

Abstract
Epstein–Barr nuclear antigen (EBNA) leader protein (EBNALP) coactivates promoters with EBNA2 and is important for Epstein–Barr virus immortalization of B cells. Investigation of the role of histone deacetylases (HDACs) in EBNALP and EBNA2 promoter regulation has now identified EBNALP and EBNA2 to be associated with HDAC4 in a lymphoblastoid cell line. Furthermore, a transcription-deficient EBNALP point mutant did not associate with HDAC4. HDAC4 and 5 overexpression repressed EBNA2 activation and EBNALP coactivation, whereas other HDACs had little effect. Moreover, EBNALP expression decreased nuclear HDAC4. Expression of 14-3-3 anchors HDAC4 in the cytoplasm, increased EBNALP effects, and reversed HDAC4 or 5 repression. HDAC4 reversal depended on the HDAC4 nuclear export sequence. Consistent with EBNALP coactivation being mediated by nuclear HDAC4 depletion, HDAC4 overexpression increased nuclear HDAC4 and specifically repressed EBNA2-dependent activation as well as EBNALP-dependent coactivation. Also, EBNALP, HDAC4, and 14-3-3 could be immunoprecipitated in a single complex. Thus, these data strongly support a model in which EBNALP coactivates transcription by relocalizing HDAC4 and 5 from EBNA2 activated promoters to the cytoplasm. The observed EBNALP effects are likely also in part through HDAC5, which is highly homologous to HDAC4.
Keywords: activator, Epstein–Barr virus, lymphocyte transformation, repressor
Epstein–Barr virus (EBV) is a γ-herpesvirus that causes lymphocyte-proliferative diseases, including posttransplant and AIDS-associated lymphoproliferative diseases, African Burkitt's lymphoma, and Hodgkin's disease. EBV also causes nasopharyngeal carcinoma (1). EBV infection of human B lymphocytes results in Latency III, characterized by expression of Epstein–Barr nuclear antigen 2 (EBNA2) and EBNA leader protein (EBNALP), which are essential for lymphoblastoid cell line (LCL) outgrowth (1–4). EBNA2 activates transcription of both host and virus genes (1, 5, 6) by associating with RBP/CBF1 (5–7) and activating transcription in part through PU.1 or AUF1 (8–10). Once positioned at promoters, EBNA2 recruits basal and activation-related transcription factors to promoters, including TFIIH, TAF40, and histone acetyl transferases p300, CBP, and p/CAF (11). Histone acetyl transferase activity is important for promoter activation.
EBNA2-mediated transcription is strongly up-regulated by EBNALP, which is encoded in the leader of the EBNA2 transcript and is expressed along with EBNA2 at the start of lymphocyte infection (12–14). EBNALP unstably interacts with EBNA2 (13, 14) and stably associates with cell proteins HA95 (15, 16), PKA, Hsp70 (15, 17), DNA-PK (15), Hax-1 (18), ERR1 (19), cdc2 (20, 21), and sp100 (22). HA95 overexpression down-regulates EBNALP effects, whereas Hax-1, ERR1, and Sp100 may up-regulate EBNALP effects. EBNALP is in the nucleus and the cytoplasm of EBV-infected lymphocytes, whereas EBNA2 is only in the nucleus (23). EBNALP partial localization to the cytoplasm may be significant because the amino acids required for cytoplasmic localization are conserved among EBNALPs of primate lymphocryptoviruses (24, 25).
EBNALP consists of 66 amino acid repeats, which are a scaffold for self-association (as well as for association with EBNA2 and other factors important for transcription), and 45 unique C-terminal amino acids, which regulate repeat domain-mediated interactions and effects (13, 24, 25). Because transcription is regulated by the balance between histone acetylation and deacetylation (26) and the EBNA2 acidic domain recruits p300/CBP and p/CAF histone acetyl transferases (11), we now investigate whether EBNALP can coactivate with EBNA2 by down-regulating class I or II histone deacetylases (HDACs) to coactivate transcription.
Results
HDAC4 and 5 Repress Activation, and EBNALP Reverses the Effect.
To test whether EBNA2 transcriptional activation can be inhibited by HDACs, the effect of HDAC overexpression on EBNA2-dependent transcription was tested. Surprisingly, only HDAC4 and HDAC5 repressed EBNA2 activation of the EBV LMP1p and Cp promoters (Fig. 1A). HDAC4 repressed the LMP1p and Cp promoters by 30–40% and HDAC5 by 50–60% (Fig. 1A). HDACs 1, 3, 6, and 7 did not effect EBNA2 activation even though these HDACs were equally well expressed (Fig. 1B). These results indicate that HDAC4 and HDAC5 can specifically down-modulate EBNA2 activation.
Fig. 1.
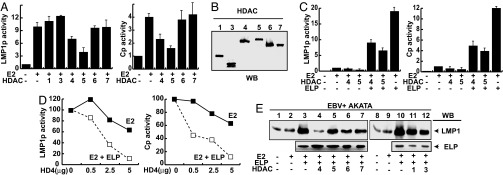
EBV LMP1p and Cp promoter activation and coactivation by EBNA2 and EBNALP is repressed by HDAC4 and HDAC5. (A) 2× LMP1p promoter-luciferase or 8× Cp promoter-luciferase reporter gene activation by EBNA2 (2.5 μg) cotransfected with the indicated HDACs (10 μg) in BJAB cells. The expression levels of EBNA2 were not affected. (B) Expression levels of different HDACs used in reporter assays by immunoblot against Flag-tag (HDAC1, 5, 7) or myc-tag (HDAC2, 4, 6). (C) Reporter gene assays of BJAB cells transfected with the indicated expression plasmids (2.5 μg for EBNA2, 5 μg for EBNALP, or 10 μg for HDACs) on the LMP1p or Cp promoters. The expression levels of EBNA2 and EBNALP were not affected. (D) Reporter assays performed on BJAB cells transfected with a constant amount of EBNA2 (2.5 μg, black lines) or EBNA2 plus EBNALP (2.5 μg and 5 μg for EBNA2 and EBNALP respectively, gray lines) and increasing amounts of HDAC4 (HD4) as indicated, for LMP1p (Left) or Cp (Right) promoters. Values for each curve were normalized to 100% of activity in the absence of HDAC4. (E) The EBV+ Akata cell line was transfected with the indicated expression plasmids (2.5 μg for EBNA2, 5 μg for EBNALP, or 10 μg for HDACs) and LMP1 induction detected by immunoblot using S12 antibodies (LMP1) at 48 h after transfection. The expression levels of EBNA2 were not affected. All reporter gene assays were performed as described in Materials and Methods. Empty vector was used to correct for the total mass of transfected DNA. β-Galactosidase was used as an internal control for transfection efficiency, and luciferase values were normalized by using β-galactosidase values. Activation values reflect the values ± SD. Each experiment was independently repeated at least three times. E2, EBNA2; ELP, EBNALP; WB, Western blot.
To investigate whether EBNALP can relieve HDAC4- or HDAC5-mediated repression of EBNA2, EBNALP was expressed in cells in which EBNA2 activity was repressed by HDAC4 or HDAC5 (Fig. 1C). EBNALP reversed HDAC4 or HDAC5 repression of the LMP1p or Cp promoters and activated the promoters 3- to 8-fold. EBNALP coactivation with EBNA2 was not as strong in the presence of higher HDAC4 or HDAC5 levels as in the absence of HDAC4 or HDAC5. These results indicate that EBNALP can reverse the repressive effects of HDAC4 or HDAC5 overexpression on EBNA2, and that HDAC4 or HDAC5 overexpression limits EBNALP coactivation. HDACs 1, 3, 6, and 7 overexpression did not effect EBNALP coactivation in reporter assays (data not shown).
The effects of HDAC4 overexpression on EBNA2 activation and EBNALP coactivation were further evaluated by expressing increasing amounts of HDAC4 in cells in which the LMP1p or Cp promoters were activated by EBNA2 or EBNA2 and EBNALP. Interestingly, low levels of HDAC4 overexpression did not inhibit EBNA2-mediated promoter activation, but inhibited EBNALP-mediated coactivation (Fig. 1D). Moreover, high levels of HDAC4 expression inhibited EBNA2 activation by 40% and inhibited EBNALP coactivation by 90% (Fig. 1D). Thus, lower HDAC4 levels are required to inhibit EBNALP LMP1p and Cp promoter coactivation than are required to inhibit EBNA2 activation.
HDAC4 also down-regulated EBNA2 and EBNALP coactivation of LMP1p in Latency I-infected Akata Burkitt's lymphoma cells (Fig. 1E). Indeed, HDAC4 had a significantly greater effect than HDACs 1, 3, 5, 6, or 7, which had a lesser effect (Fig. 1E).
HDAC4 Distribution in LCL Nucleus and Cytoplasm Is Kinase-Dependent.
HDAC4 characteristically shuttles between the nucleus and cytoplasm, and localizes predominantly to the cytoplasm (27). HDAC4 shuttling is determined by phosphorylation by specific protein kinases, including calcium/calmodulin-dependent protein kinases (28–33). In a typical LCL, HDAC4 localized almost exclusively to the cytoplasm (Fig. 2A Left). Treatment with the broad-spectrum kinase inhibitor Staurosporine (34) increased nuclear HDAC4 from 2% to 25% after 3 h of treatment, indicating that 8% of HDAC4 can shuttle to the nucleus each hour (Fig. 2A Right). As expected, p84, a nuclear marker localized to the nucleus and tubulin localized to the cytoplasm, in the presence or absence of staurosporine (Fig. 2A). Interestingly, in IB4 LCL cells, HDAC4 was partly associated with tubulin (Fig. 2A). HDAC5 antibodies were inefficient in HDAC5 immunoprecipitation and Western blot detection, although GST-14-3-3 pull-down followed by Western blot analysis enabled endogenous HDAC5 detection (data not shown).
Fig. 2.

HDAC4 shuttles in a phosphorylation-dependent manner in LCLs and is associated with EBNA2 and EBNALP. (A) SDS/PAGE of mock-treated or staurosporine-treated (Stauro) (5 μM, 3 h) IB4 LCL cells fractionated as described in Materials and Methods. Immunoprecipitation used polyclonal antibodies directed against HDAC4. Immunoprecipitated proteins were detected by Western blot analysis with a monoclonal antibody against HDAC4 (HD4). p84 (nuclear matrix marker) and tubulin (tub) were used as fractionation controls (C, cytoplasmic fraction; N, nuclear fraction; IP, immunoprecipitation proteins; HD4, HDAC4). Quantification of Western blot signal by using Kodak 400R Image Station software is shown. Numbers represent percentages of total immunoprecipitated HDAC4 (cytoplasm plus nuclear) in each experiment. (B) IB4 cells cytoplasmic and nuclear fractions were Western blotted by using polyclonal antibody against HDAC4 or IgG. Proteins were loaded on an SDS/PAGE gel and detected by Western blot analysis with monoclonal antibodies against HDAC4 (abcam Ab12171), EBNALP (JF186), or EBNA2 (PE2). Numbers represent percentages of total HDAC4 in cytoplasmic and nuclear fractions, quantified by using Kodak 400R Image Station software.
HDAC4 Associates with EBNA2 in LCL Nuclei and EBNALP in Nuclei and Cytoplasm.
Because HDAC4 could mediate repression by interaction with transcription factors at promoter site, we tested whether HDAC4 associates with EBNA2 or EBNALP in LCL nuclei or cytoplasm. EBNA2 was only in LCL nuclear extracts, whereas EBNALP was in nuclear extracts but also in the cytoplasm (23) (Fig. 2B). When HDAC4 was immunoprecipitated from nuclear or cytoplasmic extracts by using polyclonal HDAC4 antibody, EBNALP specifically immunoprecipitated with HDAC4 from cytoplasmic and nuclear extracts, whereas EBNA2 precipitated with HDAC4 only from nuclear extracts (Fig. 2B). These data indicate that HDAC4 associates with EBNA2 and EBNALP in nuclei and with EBNALP in the cytoplasm.
EBNALP Reduces HDAC4 Nuclear Localization in B Cells.
The interaction between HDAC4 and EBNALP in the nucleus and the cytoplasm prompted us to evaluate whether EBNALP effects HDAC4 partitioning. HDAC4 distribution in BJAB lymphoblasts was compared with HDAC4 distribution in BJAB lymphoblasts stably expressing EBNALP. Stable EBNALP (ELP+) expression resulted in an 4- to 5-fold decrease in nuclear HDAC4 concentration as is evident in the comparison of HDAC4 in wild-type BJAB nuclei (ELP−) with HDAC4 in EBNALP expressing BJAB nuclei (ELP+) (Fig. 3A). Moreover, cytoplasmic HDAC4 increased 2-fold in ELP+ BJAB cells. Importantly, 3 h inhibition of phosphorylation using staurosporine resulted in a 2- and 7-fold increase in nuclear HDAC4 in BJAB cells and EBNALP expressing BJAB cells, respectively (Fig. 3B, 5 μM Stauro). These data indicate that nuclear HDAC4 levels are down-regulated by EBNALP expression and that the effect is largely phosphorylation-dependent.
Fig. 3.

EBNALP decreases nuclear concentration of HDAC4 and a transcriptionally inactive EBNALP does not interact with HDAC4. (A) Wild-type (ELP−) or stably expressing EBNALP (ELP+) BJAB cells were fractionated, and endogenous HDAC4 was immunoprecipitated by using a polyclonal antibody and detected by using monoclonal HDAC4 antibody versus. Tubulin blot (tub) and p84 blot are included as loading and fractionation controls. Numbers represent quantifications of HDAC4 relative to cytoplasmic HDAC4 in wild-type BJAB, normalized to 1 by using Kodak 400R Image Station software. (B) Mock- or staurosporine-treated (Stauro) (5 μM, 3 h) ELP− or ELP+ BJAB cells were fractionated into nuclear and cytoplasmic fractions, and nuclear fractions were subjected to immunoprecipitation with polyclonal antibodies against HDAC4. Immunoprecipitates were loaded on an SDS/polyacrylamide gel, and proteins were detected by Western blotting with the indicated antibodies. Numbers represent quantifications of HDAC4 calculated as in A. (C) GST-HDAC4 from Escherichia coli was used to pull down wild-type EBNALP (ELP) or 3S/AEBNALP (3S/A ELP) from whole-cell extracts of transfected BJAB cells. Immunoprecipitates were loaded on SDS/PAGE, and Western blot analysis was performed by using JF186 antibody (anti-EBNALP). Numbers represent quantifications of pulled-down ELP, expressed as a percentage of total ELP or 3S/A ELP in each experiment.
A Coactivation-Incompetent EBNALP Mutant Is Deficient in HDAC4 Binding.
EBNALP is serine-phosphorylated and mutation of three critical serines in the EBNALP W repeats to alanine (3S/A) abrogates coactivation with EBNA2, whereas mutation to glutamic acid is wild type in coactivation (20, 24). To test whether the critical EBNALP serines are also required for HDAC4 interaction, GST-HDAC41–618 was used to pull down EBNALP from BJAB cells transfected with wild-type EBNALP or with the coactivation and phosphorylation-deficient alanine triple-point mutant EBNALP(3S/A). GST-HDAC4 pulled down 5% of EBNALP, but only 0.8% of 3S/A EBNALP. Thus, coactivation deficient EBNALP is also deficient in HDAC4 binding. Because an EBNALP triple-point mutant that is deficient in coactivation and phosphorylation is also deficient in HDAC4 binding, EBNALP phosphorylation is likely necessary for HDAC4 binding and movement by EBNALP to the cytoplasm and transcriptional coactivation.
HDAC4 or 5 Overexpression Represses EBNALP Coactivation, and 14-3-3 Relocalizes and Restores Coactivation to the LMP1p but Not Cp Promoters.
To evaluate whether EBNALP coactivation depends on HDAC4 or HDAC5 nuclear export, 14-3-3 overexpression was used to relocalize HDAC4 and HDAC5 to the cytoplasm (27, 35–37). Interestingly, 14-3-3 overexpression did not affect EBNA2 5-fold activation of the LMP1p promoter (adjusted to 1 in Fig. 4A) and doubled EBNALP coactivation from 17- to 33-fold (Fig. 4A). Furthermore, HDAC4 or HDAC5 overexpression reduced EBNALP coactivation from 27- to 11-fold or from 5- to 1-fold and 14-3-3 expression completely restored EBNALP coactivation (Fig. 4 B and C). Moreover, HDAC4 with a point mutant Nuclear Export Sequence (HD4NES) (30) repressed EBNALP coactivation from 7- to 3-fold and 14-3-3 overexpression had no effect on HD4NES repression (Fig. 4D).
Fig. 4.

14-3-3 relieves HDAC4- and HDAC5-dependent repression and enhances EBNALP coactivation with EBNA2 on LMP1p but not on Cp promoters. (A–D) Reporter gene analyses in BJAB cells transfected with the indicated expression plasmids and an LMP1p promoter-luciferase reporter plasmid. EBNA2 activation was normalized to 1. (E) Reporter gene assays performed on BJAB cells transfected with the indicated expression plasmids on the Cp promoter. (F) Consecutive coimmunoprecipitation assay from BJAB cells transfected with EBNALP, HDAC4 (myc- or Flag-tagged), and 14-3-3. Conditions for this assay are described in Materials and Methods. Proteins were coimmunoprecipitated and detected by Western blot analysis with the specified antibodies.
Surprisingly, 14-3-3 overexpression did not abrogate HDAC4- or 5-mediated repression of EBNA2 and EBNALP on Cp promoter activation (Fig. 4E). These data indicate that EBNALP coactivates transcription through different mechanisms at the Cp and LMP1p promoters and that cytoplasmic sequestration of HDAC4 or 5 is not sufficient for reversal of HDAC4 or HDAC5 repressive effects on the Cp promoter.
HDAC4 Is in a Ternary Complex with EBNALP and 14-3-3.
To test whether HDAC4, EBNALP, and 14-3-3 can be in one complex in BJAB Burkitt's lymphoma cells, Flag-tagged HDAC4 was expressed in cells with EBNALP and 14-3-3, immunoprecipitated with Flag-antibody, eluted from beads with Flag peptide, re-immune precipitated with EBNALP antibody, and analyzed by Western blotting. More than 5% of EBNALP and HDAC4 were in the EBNALP and HDAC4 complex along with <5% 14-3-3 (Fig. 4F). These data indicate that HDAC4 and EBNALP stably associate at a high level and are partly complexed with 14-3-3.
Discussion
These data implicate nuclear depletion of HDAC4 in EBNALP coactivation of the EBV LMP1p promoter with EBNA2. Consistent with this model, HDAC4 was associated with EBNA2 in LCL nuclei and with EBNALP in LCL nuclei and cytoplasm. EBNALP expression decreased endogenous nuclear HDAC4 and relocalized HDAC4 to LCL cytoplasm. Treatment of LCLs with staurosporine resulted in nuclear accumulation indicating that cytoplasmic accumulation is the result of a dynamic process of nuclear entry and kinase-dependent nuclear export and cytoplasmic retention. Importantly, EBNALP association with HDAC4 required serine residues that are essential for EBNALP coactivation of the LMP1p promoter with EBNA2, establishing a genetic linkage between EBNALP coactivation and HDAC4 association. Consistent with EBNALP coactivation of the LMP1p promoter being dependent on HDAC4 nuclear export, 14-3-3 overexpression localized HDAC4 to the cytoplasm and potentiated EBNALP coactivation of the LMP1p promoter with EBNA2. Thus, these data strongly support a model in which HDAC4 is associated with EBNA2 at the LMP1p promoter and EBNALP coactivates by decreasing HDAC4 nuclear concentration (Fig. 5).
Fig. 5.

EBNALP model of action. At the LMP1p promoter, corepressor protein complex including HDAC4 and HDAC5 and associated proteins (HP1α and SMRT/NCoR, among others) prevent EBNA2 from increasing promoter activity. EBNALP binding to HDAC4 and HDAC5 results in nuclear depletion of HDAC4 and coactivation.
The effects of HDAC4 overexpression on EBNA2- and EBNALP-mediated transcription in reporter assays and LMP1 expression assays could be due to intrinsic HDAC4 repression at EBNA2 up-regulated promoter sites or to HDAC4-dependent recruitment of a repressor (38, 39). Alternatively, HDAC4 could block EBNA2 interaction with an activator at promoters or inhibit EBNA2 binding to promoters. HDAC4 and HDAC5 associate with large protein complexes, which include transcriptional corepressors and activators at promoter sites (for review, see ref. 32). In particular, HDAC4 and HDAC5 can associate with and repress MEF2 or serum response factor-dependent transcription (28, 37, 40–42). HDAC4 can also recruit proteins with repressive effects, such as HDAC3, mSin3A, SMRT, NCoR, and HP1α (38, 39). HDAC4 and HDAC5 shuttle in and out of the nucleus in a phosphorylation-dependent manner (27, 28, 30, 31, 33, 36, 37, 40). Several protein kinases, including calcium/calmodulin protein kinases II and IV phosphorylate HDAC4 and HDAC5 on specific serine residues and promote 14-3-3 protein binding (28–33). As a result, HDAC4 and HDAC5 relocate to the cytoplasm in a Crm1-dependent manner. Cytoplasmic shuttling regulation de-represses HDAC4 and HDAC5-repressed promoters (27–29, 31, 33, 36, 37, 40).
Our data strongly support a nucleo-cytoplasmic model for EBNALP effects in LMP1p coactivation. EBNALP reduces HDAC4 nuclear concentrations, resulting in coactivation of transcription. Whether EBNALP primarily promotes HDAC4 nuclear export or cytoplasmic retention is under investigation. The shuttling protein HA95 is strongly associated with EBNALP (15, 16), and may mediate HDAC4 export in a Crm-1-independent way (43, 44). Moreover, EBNALP, HDAC4, and 14-3-3 proteins are in a ternary complex, presumably in the cytoplasm, where 14-3-3 proteins anchor HDAC4 (28, 29, 36, 37). EBNALP binding to HDAC4 next to the nuclear localization signal and phosphorylation sites necessary for 14-3-3 binding (data not shown) could also inhibit HDAC4 nuclear import and stabilize 14-3-3 binding.
Our data are consistent with a model (Fig. 5) in which EBNALP coactivates with EBNA2 by displacing HDAC4 and HDAC4 associated repressors such as HP1α from EBNA2-bound promoter sites. EBNALP association with repressors has previously been implicated in coactivation (22). EBNALP displaces Sp100 and HP1α from ND10 bodies. Importantly, an Sp100 mutant deficient in ND10 localization could replace EBNALP in coactivating EBNA2 effects on LMP1p expression; and an Sp100 mutant deficient in HP1α association was inactive in EBNA2 coactivation. These data are consistent with a model that removal of Sp100 and associated HP1α from EBNA2 promoter sites underlies EBNALP coactivation. Because HDAC4 also associates with HP1α (39), EBNALP displacement of HDAC4 from EBNA2 responsive promoters may be the mechanism underlying EBNALP effects observed here. In this way, the data presented here support the importance of repressor displacement in EBNALP coactivation and further implicate HP1α as an important repressor of EBNA2 activation at some promoters.
EBNA2 activation and EBNALP coactivation of the EBV Cp promoter were also effected by HDAC4 and HDAC5 overexpression. Interestingly, HDAC4 effects on Cp were stronger than on LMP1p and were not reversed by cotransfection with 14-3-3 proteins, suggesting a different mechanism of regulation. This data are consistent with our unpublished observations indicating that overexpression of different HDAC4 deletion mutants are required for repression of each promoter. Cp promoter-repressive HDAC4 may be sumoylated, which would result in nuclear retention (45), and hence be unaffected by 14-3-3 overexpression.
One important result from this study is that the transcriptionally incompetent 3S/A EBNALP triple-point mutant cannot interact with HDAC4. Interestingly, other transcriptionally inactive EBNALP mutants bind to HDAC4 (data not shown), suggesting that they may interfere with other aspects of coactivation or transport, further strengthening the hypothesis that EBNALP might coactivate transcription through several mechanisms.
Although they are uniquely essential HDACs (30, 46), HDAC4 and HDAC5 are likely to act through similar mechanisms given their extensive sequence conservation. Interestingly, HDAC5 was the stronger repressor of EBNA2 and EBNALP coactivation of the LMP1p or Cp promoters in reporter assays, but did not repress EBNA2 and EBNALP-dependent endogenous LMP1 expression in Latency I-infected Akata cells (Fig. 1E). HDAC5 levels are low in B cells, and could not be detected by using immunoprecipitation with HDAC5 antibodies and Western blot analysis (data not shown), although HDAC5 could be detected in similar amounts than HDAC4 by using bacterially expressed GST-14-3-3 pull-downs and Western blot analysis. The stronger HDAC5 effect on EBNA2 activation and EBNALP coactivation in reporter assays may be due to HDAC5 more avid nuclear localization (36, 46). Like HDAC4, HDAC5 shuttles to the cytoplasm upon phosphorylation by CaMKIV and 14-3-3 protein binding (28, 29). Thus, HDAC4 and HDAC5 may independently or conjointly repress EBNA2 and EBNALP coactivation.
These data substantially add to the intrinsic complexity of EBNALP coactivation with EBNA2, which appears to be responsive to multiple signals in infected cells, including cell cycle, cell contact, Protein Kinase A, and Casein Kinase II (16, 20, 21). The complexity of these regulatory effects is likely due to the need for regulation of transactivation during initial EBNA2 activation and EBNALP coactivation early in B lymphocyte infection, when EBNALP and EBNA2 are the only EBV proteins that are expressed as well as later in Latency III EBV infection when EBNA2 up-regulation is modified by EBNA3A, EBNA3B, and EBNA3C, which also regulate transcription through RBP-jK/CBF1 (1, 8, 47–52).
Materials and Methods
Expression Plasmids.
pSG5-EBNA2 (wild type) and pSG5-EBNALP (wild type carrying four W1W2 repeat copies and one Y1Y2 copy) expression plasmids were described before (12–14). pSG5-EBNALP (wild type carrying only two W1W2 copies and one Y1Y2 copy), and pSG5–3S/A EBNALP [carrying two W1W2 copies with the triple single amino acid substitution (W-S34,36,63-A) and one Y1Y2 copy], were provided by A. Bell (University of Birmingham, Birmingham, United Kingdom) (24). pBJ5-HDAC1-Flag, pBJ5-HDAC5-Flag, and pBJ5-HDAC6-Flag were kindly provided by S. L. Schreiber (Harvard University, Cambridge, MA); pcDNA3.1-HDAC3-myc, pcDNA3.1-HDAC4-myc, and pGEX2tk-HDAC41–618 were kindly provided by T. Kouzarides (University of Cambridge, Cambridge, United Kingdom); and pcDNA3.1-HDAC7-Flag was a gift from E. Verdin (University of California, San Francisco, CA). pcDNA3–14-3-3 γ-myc was a gift from A. Aitken (University of Edinburgh, Edinburgh, U.K.). pcDNA3-HDAC4-Flag was a gift from C. Brancolini (University of Udine, Udine, Italy).
Cell Lines.
All cell lines were cultured as described (13). BJAB is an EBV negative Burkitt Lymphoma cell line (53). Akata cell line is an EBV-positive strain (54). IB4 is an EBV-infected cord blood lymphocyte derived LCL, which expresses all Latency III genes (55).
Reporter Assays.
Up to 40 μg of total DNA was electroporated into 1 × 107 log phase-BJAB cells by using a Bio-Rad Gene Pulser (2.2 kV, 960 μF). After transfection, cells were incubated at 37°C for 24 h before lysis, and luciferase measurements were normalized by using β-galactosidase values from cotransfected β-galactosidase expression plasmid. Cp and LMP1p reporter constructs were as described (13, 14). Assays were done in duplicate and repeated at least three times with similar results.
Immunodetection.
Detection of proteins was performed by standard Western blot procedures. Antibodies used were as follows: mouse monoclonal anti-EBNALP (JF186) (56), mouse monoclonal anti-LMP1 (S12) (57), mouse anti-Flag (M2; Sigma), mouse anti-myc (9E10; Santa Cruz Biotechnology), rabbit polyclonal anti-HDAC4 (ab12172; Abcam), mouse monoclonal anti-HDAC4 (ab12171; Abcam), mouse anti-p84 (ab487; Abcam), and mouse anti-α-tubulin (Sigma).
Cellular Fractionation.
IB4 cells (5 × 107) were harvested and washed in cold PBS. Cells were suspended in ice-cold buffer A (hypotonic) (10 mM Hepes, pH 7.9/10 mM KCl/1.5 mM MgCl2/5 mM DTT), and protease inhibitors (Complete; Roche) for 20 min in ice. Cell swelling was monitored by microscope (×400 lens). Cells were then transferred to a pre-cooled Dounce homogenizer and homogenized with 20 strokes (type B pestle) in ice. Cell extract was centrifuged at 4°C and 400 × g for 5 min to pellet nuclei. Supernatant (cytoplasm extract) was cleared at 4°C and 9,300 × g for 15 min and transferred to a new tube. The extract was made 150 mM NaCl final by the addition of the appropriate amount of 5 M NaCl before the addition of the antibodies. Nuclei were washed in buffer A, suspended in ice-cold buffer B (hypertonic) (20 mM Hepes, pH 7.9/20% glycerol/420 mM NaCl/1.5 MgCl2/2 mM EDTA/5 mM DTT), and protease inhibitors (Complete; Roche) for 30 min in ice and centrifuged at 4°C and 9,300 × g rpm for 15 min. Supernatant (nuclear extract) was then diluted with the appropriate amount of buffer B without NaCl to make it 150 mM NaCl final, before the addition of the antibodies.
Immunoprecipitation.
Transfected BJAB or IB4 cells were lysed in lysis buffer (1% Nonidet P-40/50 mM Tris·HCl, pH 7.4/2 mM EDTA/150 mM NaCl) or as described for cellular fractionation, and antibodies were added after centrifugation at 4°C and 10,000 rpm for 15 min. Immunoprecipitations were performed at 4°C. After 2–3 h of rotation, protein A-Sepharose (Amersham Pharmacia) was added, and samples were incubated for another 30 min to 1 h. After incubation, complexes were pelleted and washed three to five times in the correspondent lysis buffer. Complexes were eluted by the addition of 2× SDS/PAGE sample buffer and loaded on the gel. Proteins were then transferred to nitrocellulose filters and detected by using the indicated antibodies.
Double Immunoprecipitation.
Conditions were the same as for single immunoprecipitations, except that immunoprecipitated proteins were eluted from the Flag-beads by incubating with 0.5 mg/ml 3× Flag peptide (Sigma) at room temperature for 30 min. Flag-beads were separated from the proteins by centrifugation through a QIAquick spin column (Qiagen). A second immunoprecipitation was then done, as for the first immunoprecipitation.
Bacterial Expressed GST-HDAC41–618 and GST Pull-Down.
Rosetta(DE3)pLysS competent cells (Novagen) were transformed with pGEX2tk-HDAC41–618 or pGEX2tk (empty vector, expressing GST). A single colony was grown in LB media, and expression was induced by the addition of 500 μM IPTG for 3 h at 37°C. Bacteria were harvested, washed, resuspended in 1% Triton X-100 in PBS with protease inhibitors (Complete; Roche), and stored at −80°C. Aliquots were thawed, sonicated, and cleared by centrifugation at 4°C and 9,300 × g for 15 min. Supernatant, containing soluble GST-HDAC41–618 or GST, were incubated with glutathione-Sepharose (Amersham Pharmacia) (50 μl of slurry in PBS) for 2 h with rotation at 4°C. Glutathione-bound GST-HDAC41–618 or GST was washed five times with lysis buffer with protease inhibitors and stored at 4°C. An aliquot of the Sepharose beads-GST-HDAC41–618 or GST was tested for expression and purification in SDS/PAGE, followed by Coomassie stain. For pull-down experiments, GST-HDAC41–618 was added to BJAB total extracts, prepared as described under immunoprecipitation, from cells expressing wild-type EBNALP or 3S/A EBNALP, and incubated at 4°C for 2–3 h. Beads were pelleted and washed five times with BJAB lysis buffer, and proteins were eluted by the addition of 2× sample buffer before loading on the gel. Proteins were visualized by Western blot analysis with antibodies against EBNALP (JF186).
Staurosporine Treatment.
Equal volume of staurosporine or vehicle (DMSO) (Calbiochem) was added to cell cultures to a final concentration of 5 μM for 3 h at 37°C. After treatment, cells were harvested, washed, and lysed as described.
Acknowledgments
We thank Drs. Eric Johannsen, Mike Calderwood, Bo Zhao, Chih-Wen Peng, and Ellen Cahir-McFarland for discussion and advice. This work was supported by National Cancer Institute Grant CA47006.
Abbreviations
- EBNA
Epstein–Barr nuclear antigen
- EBNALP
EBNA leader protein
- HDAC
histone deacetylase
- LCL
lymphoblastoid cell line.
Footnotes
The authors declare no conflict of interest.
References
- 1.Fields BN, Knipe DM, Howley PM, Griffin DE. Fields' Virology. Philadelphia: Lippincott Williams & Wilkins; 2001. [Google Scholar]
- 2.Cohen JI, Wang F, Kieff E. J Virol. 1991;65:2545–2554. doi: 10.1128/jvi.65.5.2545-2554.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen JI, Wang F, Mannick J, Kieff E. Proc Natl Acad Sci USA. 1989;86:9558–9562. doi: 10.1073/pnas.86.23.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannick JB, Cohen JI, Birkenbach M, Marchini A, Kieff E. J Virol. 1991;65:6826–6837. doi: 10.1128/jvi.65.12.6826-6837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. Proc Natl Acad Sci USA. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling PD, Hsieh JJ, Ruf IK, Rawlins DR, Hayward SD. J Virol. 1994;68:5375–5383. doi: 10.1128/jvi.68.9.5375-5383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henkel T, Ling PD, Hayward SD, Peterson MG. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 8.Robertson ES, Grossman S, Johannsen E, Miller C, Lin J, Tomkinson B, Kieff E. J Virol. 1995;69:3108–3116. doi: 10.1128/jvi.69.5.3108-3116.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR. J Virol. 1995;69:253–262. doi: 10.1128/jvi.69.1.253-262.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuentes-Panana EM, Peng R, Brewer G, Tan J, Ling PD. J Virol. 2000;74:8166–8175. doi: 10.1128/jvi.74.17.8166-8175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Grossman SR, Kieff E. Proc Natl Acad Sci USA. 2000;97:430–435. doi: 10.1073/pnas.97.1.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada S, Kieff E. J Virol. 1997;71:6611–6618. doi: 10.1128/jvi.71.9.6611-6618.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng CW, Xue Y, Zhao B, Johannsen E, Kieff E, Harada S. Proc Natl Acad Sci USA. 2004;101:1033–1038. doi: 10.1073/pnas.0307808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng CW, Zhao B, Kieff E. J Virol. 2004;78:11439–11442. doi: 10.1128/JVI.78.20.11439-11442.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han I, Harada S, Weaver D, Xue Y, Lane W, Orstavik S, Skalhegg B, Kieff E. J Virol. 2001;75:2475–2481. doi: 10.1128/JVI.75.5.2475-2481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han I, Xue Y, Harada S, Orstavik S, Skalhegg B, Kieff E. Mol Cell Biol. 2002;22:2136–2146. doi: 10.1128/MCB.22.7.2136-2146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitay MK, Rowe DT. Virology. 1996;220:91–99. doi: 10.1006/viro.1996.0289. [DOI] [PubMed] [Google Scholar]
- 18.Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M, Yokoyama A, Matsuda G, Kato K, Kanamori M, Hirai K. J Virol. 2000;74:10104–10111. doi: 10.1128/jvi.74.21.10104-10111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi M, Kawaguchi Y, Hirai K, Mizuno F. J Gen Virol. 2003;84:319–327. doi: 10.1099/vir.0.18615-0. [DOI] [PubMed] [Google Scholar]
- 20.Kato K, Yokoyama A, Tohya Y, Akashi H, Nishiyama Y, Kawaguchi Y. J Gen Virol. 2003;84:3381–3392. doi: 10.1099/vir.0.19454-0. [DOI] [PubMed] [Google Scholar]
- 21.Kitay MK, Rowe DT. J Virol. 1996;70:7885–7893. doi: 10.1128/jvi.70.11.7885-7893.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling PD, Peng RS, Nakajima A, Yu JH, Tan J, Moses SM, Yang WH, Zhao B, Kieff E, Bloch KD, Bloch DB. EMBO J. 2005;24:3565–3575. doi: 10.1038/sj.emboj.7600820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petti L, Sample C, Kieff E. Virology. 1990;176:563–574. doi: 10.1016/0042-6822(90)90027-o. [DOI] [PubMed] [Google Scholar]
- 24.McCann EM, Kelly GL, Rickinson AB, Bell AI. J Gen Virol. 2001;82:3067–3079. doi: 10.1099/0022-1317-82-12-3067. [DOI] [PubMed] [Google Scholar]
- 25.Peng R, Tan J, Ling PD. J Virol. 2000;74:9953–9963. doi: 10.1128/jvi.74.21.9953-9963.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKinsey TA, Zhang CL, Olson EN. Curr Opin Genet Dev. 2001;11:497–504. doi: 10.1016/s0959-437x(00)00224-0. [DOI] [PubMed] [Google Scholar]
- 27.Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, Bazett-Jones DP, Yang XJ. Mol Cell Biol. 2000;20:6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, McKinsey TA, Zhang CL, Olson EN. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 29.McKinsey TA, Zhang CL, Lu J, Olson EN. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKinsey TA, Zhang CL, Olson EN. Mol Cell Biol. 2001;21:6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miska EA, Langley E, Wolf D, Karlsson C, Pines J, Kouzarides T. Nucleic Acids Res. 2001;29:3439–3447. doi: 10.1093/nar/29.16.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang XJ, Gregoire S. Mol Cell Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Ito A, Kane CD, Liao TS, Bolger TA, Lemrow SM, Means AR, Yao TP. J Biol Chem. 2001;276:35042–35048. doi: 10.1074/jbc.M105086200. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Randall WR, Schneider MF. J Cell Biol. 2005;168:887–897. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertos NR, Wang AH, Yang XJ. Biochem Cell Biol. 2001;79:243–252. [PubMed] [Google Scholar]
- 36.Grozinger CM, Schreiber SL. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youn HD, Grozinger CM, Liu JO. J Biol Chem. 2000;275:22563–22567. doi: 10.1074/jbc.C000304200. [DOI] [PubMed] [Google Scholar]
- 38.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhang CL, McKinsey TA, Olson EN. Mol Cell Biol. 2002;22:7302–7312. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis FJ, Gupta M, Camoretti-Mercado B, Schwartz RJ, Gupta MP. J Biol Chem. 2003;278:20047–20058. doi: 10.1074/jbc.M209998200. [DOI] [PubMed] [Google Scholar]
- 41.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. J Biol Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 42.Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westberg C, Yang JP, Tang H, Reddy TR, Wong-Staal F. J Biol Chem. 2000;275:21396–21401. doi: 10.1074/jbc.M909887199. [DOI] [PubMed] [Google Scholar]
- 44.Yang JP, Tang H, Reddy TR, Wong-Staal F. J Biol Chem. 2001;276:30694–30700. doi: 10.1074/jbc.M102809200. [DOI] [PubMed] [Google Scholar]
- 45.Kirsh O, Seeler JS, Pichler A, Gast A, Muller S, Miska E, Mathieu M, Harel-Bellan A, Kouzarides T, Melchior F, Dejean A. EMBO J. 2002;21:2682–2691. doi: 10.1093/emboj/21.11.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H. J Neurochem. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- 47.Cooper A, Johannsen E, Maruo S, Cahir-McFarland E, Illanes D, Davidson D, Kieff E. J Virol. 2003;77:999–1010. doi: 10.1128/JVI.77.2.999-1010.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Radkov SA, Bain M, Farrell PJ, West M, Rowe M, Allday MJ. J Virol. 1997;71:8552–8562. doi: 10.1128/jvi.71.11.8552-8562.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radkov SA, Touitou R, Brehm A, Rowe M, West M, Kouzarides T, Allday MJ. J Virol. 1999;73:5688–5697. doi: 10.1128/jvi.73.7.5688-5697.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson ES, Lin J, Kieff E. J Virol. 1996;70:3068–3074. doi: 10.1128/jvi.70.5.3068-3074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosendorff A, Illanes D, David G, Lin J, Kieff E, Johannsen E. J Virol. 2004;78:367–377. doi: 10.1128/JVI.78.1.367-377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waltzer L, Perricaudet M, Sergeant A, Manet E. J Virol. 1996;70:5909–5915. doi: 10.1128/jvi.70.9.5909-5915.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menezes J, Leibold W, Klein G, Clements G. Biomedicine. 1975;22:276–284. [PubMed] [Google Scholar]
- 54.Takada K, Horinouchi K, Ono Y, Aya T, Osato T, Takahashi M, Hayasaka S. Virus Genes. 1991;5:147–156. doi: 10.1007/BF00571929. [DOI] [PubMed] [Google Scholar]
- 55.Sample J, Hummel M, Braun D, Birkenbach M, Kieff E. Proc Natl Acad Sci USA. 1986;83:5096–5100. doi: 10.1073/pnas.83.14.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finke J, Rowe M, Kallin B, Ernberg I, Rosen A, Dillner J, Klein G. J Virol. 1987;61:3870–3878. doi: 10.1128/jvi.61.12.3870-3878.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mann KP, Thorley-Lawson D. J Virol. 1987;61:2100–2108. doi: 10.1128/jvi.61.7.2100-2108.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]