain, 25 animals were scored.
Adult body size.
L4s were grown for 24 h and measured for size by multiplying length and width to obtain area as a rough estimate of organism size. For each strain, 10 animals were scored.
Gonadal width.
Mid-L4s were scored as rescued if the proximal gonadal arm spanned less than two thirds of the width of the body cavity. For each strain, 20 animals were scored.
Sterility.
Mid-L4s were grown for 48 h and scored for embryo production. For each strain, 20 animals were scored.
Image acquisition
Nomarski micrographs were obtained on a microscope (Imager.D1; Carl Zeiss MicroImaging, Inc.) with either a 63× NA 1.4 or 10× NA 0.3 objective lens. Images were captured in Openlab 4.0.4 (Improvision) with a camera (C4742-95; Hamamatsu).
Online supplemental material
Fig. S1 shows confocal localization and Western blots for transgenes that are not depicted in Fig. 5. Supplemental methods provide information on plasmids, extrachromosomal arrays, and strains used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200608061/DC1.
Supplementary Material
Acknowledgments
We thank Dana Byrd, Mike Chesney, Stephanie Hesselson, and Bryan Phillips for comments on the manuscript.
D. Hesselson is supported by the Canadian Institutes of Health Research. J. Kimble is supported by the National Institutes of Health and is an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper: AT, anaphylatoxin; FBL, FIBULIN; FIB, fibulin type.
References
- Argraves, W.S., H. Tran, W.H. Burgess, and K. Dickerson. 1990. Fibulin is an extracellular matrix and plasma glycoprotein with repeated domain structure. J. Cell Biol. 111:3155–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth, J.L., K.M. Argraves, E.F. Roark, C.D. Little, and W.S. Argraves. 1998. Identification of chicken and C. elegans fibulin-1 homologs and characterization of the C. elegans fibulin-1 gene. Matrix Biol. 17:635–646. [DOI] [PubMed] [Google Scholar]
- Blelloch, R., and J. Kimble. 1999. Control of organ shape by a secreted metalloprotease in the nematode Caenorhabditis elegans. Nature. 399:586–590. [DOI] [PubMed] [Google Scholar]
- Debeer, P., E.F. Schoenmakers, W.O. Twal, W.S. Argraves, L. De Smet, J.P. Fryns, and W.J. Van De Ven. 2002. The fibulin-1 gene (FBLN1) is disrupted in a t(12;22) associated with a complex type of synpolydactyly. J. Med. Genet. 39:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli, G., J. Falk-Marzillier, O. Schiraldi, W.G. Stetler-Stevenson, and V. Quaranta. 1997. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 277:225–228. [DOI] [PubMed] [Google Scholar]
- Grassel, S., F.X. Sicot, S. Gotta, and M.L. Chu. 1999. Mouse fibulin-2 gene. Complete exon-intron organization and promoter characterization. Eur. J. Biochem. 263:471–477. [DOI] [PubMed] [Google Scholar]
- Hesselson, D., C. Newman, K.W. Kim, and J. Kimble. 2004. GON-1 and fibulin have antagonistic roles in control of organ shape. Curr. Biol. 14:2005–2010. [DOI] [PubMed] [Google Scholar]
- Kang, S.H., and J.M. Kramer. 2000. Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans. Mol. Biol. Cell. 11:3911–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, C.A., B.D. Dimitroff, J.M. Rawson, and S.B. Selleck. 2004. Spatial regulation of Wingless morphogen distribution and signaling by Dally-like protein. Dev. Cell. 7:513–523. [DOI] [PubMed] [Google Scholar]
- Kohl, J. 2001. Anaphylatoxins and infectious and non-infectious inflammatory diseases. Mol. Immunol. 38:175–187. [DOI] [PubMed] [Google Scholar]
- Kostka, G., R. Giltay, W. Bloch, K. Addicks, R. Timpl, R. Fassler, and M.L. Chu. 2001. Perinatal lethality and endothelial cell abnormalities in several vessel compartments of fibulin-1-deficient mice. Mol. Cell. Biol. 21:7025–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer, B., S. Crittenden, M. Gallegos, G. Moulder, R. Barstead, J. Kimble, and M. Wickens. 1999. NANOS-3 and FBF proteins physically interact to control the sperm-oocyte switch in Caenorhabditis elegans. Curr. Biol. 9:1009–1018. [DOI] [PubMed] [Google Scholar]
- Kreuger, J., L. Perez, A.J. Giraldez, and S.M. Cohen. 2004. Opposing activities of Dally-like glypican at high and low levels of Wingless morphogen activity. Dev. Cell. 7:503–512. [DOI] [PubMed] [Google Scholar]
- Kubota, Y., R. Kuroki, and K. Nishiwaki. 2004. A fibulin-1 homolog interacts with an ADAM protease that controls cell migration in C. elegans. Curr. Biol. 14:2011–2018. [DOI] [PubMed] [Google Scholar]
- Lecka-Czernik, B., C.K. Lumpkin Jr., and S. Goldstein. 1995. An overexpressed gene transcript in senescent and quiescent human fibroblasts encoding a novel protein in the epidermal growth factor-like repeat family stimulates DNA synthesis. Mol. Cell. Biol. 15:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. 1983. Epidermal growth factor-like transforming growth factor. I. Isolation, chemical characterization, and potentiation by other transforming factors from feline sarcoma virus-transformed rat cells. J. Biol. Chem. 258:13606–13613. [PubMed] [Google Scholar]
- Muriel, J.M., C. Dong, H. Hutter, and B.E. Vogel. 2005. Fibulin-1C and Fibulin-1D splice variants have distinct functions and assemble in a hemicentin-dependent manner. Development. 132:4223–4234. [DOI] [PubMed] [Google Scholar]
- Muriel, J.M., X. Xu, J.M. Kramer, and B.E. Vogel. 2006. Selective assembly of fibulin-1 splice variants reveals distinct extracellular matrix networks and novel functions for perlecan/UNC-52 splice variants. Dev. Dyn. 235:2632–2640. [DOI] [PubMed] [Google Scholar]
- Nikolova, G., N. Jabs, I. Konstantinova, A. Domogatskaya, K. Tryggvason, L. Sorokin, R. Fassler, G. Gu, H.P. Gerber, N. Ferrara, et al. 2006. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev. Cell. 10:397–405. [DOI] [PubMed] [Google Scholar]
- Nishiwaki, K., N. Hisamoto, and K. Matsumoto. 2000. A metalloprotease disintegrin that controls cell migration in Caenorhabditis elegans. Science. 288:2205–2208. [DOI] [PubMed] [Google Scholar]
- Prigent, S.A., and N.R. Lemoine. 1992. The type 1 (EGFR-related) family of growth factor receptors and their ligands. Prog. Growth Factor Res. 4:1–24. [DOI] [PubMed] [Google Scholar]
- Qing, J., V.M. Maher, H. Tran, W.S. Argraves, R.W. Dunstan, and J.J. McCormick. 1997. Suppression of anchorage-independent growth and matrigel invasion and delayed tumor formation by elevated expression of fibulin-1D in human fibrosarcoma-derived cell lines. Oncogene. 15:2159–2168. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., G. Kostka, W. Gohring, H. Wiedemann, K. Mann, M.L. Chu, and R. Timpl. 1995. Structural characterization of two variants of fibulin-1 that differ in nidogen affinity. J. Mol. Biol. 245:241–250. [DOI] [PubMed] [Google Scholar]
- Timpl, R., T. Sasaki, G. Kostka, and M.L. Chu. 2003. Fibulins: a versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 4:479–489. [DOI] [PubMed] [Google Scholar]
- Tran, H., W.J. VanDusen, and W.S. Argraves. 1997. The self-association and fibronectin-binding sites of fibulin-1 map to calcium-binding epidermal growth factor-like domains. J. Biol. Chem. 272:22600–22606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}