

Abstract
On passaging synthetic prions, two isolates emerged with incubation times differing by nearly 100 days. Using conformational-stability assays, we determined the guanidine hydrochloride (Gdn·HCl) concentration required to denature 50% of disease-causing prion protein (PrPSc) molecules, denoted as the [Gdn·HCl]1/2 value. For the two prion isolates enciphering shorter and longer incubation times, [Gdn·HCl]1/2 values of 2.9 and 3.7 M, respectively, were found. Intrigued by this result, we measured the conformational stabilities of 30 prion isolates from synthetic and naturally occurring sources that had been passaged in mice. When the incubation times were plotted as a function of the [Gdn·HCl]1/2 values, a linear relationship was found with a correlation coefficient of 0.93. These findings demonstrate that (i) less stable prions replicate more rapidly than do stable prions, and (ii) a continuum of PrPSc structural states enciphers a multitude of incubation-time phenotypes. Our data argue that cellular machinery must exist for propagating a large number of different PrPSc conformers, each of which enciphers a distinct biological phenotype as reflected by a specific incubation time. The biophysical explanation for the unprecedented plasticity of PrPSc remains to be determined.
Keywords: conformational stability, memory, strain, synthetic prions, tertiary structure
Prion diseases are neurodegenerative disorders that afflict humans and other mammals. In humans, Creutzfeldt–Jakob disease generally presents with a rapidly progressive dementia, with memory impairment and other cognitive deficits (1). The prion diseases are unique in that a single pathologic process may present as a sporadic, genetic, or infectious illness (2). Despite a wealth of data arguing that prions are composed solely of isoforms of the disease-causing prion protein, designated PrPSc, the composition of the infectious prion particle continues to be debated (3–5), largely because prions exist in many different strains.
For many years, it was postulated that prions must carry a small nucleic acid to encode strain-specific information. Studies of the protease-resistant, C-terminal fragment of PrPSc (PrP 27–30) revealed that some strains exhibit differences in the resistance of PrPSc to limited proteolysis (6–8), but this procedure, similar to glycoform analyses, could not always distinguish one strain from many others (9–11). In search of better methods to study prion strains, we first developed the conformation-dependent immunoassay (12) and later conformational-stability assays (see Supporting Text, which is published as supporting information on the PNAS web site) (13, 14).
The conformational-stability assay combines guanidine hydrochloride (Gdn·HCl)-mediated denaturation of PrPSc with limited proteolysis using proteinase K (PK; ref. 13). Using this approach, many different prion strains exhibited distinct denaturation profiles. The denaturation profile of a new strain of prions derived by passage through mice expressing a chimeric PrP transgene was sufficiently different to allow us to discriminate a new prion isolate from many naturally occurring strains (14). A prion strain is generally defined by its incubation time in a host, the neuropathologic lesion profile, and the pattern of PrPSc deposition in the CNS (15, 16). Moreover, these properties remain constant when the strain is repeatedly passaged in an inbred host. Strains have often been isolated by repeated passage at limiting dilution; this process has been termed “biological cloning” (17).
Because the cloning of prion strains requires repeated passaging in inbred mice, we chose to study prions in the brains of individual mice that in many cases had not been cloned. In the studies reported here, many of the prion inocula are derived from mice initially inoculated with synthetic prions; none of these inocula were cloned and thus, they are referred to as isolates.
We describe here the conformational stabilities of 30 prion isolates from synthetic and naturally occurring sources. Using conformational-stability assays, we determined the Gdn·HCl concentration required to denature 50% of the PrPSc molecules, denoted the [Gdn·HCl]1/2 value. When the incubation times were plotted against the [Gdn·HCl]1/2 values, a linear relationship was found, indicating that less stable prions replicate more rapidly than do stable prions. Remarkably, we found that a continuum of structural states of PrPSc enciphers a multitude of incubation-time phenotypes.
Results
In earlier studies, we reported that polymerization of N-terminally truncated recombinant (rec)PrP composed of mouse residues 89–230 into amyloid fibrils rendered the protein “infectious” (18). Transgenic (Tg) mice expressing truncated PrP denoted MoPrP(Δ23–88) were designated Tg9949 and inoculated with the recMoPrP(89–230) amyloid fibrils. These mice exhibited signs of neurologic dysfunction after prolonged incubation times ranging from 379 to 523 days (Table 1; ref. 18). The PrP expressed in the Tg9949 mice corresponds in length and sequence to that used to produce the amyloid fibrils.
Table 1.
Transmission of synthetic prions to three different lines of mice
| MoSP1 inoculum* | Incubation period days* |
Tg9949 mice |
FVB mice |
Tg4053 mice |
|||
|---|---|---|---|---|---|---|---|
| Incubation period,days ± SEM | (n/n0)† | Incubation period,days ± SEM | (n/n0)† | Incubation period,days ± SEM | (n/n0)† | ||
| MK4977 | 379 | 258 ± 25 | 7/7 | 154 ± 4 | 9/9 | 90 ± 1 | 10/10 |
| MK4973 | 505 | 237 ± 19 | 12/12 | 530 ± 82 | 6/6 | 306 ± 9 | 4/4 |
| MK4974 | 505 | 266 ± 20 | 10/10 | 623 ± 19 | 5/5 | 320 ± 16 | 4/4 |
| MK4979 | 522 | 332 ± 23 | 10/10 | 586 ± 31 | 5/5 | 392 ± 22 | 5/5 |
| MK4985 | 523 | 240 ± 22 | 10/10 | 522 ± 29 | 7/7 | 324 ± 6 | 7/7 |
*Inoculum was a 10-fold dilution of a 10% crude brain homogenate prepared from a Tg9949 mouse. The experimental identification number of each mouse is indicated in the first column. After inoculation with amyloid fibrils composed of recMoPrP(89–230), each mouse developed signs of neurologic dysfunction after a prolonged incubation period as indicated in the second column.
†n, number of animals developing clinical signs of prion disease; n0, number of animals inoculated. Animals dying atypically after inoculation were excluded (55).
Brain extracts from each of the five Tg9949 mice were passaged into Tg9949/FVB, WT FVB, and Tg4053/FVB mice overexpressing WT MoPrP (Table 1). The prions in the five Tg9949 brain extracts produced similar incubation times of ≈250 days upon second passage in Tg9949 mice (Table 1). Unexpectedly, passage of the five Tg9949 brain extracts into FVB and Tg4053 mice indicated the presence of at least two different prion isolates (Table 1). The Tg9949 mouse MK4977 developed neurologic dysfunction 379 days after inoculation with recMoPrP(89–230) amyloid; the prions in its brain produced incubation times of 154 and 90 days in FVB and Tg4053 mice, respectively (Table 1). In contrast, the MK4973 mouse developed neurologic dysfunction after 505 days; prions in its brain gave incubation times of 530 and 306 days when passaged in FVB and Tg4053 mice, respectively (Table 1). The prions found in the brains of the MK4974, MK4979, and MK4985 mice were similar to those in the MK4973 mouse based on passaging in FVB and Tg4053 mice (Table 1). Incubation times in FVB mice ranged from 522 to 623 days and in Tg4053 mice from 320 to 392 days.
When the brains of Tg9949 mice inoculated with the MK4977 extract were compared with those inoculated with the MK4985 extract, the neuropathology was indistinguishable, in accord with the initial transmission of synthetic prions to Tg9949 mice (18).
In contrast to the similar neuropathology in Tg9949 mice, the neuropathologies in FVB and Tg4053 mice inoculated with the MK4977 isolate differed dramatically from those found in mice inoculated with the MK4985 isolate. Interestingly, the indistinguishable neuropathology in Tg9949 mice inoculated with the two extracts parallels the similarity in incubation times (Table 1), and the different neuropathologies in FVB and Tg4053 mice (Fig. 1) parallel the very different incubation times observed for the two extracts.
Fig. 1.

Neuropathologic analysis of FVB and Tg4053 mice inoculated with two isolates from Tg9949 mice that had received synthetic prions (see Table 1). FVB mice inoculated with the MK4977 isolate show moderate vacuolation in the thalamus (A) and diffuse PrPSc deposits in the CA1 region (B) and cerebellar cortex (C). In contrast, FVB mice inoculated with the MK4985 isolate show reduced vacuolation in the thalamus (D) and PrPSc clustering in the dentate gyrus (E) and deep cerebellar nuclei (F). Tg4053 mice inoculated with the MK4977 isolate show moderate vacuolation in the thalamus (G) but more in the hippocampus where it is associated with intense PrPSc deposits (H). Minimal vacuolation and PrPSc deposition in the cerebellar cortex (I). The MK4985 isolate caused more intense vacuolation of the thalamus (J) and other regions, except for the CA1 region, which contained sparse PrPSc deposits (K). In the cerebellar cortex, abundant vacuoles were associated with abundant plaque-like PrPSc deposits (L). Dt, dentate gyrus of the hippocampus; g, cerebellar granule cell layer; Pk, Purkinje cell; Py, pyramidal cell layer of the CA1 region. [Scale bar: J, 40 μm (and applies to A, G, and D); L, 30 μm (and applies to B, C, E, F, H, I, and K).]
In two FVB mice inoculated with the MK4977 isolate, we found widespread moderately intense vacuolar degeneration (Fig. 1A) and moderately severe reactive astrocytic gliosis (not shown). Immunohistochemistry showed widespread granular deposits of PrPSc (Fig. 1 B and C). In contrast, FVB mice inoculated with the MK4985 isolate showed aggregates of PrPSc and little vacuolar degeneration (Fig. 1D). In the hippocampus, PrPSc accumulated exclusively in the molecular layer of the dentate gyrus where they appeared to be within astrocytes (Fig. 1E). No PrPSc deposits were located in the cerebellar cortex, as was the case with the MK4977 inoculum; rather, they formed aggregates between nerve cell bodies within deep cerebellar nuclei where they appeared to be located within reactive glia (Fig. 1F). In contrast to FVB mice, two Tg4053 mice inoculated with the MK4977 isolate exhibited widespread vacuolar degeneration in multiple brain regions (Fig. 1G) but not in the cerebellar cortex. Reactive astrocytic gliosis (not shown) and the fine granular PrPSc deposits were consistent with the degree of vacuolation, which was intense in the hippocampal CA1 region (Fig. 1H) and less so in the cerebellar cortex (Fig. 1I). In contrast to the MK4977 isolate, the MK4985 isolate provoked minimal neuropathology in the two Tg4053 mice. The exceptions were the thalamus (Fig. 1J) and the cerebellar granule cell layer, where the number and size of vacuoles were substantially greater than those produced by the MK4977 isolate. Large numbers of vacuoles in the cerebellum were associated with large aggregates of PrPSc between Purkinje and granule cells (Fig. 1L). The small number of vacuoles in the hippocampal CA1 region was associated with small numbers of punctate PrPSc deposits (Fig. 1K), in contrast to the intense PrPSc deposits in mice inoculated with the MK4977 isolate. The marked differences in the neuropathologic and PrPSc deposition phenotypes in FVB and Tg4053 mice support the hypothesis that the MK4977 and MK4985 isolates are distinct.
To assess whether the brain of the MK4977 mouse contained more than one prion isolate, we inoculated its brain extract into Tg9949 mice (Fig. 2A). The second-passage survival curve was biphasic, suggesting that at least two different prion isolates were present in the MK4977 inoculum. This notion was confirmed on third passage: Prions from a Tg9949 mouse (MM6055) developing neurologic dysfunction at 204 days caused disease in 172 ± 8 days, and prions from another Tg9949 mouse (MM6060) developing neurologic dysfunction at 270 days caused disease in 335 ± 47 days (Fig. 2B and Table 2).
Fig. 2.

Survival curves of Tg9949 mice after inoculation with synthetic prions. (A) First passage (filled triangles) of synthetic prions in Tg9949 mice. For the second passage (filled squares), homogenate from the brain of one Tg9949 mouse that died at 382 days (MK4977, indicated by the filled triangle symbol inside the square box) was inoculated into Tg9949 mice. (B) For the third passage of synthetic prions, homogenates were prepared from the brains of two ill Tg9949 mice, MM6055 (diamond in A) and MM6060 (circle in A), and inoculated into other Tg9949 mice. Tg9949 mice inoculated with the MM6055 homogenate developed neurologic signs with a mean incubation period of 172 ± 8 days (open diamonds). Tg9949 mice inoculated with the MM6060 homogenate exhibited a mean incubation of 335 ± 47 days (open circles).
Table 2.
Conformational stabilities of synthetic and naturally occurring prion isolates
| Inoculum* | Recipient mouse | Mouse ID | [Gdn·HCl]1/2,M ± SEM | Incubation period,days ± SEM | (n/n0)† |
|---|---|---|---|---|---|
| RecMoPrP (89–230) | Tg9949 | MK4985 | 4.9 ± 0.1 | 523 | |
| Tg9949 | MK4979 | 5.1 ± 0.1 | 522 | ||
| Tg9949 | MK4977 | 3.3 ± 0.3 | 379 | ||
| Tg9949 | MK4974 | 5.0 ± 0.1 | 505 | ||
| MK4977 | Tg9949 | MM6060 | 3.7 ± 0.1 | 270 | |
| Tg9949 | MM6059 | 3.7 ± 0.1 | 270 | ||
| Tg9949 | MM6055 | 2.9 ± 0.1 | 204 | ||
| Tg9949 | MM6054 | 4.0 ± 0.1 | 327 | ||
| Tg9949 | MM6050 | 2.4 ± 0.1 | 190 | ||
| MK4985 | Tg9949 | MN19826 | 3.8 ± 0.1 | 312 | |
| Tg9949 | MN19822 | 3.2 ± 0.3 | 196 | ||
| Tg4053 | MN10362 | 3.8 ± 0.1 | 323 | ||
| FVB | MN70149 | 4.4 ± 0.1 | 453 | ||
| MK4973 | Tg4053 | MN9060 | 3.8 ± 0.1 | 299 | |
| MM6060 | Tg9949 | MN12910 | 3.7 ± 0.2 | 267 | |
| Tg9949 | MN12916 | 3.4 ± 0.1 | 301 | ||
| Tg9949 | MO516 | 3.8 ± 0.2 | 302 | ||
| MK4977 | Tg4053 | MN11316 | 1.7 ± 0.1 | 88 | |
| FVB | MM72698 | 1.7 ± 0.1 | 145 | ||
| Me7 | Tg9949 | MJ8291 | 2.9 ± 0.1 | 229 | |
| CD-1 | N/A | 1.6 ± 0.1 | 145 ± 2 | 24/24 | |
| 22L | C57Bl6 | N/A | 2.4 ± 0.1 | 150 | |
| RML | Tg9949 | N/A | 1.7 ± 0.1 | 161 ± 4 | 11/11 |
| Tg4053 | MN8064 | 1.7 ± 0.1 | 60 | ||
| CD-1 | N/A | 1.7 ± 0.1 | 133 ± 2 | 29/29 | |
| FVB | N/A | 1.7 ± 0.1 | 117 ± 3 | 10/10 | |
| 301V | CD-1 | N/A | 2.2 ± 0.1 | 230 ± 3 | 10/10 |
| 139A | C57Bl6 | N/A | 2.0‡ | 147 ± 1 | 14/14 |
| BSE | Tg4092 | MI1909, MI1907 | 2.8 ± 0.1 | 240 | |
| Scrapie | Tg4092 | MI14146, MI14141 | 2.2 ± 0.1 | 220 |
*Inocula are synthetic prions formed from recMoPrP(89–230) polymerized into amyloid; Tg9949 mouse brains MK4973, MK4977, MK4985, and MM6060; mouse prion strains Me7, 22L, RML, 301V, and 139A; bovine spongiform encephalopathy (BSE) from cattle and scrapie from sheep.
†n, number of animals developing clinical signs of prion disease; n0, number of animals inoculated. Animals dying atypically after inoculation were excluded (55).
‡SEM for the [Gdn·HCl]1/2 value for prion strain 139A was not determined because the conformational-stability curves could not be fitted for statistical analysis (see Fig. 6C).
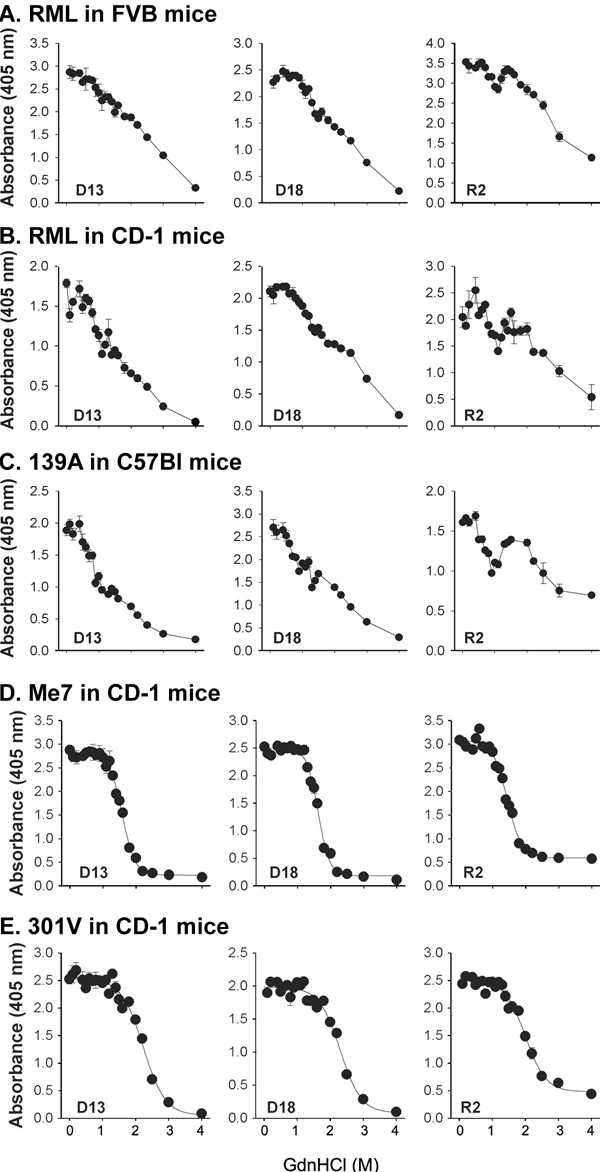
Next, we analyzed the conformational stability of these prion isolates. We tested the ability of the conformational-stability assay to discriminate among naturally occurring prion strains passaged in mice. In initial studies, we used an ELISA to quantify the amount of PrP 27–30 after exposure to Gdn·HCl (Fig. 5, which is published as supporting information on the PNAS web site; see ref. 13).
Encouraged by the ELISA studies to measure the conformational stability of PrPSc in pools prepared from the brains of several mice, we undertook similar studies on the brains of Tg9949 mice inoculated with synthetic prions. To analyze the prions present in a small portion of a single brain, we developed a protocol using Western blotting and densitometry (, which are published as supporting information on the PNAS web site). Although the [Gdn·HCl]1/2 of PrPSc for a particular prion strain (19) was similar by the two methods, the Western blot protocol did not reproduce the biphasic curves found for Rocky Mountain Laboratory (RML) and 139A using ELISA (Fig. 5 and Table 3, which are published as supporting information on the PNAS web site). The [Gdn·HCl]1/2 value is the concentration of Gdn·HCl required to denature 50% of PrPSc molecules, as judged by sensitivity of PK digestion.
We compared the conformational stabilities of the prions in the brains of two Tg9949 mice (MM6055 and MM6060) inoculated with prions from the MK4977 mouse (Fig. 2A). The prions in the MM6055 mouse that became ill at 204 days after inoculation exhibited a [Gdn·HCl]1/2 value of 2.9 M, whereas prions in the MM6060 mouse that became ill at 270 days after inoculation displayed a [Gdn·HCl]1/2 value of 3.7 M (Fig. 3).
Fig. 3.

Conformational-stability assays of synthetic prions on second passage in Tg9949 mice. Brain homogenates from one Tg9949 mouse (MK4977) that showed signs of neurologic dysfunction at 379 days after inoculation with synthetic prions were inoculated into two other Tg9949 mice. (A and C) Western blots of PrPSc in brain extracts of mouse MM6055 that was killed after developing signs of neurologic dysfunction at 204 days after inoculation (A) and mouse MM6060 that showed signs of neurologic dysfunction at 270 days after inoculation (C). Lanes 0–6, samples were exposed to increasing concentrations of GdnHCl (0–6 M, as indicated), then subjected to digestion with 20 μg/ml PK for 1 h at 37°C. Lane C, undigested control sample that was not exposed to Gdn·HCl. Molecular weight markers based on the migration of protein standards are indicated in kilodaltons. (B and D) Denaturation curves obtained by scanning the PrP 27–30 signals in the Western blots shown in A and C, respectively, show the [Gdn·HCl]1/2 values.
That these two isolates are distinct is supported by the differences in the incubation times found on subsequent passage (Fig. 2B). Intrigued by this finding, which seemed counterintuitive, we measured the conformational stabilities of 30 prion isolates from synthetic and naturally occurring sources (Figs. 8 and 9, which are published as supporting information on the PNAS web site.)
Using Western blotting and densitometry, we measured the conformational stabilities of synthetic prions upon first passage in four Tg9949 mice (Fig. 8). The denaturation curves were monophasic, but the slopes varied. The [Gdn·HCl]1/2 values calculated from the denaturation curves ranged from 3.3 to 5.1 M (Table 2). Many synthetic prions passaged in either WT or Tg mice exhibited much higher conformational stabilities than those observed for naturally occurring prions (13, 14). Upon second passage of the synthetic prions into Tg9949 mice, the conformational stabilities decreased, with [Gdn·HCl]1/2 values between 2.4 and 4.0 M (Fig. 3 and 9; Table 2).
When we plotted the incubation periods as a function of the [Gdn·HCl]1/2 values for 14 synthetic prion isolates passaged in Tg9949 mice, we found a linear relationship between these two parameters (R = 0.91; Fig. 4A). The brains of Tg9949 mice with incubation periods >500 days displayed extremely stable PrPSc conformers, with [Gdn·HCl]1/2 values of ≈5 M. Tg9949 mice succumbing to disease in ≈200 days had less-stable PrPSc molecules that exhibited [Gdn·HCl]1/2 values between 2.4 and 2.9 M. Generally, mice with identical incubation times exhibited similar often superimposable [Gdn·HCl]1/2 values.
Fig. 4.

The conformational stability of prions is directly proportional to the length of the incubation time in mice. The [Gdn·HCl]1/2 values for prions were plotted as a function of the incubation times. (A) Prions in the brains of Tg9949 mice infected with synthetic prions showed an excellent correlation (R = 0.91) between these parameters. (B) Naturally occurring prions passaged in both non-Tg and Tg mice showed a strong correlation (R = 0.77). (C) Synthetic prions (circles) in the brains of Tg9949, Tg4053, and non-Tg mice were plotted with many naturally occurring prions passaged (squares) in both non-Tg and Tg mice. R > 0.93.
Impressed by the excellent correlation between the [Gdn·HCl]1/2 values and length of the incubation time in Tg9949 mice inoculated with synthetic prions, we asked whether the same relationship might hold for naturally occurring prions. We determined the [Gdn·HCl]1/2 values for (i) Rocky Mountain Laboratory prions passaged in WT FVB, WT CD-1, Tg4053, and Tg9949 mice; (ii) Me7 prions in WT CD-1 and Tg9949 mice; (iii) 301V prions in WT CD-1 mice; (iv) bovine spongiform encephalopathy and scrapie prions in Tg4092 mice expressing the bovine PrP gene; and (v) 139A and 22L prions in WT C57Bl6 mice (Table 2). When the [Gdn·HCl]1/2 values for these naturally occurring prions were plotted as a function of the incubation times in Tg and non-Tg mice, the data could be fitted to a straight line (Fig. 4B). The slope of the line was slightly diminished compared with the line in Fig. 4A, and the correlation coefficient decreased to 0.77.
Buoyed by finding that both synthetic and naturally occurring prions exhibited the same relationship between conformational stability and the incubation time, we plotted all of the data points in Figs. 4 A and B together with the [Gdn·HCl]1/2 values for synthetic prions passaged in WT and Tg4053 mice. In this plot (Fig. 4C), a total of 30 points for synthetic and naturally occurring prions are shown; the correlation coefficient improved to 0.93. Thus, an excellent correlation exists between the conformational stability of PrPSc and the length of the incubation time over a range from 60 to >500 days.
Discussion
Our finding that the length of the incubation time in mice is directly proportional to the conformational stability of the prion isolate creates an approach for the rapid characterization of prion strains. It may now be possible to replace many incubation time determinations with Gdn·HCl-melting curves. In those cases for which such an approach is applicable, we can expect a >100-fold acceleration in the studies.
Continuum of Prions.
The availability of 30 different isolates with incubation times ranging from 60 to 523 days in mice allowed us to ask whether the conformational-stability assay could be related to the incubation-time phenotype. We found that the [Gdn·HCl]1/2 value is directly proportional to the length of the incubation time (Fig. 4). This relationship seems to hold whether the prion isolate has been passaged many times and the incubation time is constant, as is the case for RML, or the incubation time has changed with successive passaging, as is the case for some synthetic prion isolates (Table 2). In other words, the [Gdn·HCl]1/2 value reflects the incubation time that is specified by the prions being replicated in the brain.
It may be important to emphasize that we did not analyze prion isolates from mice where prolonged incubation times resulted from a mismatch between the sequence of PrPSc in the inoculum and that of the cellular PrP isoform (PrPC) expressed by the host. Such extended incubation times are generally because of a species transmission barrier and diminish on second passage in the homologous host (20, 21).
In our studies, no prion isolates with [Gdn·HCl]1/2 values of <1.5 M were found (Table 2). It is reasonable to hypothesize that a lower limit is set by the inability of very unstable prions to maintain their high β-sheet-rich structure and thus survive. Antithetically, prion isolates with [Gdn·HCl]1/2 values of >5 M may replicate so slowly that they do not reach a sufficient concentration in the brain to cause disease within the lifetimes of the mice.
Mechanism of PrPSc Denaturation.
The mechanism by which Gdn·HCl denatures PrPSc and renders it sensitive to limited proteolysis is unknown. Whether Gdn·HCl renders PrPSc susceptible to proteolytic digestion by disrupting tertiary or quaternary structure (or both) remains to be established. Interestingly, sonication of Syrian hamster Sc237 prions did not alter the incubation time, even though the prion rods fragmented into spherical particles (22).
Notably, Syrian hamster (SHa) Sc237 prions are more readily inactivated by acidic SDS than human sporadic Creutzfeldt–Jakob disease prions by a factor of ≈105 (23); in contrast, conformational-stability assays were unable to distinguish these two isolates (13, 14, 24). A limitation of the methodology used in our studies is the need for limited PK digestion, which precludes analysis of protease-sensitive PrPSc (12, 25). In earlier studies of SHa prion strains, the incubation time was found to be directly proportional to the level of protease-sensitive PrPSc (12). Moreover, a limited study of the [Gdn·HCl]1/2 values for several SHa prion strains failed to show a correlation with the incubation times (14).
Synthetic Prion Isolates.
It is noteworthy that a single isolate as characterized by a specific incubation time and a particular [Gdn·HCl]1/2 value may, in fact, be a mixture of two or more isolates. The synthetic prions in the brain of the Tg9949 mouse (MK4977) display an incubation time of 379 days and a [Gdn·HCl]1/2 value of 3.3 M. Yet, on second passage in Tg9949 mice, at least two different prion isolates emerged (Fig. 2 and 3; Table 2).
It is uncertain whether the studies reported here could have been performed with naturally occurring prion strains alone. The availability of different isolates derived from synthetic prions seems crucial in establishing the relationship between conformational stability and incubation time. Only synthetic prions have incubation times of ≈500 days with [Gdn·HCl]1/2 values between 4 and 5 M.
Distinct from yeast prions, mammalian prions require N-terminal truncation of PrP for amyloid formation to occur (26). Different strains of synthetic yeast prions were formed from the N-terminal domain of Sup35 by varying the temperature for amyloid fibril formation (27, 28). The fibrils polymerized at 4°C were less stable than those formed at 37°C. Similar to our results, the [PSI+] prions initiated by the 4°C fibrils were less stable and reproduced more rapidly than those induced by the 37°C fibrils (29).
Investigations of Other Degenerative Diseases.
Investigations of amyotrophic lateral sclerosis (ALS) seem notable with respect to our findings. The progression of familial ALS was found to correlate with the stability of the enzyme, superoxide dismutase (SOD). Patients with familial ALS due to mutations in SOD survived longer when the mutations resulted in a more stable enzyme, whereas the clinical course was shorter if the SOD molecule was less stable (30).
It seems reasonable to ask whether investigations of degenerative diseases of unknown etiology such as multiple sclerosis, schizophrenia, and bipolar disorders might benefit from our findings. Attempts to identify mutant genes in familial forms of these diseases have been disappointing and studies of gene expression levels using microarrays unrewarding. Perhaps analyses of the structural states of proteins like those reported here might prove more fruitful. Structural variants of proteins might be responsible for a disease process that cannot be identified by either molecular genetic or gene expression approaches.
A Physiological Function for PrPSc?
The multitude of structures that PrP 27–30 can adopt raises the possibility that PrPSc is an alternatively folded protein that performs a physiological function (31, 32). Whether PrPC can also adopt a continuum of structures is unknown but the large, flexible, N-terminal region might be a source of structural variation (33). In contrast, the structural variation in PrPSc occurs in the C-terminal, protease-resistant portion of the molecule denoted PrP 27–30. PrPSc but not PrPC accumulation stimulates the Notch and Hes pathways, resulting in the loss of dendritic processes (34, 35). PrPC seems to function in signal transduction and neurite outgrowth in conjunction with the neural cell adhesion molecule, N-CAM (36, 37). Moreover, PrPC-deficient mice exhibit defects in spatial memory tasks (38).
That PrPSc can exist in so many distinct structural states raises the possibility that this protein or other prion-like neuronal proteins function in information storage within the CNS. In such a scenario, PrPSc would exist in the CNS at levels well below those that are infectious (39, 40). The slow turnover and multitude of possible structures make PrPSc an interesting candidate for information storage in short-term memory. Recent studies of the cytoplasmic polyadenylation element binding protein have raised the possibility of a prion-like protein controlling the localized synthesis of new proteins in long-term memory within a single dendrite (41–43). Our results raise the possibility of an alternative role for prion proteins in short-term or working memory where protein synthesis is not required and transient information storage might be encrypted in a large array of macromolecular structural states.
Future Studies.
Our discovery that the [Gdn·HCl]1/2 value for a particular mouse isolate is directly proportional to the incubation time has several implications. First, it may provide a rational approach for the production of synthetic prions with short incubation times. We might be able to produce infectious prions that replicate rapidly by identifying conditions for polymerization of recPrP into fibrils with relatively low [Gdn·HCl]1/2 values. Our findings predict that less-stable recMoPrP amyloids will cause disease in mice much sooner than more stable polymers. Second, in an optimistic view, we have begun to understand how strain-specific phenotypes are enciphered within the conformation of PrPSc. Whether this discovery will prove sufficient to begin unraveling the molecular “language” used to encipher strain-specified properties is uncertain. Third, determining the atomic structures of PrPSc molecules enciphering short, intermediate, and prolonged incubation times is of utmost importance. Although the insolubility of native PrPSc continues to hinder high-resolution structural studies, fiber diffraction of infectious amyloid fibrils formed from recMoPrP might circumvent this problem.
Materials and Methods
Materials.
The isolation of 301V (44), Me7 (45), RML (46), and synthetic prions (18) has been described. Tg9949 and Tg4053 mice have been described (47, 48). CD-1 and FVB mice were purchased from Charles River Breeding Laboratories (Wilmington, MA).
Brain homogenates [10% (wt/vol)] were prepared and inoculated into mice as described (49). After inoculation, mice were examined daily for signs of neurologic dysfunction (50, 51). As neurologic deficits progressed, the mice were killed, and their brains were removed for histological and biochemical analysis.
Preparation of HuM-Fabs was performed as described (31).
Neuropathology.
Brains were removed, immersion-fixed in 10% buffered formalin, and embedded in paraffin. Sections (8 μm) were stained with hematoxylin/eosin. Peroxidase immunohistochemistry with antibodies to glial fibrillary acidic protein was used to evaluate reactive astrocytic gliosis. Hydrolytic autoclaving was performed as described (52), using HuM-R2 to detect PrPSc.
PrPSc Detection.
PrPSc was measured by limited PK digestion, SDS/PAGE, and Western blotting, as described (49, 53).
Conformational-Stability Assay.
The P2 fraction was prepared from brain homogenates and analyzed by ELISA, as described (13, 54). Alternatively, 50 μl of 10% brain homogenate was mixed with 50 μl of Gdn·HCl, varying from 0 to 8 M; for final concentrations of Gdn·HCl >4 M, a smaller proportion of brain homogenate was used. The details of this Western blot procedure have been described (19).
Densitometry.
Densitometry was performed by using Kodak Digital Science Image Station 440CF equipped with Kodak 1D Image Analysis software (Eastman Kodak, Rochester, NY). Data were analyzed and best-fitted by using the four-parameter sigmoid algorithm of the SigmaPlot software package (SPSS, Chicago, IL).
Supplementary Material
Acknowledgments
We thank Jonathan Weissman for valuable discussions, Charles Yanofsky for thoughtful critique of the manuscript, Suzette Priola (Rocky Mountain Laboratories, Hamilton, MT) for the 22L prion strain, and the staff at the Hunters Point Animal Facility for their assistance with the animal experiments. This work was supported by grants from the National Institutes of Health (AG02132, AG10770, and AG021601) as well as by gifts from the G. Harold and Leila Y. Mathers Charitable Foundation and the Sherman Fairchild Foundation.
Abbreviations
- Gdn·HCl
guanidine hydrochloride
- PrP
prion protein
- PrPC
cellular PrP isoform
- PrPSc
disease-causing alternative PrP isoform
- PrP 27–30
infectious protease-resistant C-terminal fragment of PrPSc
- rec
recombinant
- Tg
transgenic.
Footnotes
Conflict of interest statement: G.L., D.P., S.J.D., and S.B.P. have a financial interest in InPro Biotechnology.
References
- 1.Masters CL, Richardson EP., Jr Brain. 1978;101:333–344. doi: 10.1093/brain/101.2.333. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 3.Chesebro B. Science. 2004;305:1918–1921. doi: 10.1126/science.1103581. [DOI] [PubMed] [Google Scholar]
- 4.Nishida N, Katamine S, Manuelidis L. Science. 2005;310:493–496. doi: 10.1126/science.1118155. [DOI] [PubMed] [Google Scholar]
- 5.Jeffrey M, Gonzalez L, Espenes A, Press C, Martin S, Chaplin M, Davis L, Landsverk T, Macaldowie C, Eaton S, McGovern G. J Pathol. 2006;209:4–14. doi: 10.1002/path.1962. [DOI] [PubMed] [Google Scholar]
- 6.Bessen RA, Marsh RF. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 8.Mastrianni J, Nixon F, Layzer R, DeArmond SJ, Prusiner SB. Neurology. 1997;48(Supp):A296. [Google Scholar]
- 9.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 10.Somerville RA, Chong A, Mulqueen OU, Birkett CR, Wood SC, Hope J. Nature. 1997;386:564. doi: 10.1038/386564a0. [DOI] [PubMed] [Google Scholar]
- 11.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, et al. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 12.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 13.Peretz D, Scott M, Groth D, Williamson A, Burton D, Cohen FE, Prusiner SB. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton D, DeArmond SJ, Prusiner SB, Scott MR. Neuron. 2002;34:921–932. doi: 10.1016/s0896-6273(02)00726-2. [DOI] [PubMed] [Google Scholar]
- 15.Bruce ME, McBride PA, Farquhar CF. Neurosci Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-x. [DOI] [PubMed] [Google Scholar]
- 16.Taraboulos A, Jendroska K, Serban D, Yang S.-L, DeArmond SJ, Prusiner SB. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickinson AG. Genetics. 1975;79:387–395. [PubMed] [Google Scholar]
- 18.Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 19.Legname G, Nguyen H-OB, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. Proc Natl Acad Sci USA. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattison IH. Vet Rec. 1988;123:661–666. [PubMed] [Google Scholar]
- 21.Scott MR, Peretz D, Nguyen H-OB, DeArmond SJ, Prusiner SB. J Virol. 2005;79:5259–5271. doi: 10.1128/JVI.79.9.5259-5271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKinley MP, Braunfeld MB, Bellinger CG, Prusiner SB. J Infect Dis. 1986;154:110–120. doi: 10.1093/infdis/154.1.110. [DOI] [PubMed] [Google Scholar]
- 23.Peretz D, Supattapone S, Giles K, Vergara J, Freyman Y, Lessard P, Safar JG, Glidden DV, McCulloch C, Nguyen H-OB, et al. J Virol. 2006;80:322–331. doi: 10.1128/JVI.80.1.322-331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Proc Natl Acad Sci USA. 2004;101:1380–1385. doi: 10.1073/pnas.0307825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, et al. Proc Natl Acad Sci USA. 2005;102:3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKinley MP, Meyer RK, Kenaga L, Rahbar F, Cotter R, Serban A, Prusiner SB. J Virol. 1991;65:1340–1351. doi: 10.1128/jvi.65.3.1340-1351.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka M, Chien P, Yonekura K, Weissman JS. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka M, Collins SR, Toyama BH, Weissman JS. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 30.Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M. Proc Natl Acad Sci USA. 2005;102:9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, et al. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 32.Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. J Gen Virol. 2005;86:2913–2923. doi: 10.1099/vir.0.80947-0. [DOI] [PubMed] [Google Scholar]
- 33.Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. Proc Natl Acad Sci USA. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogan RN, Baringer JR, Prusiner SB. J Neuropathol Exp Neurol. 1987;46:461–473. doi: 10.1097/00005072-198707000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Ishikura N, Clever JL, Bouzamondo-Bernstein E, Samayoa E, Prusiner SB, Huang EJ, DeArmond SJ. Proc Natl Acad Sci USA. 2005;102:886–891. doi: 10.1073/pnas.0408612101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. J Neurochem. 2005;95:1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x. [DOI] [PubMed] [Google Scholar]
- 38.Criado JR, Sanchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, Race R, Manson JC, Chesebro B, Oldstone MB. Neurobiol Dis. 2005;19:255–265. doi: 10.1016/j.nbd.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- 40.Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. J Virol. 2005;79:10796–10806. doi: 10.1128/JVI.79.16.10796-10806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Si K, Lindquist S, Kandel ER. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 42.Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- 43.Kandel ER. In Search of Memory: The Emergence of a New Science of Mind. New York: Norton; 2006. [Google Scholar]
- 44.Fraser H, Bruce ME, Chree A, McConnell I, Wells GAH. J Gen Virol. 1992;73:1891–1897. doi: 10.1099/0022-1317-73-8-1891. [DOI] [PubMed] [Google Scholar]
- 45.Dickinson AG, Meikle VM. Genet Res. 1969;13:213–225. doi: 10.1017/s0016672300002895. [DOI] [PubMed] [Google Scholar]
- 46.Chandler RL. Lancet. 1961;1:1378–1379. doi: 10.1016/s0140-6736(61)92008-6. [DOI] [PubMed] [Google Scholar]
- 47.Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Genes Dev. 1996;10:1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 49.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Wälchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, et al. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 50.Carlson GA, Kingsbury DT, Goodman PA, Coleman S, Marshall ST, DeArmond S, Westaway D, Prusiner SB. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 51.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Ann Neurol. 1982;11:353–358. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 52.Muramoto T, Kitamoto T, Tateishi J, Goto I. Am J Pathol. 1992;140:1411–1420. [PMC free article] [PubMed] [Google Scholar]
- 53.Supattapone S, Nguyen H-O B, Cohen FE, Prusiner SB, Scott MR. Proc Natl Acad Sci USA. 1999;96:14529–14534. doi: 10.1073/pnas.96.25.14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Cell. 1984;38:127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- 55.Prusiner SB, McKinley MP. Prions: Novel Infectious Pathogens Causing Scrapie and Creutzfeldt–Jakob Disease. FL: Academic, Orlando; 1987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}