

Abstract
The variety of factors and events involved in neurodegeneration renders the subject a major challenge. Neurodegenerative disorders include a number of different pathological conditions, which share similar critical metabolic processes, such as protein aggregation and oxidative stress, both of which are associated with the involvement of metal ions. In this review, Alzheimer's disease, Parkinson's disease and prion disease are discussed, with the aim of identifying common trends underlying these devastating neurological conditions. Chelation therapy could be a valuable therapeutic approach, since metals are considered to be a pharmacological target for the rationale design of new therapeutic agents directed towards the treatment of neurodegeneration.
Keywords: Neurodegeneration, protein aggregation, oxidative stress, Aβ-amyloid, α-synuclein, prion protein, chelation therapy, iron, copper
Introduction
Neurodegeneration is a complex and multifaceted process leading to many chronic disease states. A conventional definition implies a progressive neuronal death, which usually affects a specific population of nerve cells, the vulnerability of which determines the clinical manifestations of a particular neurodegenerative disease. Owing to the prevalence, morbidity and mortality, as well as social, ethical and personal burden of neurodegenerative disorders, considerable effort has been directed towards the identification of a rational strategy to treat these devastating brain pathological conditions. A substantial challenge is to discern phenomena that may represent causes from those that may represent effects. The neurodegenerative process is associated with many events, of which each one may correspond to a typical feature of a specific disease.
The factors that underlie neurodegeneration can be classified as genetic, environmental (extrinsic neurotoxins, e.g. metals, infective damage), biological (aggregation, abnormal folding and accumulation of proteins), metabolic (oxidative stress), excitotoxic (intrinsic neurotoxins, e.g. metals and excitatory amino acids), autoimmunity and ageing. Each of these factors can play a crucial role in the aetiology of neuronal degeneration, but with an influence that varies between the different pathologies.
The major neurodegenerative disorders are the object of intense study since they are characterised by a comprehensive pattern of findings, the interpretation of which can be wide ranging. Striking similarities between different pathologies render investigation complicated, as it is difficult to associate every disorder with specific neuropathological characteristics.
The neurodegenerative disorders discussed in detail in this review are Alzheimer's disease (AD), Parkinson's disease (PD) and prion disease. Reference is made to other neurodegenerative disorders, in order to better understand the general mechanisms of neurodegeneration and to classify the different pathologies. A clear classification can be achieved on the basis of the principal neuropathological changes:
neurodegenerative disorders characterised by the presence of abnormal protein components (Butterfield & Kanski, 2001; Shastry, 2003), which accumulate in the brain leading to a selective loss of neurons in an age-dependent manner such as AD (Aβ-amyloid neuritic plaques and neurofibrillary tangles), PD (α-synuclein, Lewy bodies), prion disease (amyloid plaque core surrounded by ‘petals' of sponge-like tissue, spongiosis) (Prusiner, 2001), Huntington disease (huntingtin protein aggregates) (Bonilla, 2000) and Pick's disease (Pick bodies) (Wisniewsky et al., 1972; Brion et al., 1973);
neurodegenerative disorders resulting from dysfunction/degeneration of motor neurons, which can be characterised by different pathological hallmarks as exemplified in multiple sclerosis (demyelinisation), amyotrophic lateral sclerosis, Friedreich's ataxia and progressive supranuclear palsy.
This simple division does not take into consideration all the factors that play a role in the development of the disease. Thus, the genetic factor has a primary influence on the onset of Huntington disease (one in every 10,000 persons – nearly 30,000 in the US – have Huntington disease, Juvenile Huntington occurs in approximately 16% of all cases), an autosomal dominant polyglutamine disorder that causes involuntary jerky movements and progressive mental impairment, including memory loss. This is caused by a mutation in gene IT15 on chromosome 4 resulting in expression of a glutamine-rich protein called ‘huntingtin', which aggregates into long fibrillar strands that are localised primarily to the nucleus, but may be located elsewhere in striatal neuron. The pathology usually starts at a young age. Neuronal loss occurs in the globus pallidus and in the cerebral cortex and may also be evident, usually to a lesser extent, in the thalamus, subthalamic nucleus and substantia nigra (Adams et al., 1984). Friedreich's ataxia, the most common hereditary ataxia accounting for approximately 50% of all cases of hereditary ataxia (incidence is about one in 50,000, with most studies yielding an incidence among Europeans and North Americans of European descent of approximately 1.5 per 100,000 per year), is an autosomal recessive degenerative disease, characterised by the development of muscle weakness (Alper & Narayanan, 2003). It is caused by the expansion of a GAA triplet located within the first intron of the frataxin gene on chromosome 9q13 (Campuzano et al., 1996). Frataxin is a mitochondrial protein that plays a role in iron homeostasis (Babcock et al., 1997). Deficiency of frataxin results in mitochondrial iron accumulation, defects in specific mitochondrial enzymes, enhanced sensitivity to oxidative stress and eventually free-radical-mediated cell death (Bradley et al., 2000). In this disease, coordination problems are caused by degeneration, at the spinal cord level, of nerves that control muscle movement in the arms and legs. The spinal cord becomes thinner and nerve cells undergo demyelination. In contrast, the immune system is mainly involved in multiple sclerosis (MS), a demyelinating degenerative disease of the central nervous system (CNS), which can be in progressive or relapsing-remitting form, both resulting in permanent neurological damage and disability (Weinshenker, 1995; Compston & Coles, 2002; Keegan & Noseworthy, 2002). The average age of onset for MS is relatively early (20–40 years), with 2 : 1 predominance in women over men. Geographical trends of the incidence of MS have also been noted, with northern and central Europe, northern U.S.A., Canada, south-eastern Australia, parts of the former Soviet Union and New Zealand displaying the highest levels of prevalence (>30 per 100,000 population). The onset of MS is often gradual, and the wide variety of symptoms that can occur overlap to some extent with those of other neurological disorders (Confavreux et al., 1980; Weinshenker et al., 1989). The formation of lesions or plaques (areas of inflammation), demyelination and gliosis, within the white matter of the CNS, are all hallmarks of the pathology (Macchi & Cioffi, 1992). Both genetic and environmental factors contribute to the occurrence of MS and the associated deregulation of the immune system results from several cumulative causes. The loss of neurones in amyotrophic lateral sclerosis (ALS), which is one of the most common neurodegenerative disorders (incidence of five in every 100,000 people (Piemonte & Valle d'Aosta Register for Amyotrophic Lateral Sclerosis (PARALS), 2001)) after AD (2.8 per 1000 person years in the 65–69 years age group to 56.1 per 1000 person years in the older than 90 years age group (Kukull et al., 2002)) and PD (12 and 20 cases per 100,000 per year in the most developed countries (Twelves et al., 2003)), results from a complex interplay of oxidative injury, excitotoxic stimulation, aggregation and/or dysfunction of critical proteins and genetic factors, oxidative stress possessing a major role in the pathogenesis of ALS. Degeneration of cortical and spinal motor neurones is the typical feature of ALS occurring both sporadically and as a familial disorder, with inherited cases accounting for about 10% of patients. On average, the 1990s incidence is close to 1.9 per 100,000 persons year (in southern and northern European countries and in North America). The typical age of onset is around 50 years, but juvenile cases have also been observed (Carrì et al., 2003). Transmissible spongiform encephalopathies (TSEs) or prion diseases form a group of fatal neurodegenerative disorders that have the unique property of being either infectious or sporadic or genetic in origin. They share many pathologic features with the disorders mentioned above, including the importance of oxidative damage of the brain, accumulation of aggregated proteins and neuronal cell loss. Over the past 5 years, a growing interest in the role of metal ions in TSEs has emerged. The exact pathogenic mechanism remains uncertain, but it is believed that oxidative stress plays a central role. The group is represented in humans by the sporadic or hereditary form of Creutzfeldt–Jakob disease, which includes at least 85% of all cases, and by Gerstmann–Sträussler syndrome and fatal familial insomnia, which are linked to the inheritance of point mutations in the prion protein (PrP) gene (Ghetti et al., 1996). The agent responsible for transmission of the disease is thought to be a protein called PrPSc (scrapie isoform of the PrP). PrPSc represents a conformational variant of a normal cellular isoform of the PrP of the host (PrPC). The transition from PrPC to PrPSc is a crucial pathogenic event, and the protein itself can act as the infectious agent (Prusiner, 1982).
Among the causes and the effects observed in all the pathologies listed above, protein aggregation and oxidative stress-induced damage represent a recurring phenomenon. In this review we will focus on both, emphasising functions induced and mediated by metals in the complex process that, together with other factors, eventually leads to neurodegeneration in AD, PD and prion disease.
Protein aggregation in neurodegeneration
There is extensive evidence for the association of protein aggregation and neurodegeneration in many disorders. Interestingly, metals such as iron and copper appear to play an important role in protein aggregation and therefore are likely to provide a link between the two pathological processes of protein aggregation and oxidative damage.
The cause of abnormal protein folding (misfolding) and consequent protein accumulation in the brain is still unclear. Nevertheless, genetic and environmental factors as well as age are all involved. For instance, pathogenic mutations in the genes encoding aggregating proteins, such as Aβ-amyloid, α-synuclein and PrP, are responsible for inherited forms of AD, PD and prion disease, respectively. The mutant proteins, in these examples, show an increased tendency to form so-called amyloid-like fibrils (amyloidogenic activity), the formation of which is pathogenic.
Amyloidogenic proteins can be different in terms of amino-acid sequence and/or native fold, but their corresponding fibrils are structurally similar. Electron microscopy, solid-state NMR and X-ray diffraction techniques indicate the following common features (Antzutkin et al., 2002; Torok et al., 2002; Dobson, 2003a; Sikorski et al., 2003):
unbranched and twisted structures rich in β-sheet;
fibril diameter between 5 and 13 nm;
rigid structures;
so-called ‘cross-β' structure, with β strands perpendicular to the fibril axis and backbone hydrogen bonds parallel to fibril axis;
the presence of a fibril core, which is stabilised by hydrogen bond interactions associated with the polypeptide chain.
It is still a controversial issue whether amyloid fibril formation occurs via self-assembly of parallel or antiparallel β-sheets or indeed β-helices. Both parallel and antiparallel β-sheets have been observed in amyloid fibrils using solid state NMR (Tycko, 2004) (Figure 1).
Figure 1.

Example of short β-sheets arrangements. Aβ-42 parallel (a) and antiparallel (b) possible β-strands. In the parallel β-strand, the C-terminal hydrophobic sequences (in red) are aligned, maybe promoting the aggregation process, which starts the fibril formation cascade. See online for colour figure.
The fibril construction is a cascade process, which involves the formation of intermediate structures (Figure 2). The proteins first coalesce to form small soluble oligomers that aggregate to produce high molecular weight assemblies, so called ‘protofibrils'. The protofibrils eventually exceed solubility limits and are deposited as fibrils (Rochet & Lansbury, 2000; Dobson, 2003b). The current dominant opinion is that the plaques, which deposit surrounding the neurons, are intrinsically toxic (Bence et al., 2001; Bucciantini et al., 2002). However, it is still unclear how their formation leads to cell death. Indeed, it is difficult to propose a common mechanism of toxicity consistent with each different disease. Studies with Aβ-amyloid and α-synuclein have demonstrated that small oligomeric aggregates are more likely to be toxic than the final fibrils (Lambert et al., 1998; Walsh et al., 1999; Hardy & Selkoe, 2002). This concept is consistent with the fact that fibril formation represents the final step of a cascade process, thereby providing a biomarker of neurodegeneration. The following sections focus on three proteins and their relative implications in neurodegenerative diseases: Aβ-amyloid, α-synuclein, PrP.
Figure 2.

Amyloidogenesis: fibril formation. (a) Aβ accumulation; (b) Aβ aggregation (oligomerisation), through (i) parallel or (ii) antiparallel alignment; (c) protofibrils formation; (d) mature fibril formation.
Aβ-amyloid
The essential feature of the AD brain is the presence of extracellular plaques constituted of Aβ-42 peptide deposits. The Aβ isoforms are 39–42 residue peptides, which are formed proteolytically in the cell from a large trans-membrane glycoprotein called amyloid precursor protein (APP). Interestingly, these peptides have been found also in healthy individuals, suggesting a physiological role of Aβ, which is unclear (Vigo-Pelfrey et al., 1993). The toxicity seems to be mainly associated with Aβ-42 (Figure 3).
Figure 3.

APP protein and Aβ-42 peptide amino-acidic sequence. APP Cu-binding site is described. Amino acids involved in the binding are highlighted. Aβ-peptide sequence is presented; key amino acids are indicated in red. His6, Tyr10, His13 and His14, located at the hydrophilic N-terminal, constitute the metal-binding site; Met35 at the lipophilic C-terminal corresponds to the metal-reducing site.
The observation of plaques as end-stage lesions in AD post-mortem brain tissue has led to the assumption that the accumulation of fibrils is responsible for the progression of the disease. Nevertheless, whether the oligomeric form or the fibrillated form (plaques) of Aβ is toxic still remains the object of debate (Lue et al., 1999; Mclean et al., 1999). In fact, the soluble oligomeric forms seem to play a fundamental role in the preclinical and early progression of AD (Klein et al., 2001; Wang et al., 2002). These species are formed soon after the generation of the peptide within specific intracellular vesicles and are subsequently secreted from the cell. After their formation in the cell, Aβ monomers form dimers, trimers and maybe higher oligomers. The monomeric class is the predominant water-soluble fraction and it is typically present in AD brains at six times higher concentration than that detected in control brains (Lue et al., 1999; Mclean et al., 1999). The secreted oligomers can interact with neurons in vivo, affecting their normal function. Correlation between soluble Aβ levels and the extent of synaptic loss and cognitive impairment is strong; even small doses of monomeric or dimeric Aβ-42 can determine a dramatic loss of viability in neurones (Terry et al., 1991; Dickson et al., 1995; Roher et al., 1996). It has been demonstrated that naturally secreted human Aβ alters hippocampal synaptic efficacy at physiological levels (Walsh et al., 2002). The Aβ-protofibrillar metastable forms are also strongly involved in neurotoxicity, especially acting through rapid electrophysiological changes (membrane depolarisation and increase in action potentials), which eventually cause neuronal death (Hartley et al., 1999). The neuronal dysfunction is then initiated by the formation of oligomeric and protofibrillar species.
α-Synuclein
α-Synuclein is the main component of the abnormal protein depositions constituting the Lewy bodies, intracytoplasmic inclusions (5–25 μm diameter) recognized as the hallmark of PD (Duffy & Tennyson, 1965). Little is known about the functions of α-synuclein either in normal or neurodegenerative conditions. α-Synuclein (Figure 4) is a relatively unfolded protein, possessing a random coil secondary structure. The strong electrostatic repulsion associated with the structure at neutral pH is responsible for the lack of the protein folding (Uversky et al., 2001). The nonamyloidogenic core (NAC), the central hydrophobic/amyloidogenic part of the protein, is responsible for the conformational change from random coil to β-sheet (protofibril and fibril formation) (Serpell et al., 2000). Both genetic mutations and exposure to metals accelerate the rate of α-synuclein fibril formation. Significantly high levels of Fe3+ have been found in Lewy bodies. Disruption of α-synuclein membrane-binding ability is related to aggregation process (Paik et al., 1999; Miranda et al., 2000). In accordance with this hypothesis, α-synuclein is present in random coil conformation in cytoplasm and, after translation, becomes associated with the plasma membrane and the vesicular membrane, which both represent its functional sites. At these sites, the protein is in α-helix conformation. The misfolded isoform of the protein may lose the ability to bind to membranes after the translation and thus accumulates as free α-synuclein in the cell. These events are believed to lead to oligomerisation and aggregation in vivo (Hedge & Jagannatha Rao, 2003).
Figure 4.

α-Synuclein schematic representation. α-Synuclein contains three major regions: N-terminal amphipathic α-helical domain, the NAC (nonamyloidogenic core) and the acidic C-terminal region. The sites of mutations responsible for early onset PD are indicated.
It is reported that soluble α-synuclein complexes are more likely to be mediators of neurotoxicity and the accumulation of α-synuclein in cultured human dopaminergic neurons results in reactive oxygen species-mediated apoptosis (Xu et al., 2002). In contrast, α-synuclein is not toxic in nondopaminergic human cortical neurons, where it is reported to exhibit a neuroprotective activity. Apparently, soluble α-synuclein neurotoxicity is dopamine dependent, which might offer an explanation for the selective neuronal loss, observed in PD. It has been proposed that α-synuclein may play a role in the regulation of dopamine biosynthesis by reducing the activity of tyrosine hydroxylase (Perez et al., 2002).
PrP
Prion diseases provide a fascinating example of the relation between protein folding and neurodegenerative disease. From a clinical point of view, prion diseases represent a variety of neurodegenerative states characterised by the presence of spongiform degeneration and astrocytic gliosis in the CNS, associated with rapidly progressive dementia and cerebellar ataxia (Prusiner, 1998). The neuroanatomic distribution of the lesions varies with the specific type of prion disease. Irregular rods of protein aggregates and amyloid plaques, which are resistant to proteolytic degradation, accumulate in plasmalemma, Golgi and intracytoplasmic organelles of neurons. These species, which are formed of two or four subfilaments helically wound around each other, are mainly composed of PrP 27-30 (PrP protease-resistant core, with an apparent molecular mass of 27–30 kDa) (Merz et al., 1981; Cohen et al., 1982; Prusiner et al., 1983; Jeffrey et al., 1994; Laine et al., 2001).
While studying the molecular basis of the disease, it has become clear that protein conformation plays a critical role in the pathogenic process. The conversion of the cellular, primarily α-helical PrP isoform (PrPC) to a β-sheet-rich conformation results in the accumulation of a protease-resistant disease-associated oligomeric isoform called scrapie (PrPSc). Interactions between ‘normal' and ‘abnormal' isoforms of the same proteins are well-known facilitators of aggregation (Prusiner, 1991; Pan et al., 1993). PrPC (Figure 5) is characterised by a flexible and unstructured region of 100 residues at the N-terminal tail, a globular domain of nearly identical size (120–231) containing two short antiparallel β-strands and three α-helices. The protein is present at pre- and postsynaptic level, heterogeneously distributed in healthy adult brain; it is attached to the cell surface (synaptic plasma membrane) via a glycosyl phosphatidylinositol anchor (Stahl et al., 1987). This localisation suggests an involvement in functions such as cell adhesion, ligand uptake and transmembrane signalling (Harris, 2001; Mangé et al., 2002). Since the first structural information was obtained, it has become evident that copper has a key role in the biological function of PrPC. The protein binds copper in a specific manner and apparently this binding induces a conformational transition, which in its turn modulates the protein aggregation (Brown, 2001). In particular, the formation of the disease causing isoform PrPSc involves the refolding of a specific amino-acidic region (90–140) into a β-sheet, and the metal binding might be crucial in this conformational switching (Zecca et al., 2002). Finally, an interesting general theory on diseases characterised by protein deposition finds an appropriate application in prion disease. According to the model proposed, PrPSc provides a template to assist the conversion of nascent PrPC molecules, by lowering the activation energy barrier for the conformational change (Cohen, 1999; Hijazi et al., 2003). If true, the disease-associated isoform would also be disease causing, because its presence would dramatically enhance the conversion of the normal cellular isoform. As observed in AD and PD, the toxic oligomeric protein formation is the first step of a cascade process which leads to the development of β-rich multimeric aggregates. The minimal size for the multimer has not been yet clearly established, but the target size has been indicated as 55 kDa for infectious particle, which corresponds to a dimer (Bellinger-Kawahara et al., 1988).
Figure 5.

Schematic representation of human PrP. Key amino-acidic sequences (51–91 and 92–126) in human PrP are described. In sequence (a), the histidine residues (red) at positions 96 and 111 and the toxic core sequence (blue) are highlighted. In (b), the octa-repeat region is shown, histidine residues (red) are involved in the metal binding. See online for colour figure.
AD, PD, prion disease and oxidative stress
The main agent of risk in most neurodegenerative disorders is age and this may be directly linked to oxidative stress (lipid peroxidation, protein oxidation, DNA and RNA oxidation), which increases in the brain with age and plays a central role in the pathogenic mechanisms of neurodegeneration.
Oxidative stress may be defined as an imbalance between the production of free radicals and the ability of the cell to defend against them through a set of antioxidants and detoxifying enzymes that include superoxide dismutase, catalase and glutathione. When this imbalance occurs, oxidatively modified molecules (lipids, proteins, nucleotides) accumulate in the cellular compartment causing dysfunction (Floyd & Hensley, 2002). In the case of very sensitive cells such as neurons, the lack of control of defence systems may eventually lead to cell death. Under physiological conditions, free radicals are by-products of cellular oxygen metabolism, with superoxide (O2•−), hydroxyl (OH•) and nitric oxide (NO•) species being prevalent, while hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), not radicals themselves, contributing to the cellular redox state and eventually produce radicals through various chemical reactions. All these molecules are referred to as reactive oxygen species (ROS). Mitochondrial oxidative metabolism (the major portion of the total ROS produced during aerobic metabolism comes from by-products of the electron transport chain of mitochondria), nitric oxide, phospholipid metabolism, proteolytic pathways and metal ions are all potential sources of intracellular free radicals. The reactions leading to ROS production occur at all times within the cell. However, under certain conditions, such as stroke, very high levels are produced (Floyd, 1999; Sherki et al., 2001). The brain is at risk from oxidative damage because of the following specific characteristics:
high oxygen consumption (20% of the total body basal O2 consumption);
critically high levels of both iron and ascorbate (crucial in causing membrane lipid peroxidation);
relatively low levels of antioxidant protective agents are present;
tendency to accumulate metals.
The oxidative damage induced by the redox activity of a target protein, which interacts with free radicals and metal ions, has been found as a typical hallmark in the majority of neurodegenerative disorders such as AD, PD and prion disease. This hallmark could in principle be either the primary cause or the consequence of disease progression.
Oxidative stress and AD
Oxidative stress is believed to play a major role in the dysfunction and degeneration occurring in AD, as one of the earliest events that takes place in the cytoplasm of vulnerable neurons, which are restrictively damaged in the pathology.
The chief pathological hallmark of AD is the presence of senile plaques constituted by a highly dense core formed of a mixture of 39–43 residue polypeptides derived from APP that accumulate in the cortical interstitium and cerebrovasculature in a characteristic manner. One such peptide, amyloid β-peptide 1-42 (Aβ1-42) is a minor soluble species but possesses a fibrillogenic activity that renders it central to the pathogenesis and to be particularly toxic to cells in the early stage of the peptide aggregation process (Klein et al., 2001; Wang et al., 2002).
There is strong evidence of a relationship between oxidative stress and Aβ cortical deposits. Interestingly, this correlation seems to depend on the Aβ physicochemical properties, which are consistent with both antioxidant and prooxidant activities. This characteristic of Aβ is likely to derive mostly from its ability to bind metals and, as a consequence, to mediate redox reactions (Butterfield & Kanski, 2001; Butterfield et al., 2001). In fact, Aβ1-42 is a metallo-binding peptide with binding sites for Zn(II), Cu(II) and Fe(III) (Huang et al., 1999) (Figure 3). Metal homeostasis is altered during AD, and as a consequence, metals are reported to accumulate in the neurophil with concentrations that are 3–5-fold increased compared to age-matched controls (Lovell et al., 1998). Three histidine residues (His6, His13 and His14) located in the hydrophilic N-terminal part of the peptide and a methionine (Met35) residue in the lipophilic C-terminal region have been identified as the crucial section involved with metal ion binding. Particularly, the histidine residues identify the site, which binds redox active Cu or Fe, while the methionine residue identifies the second site suggested to be involved in the reduction of Cu(II) and the generation of H2O2.
Based on these findings, an Aβ-induced oxidative stress model has been developed (Atwood et al., 2003). However, it is still unclear whether Aβ generation is a cause or an effect of the oxidative damage observed in the process of neurodegeneration in the AD brain.
Interestingly, metal ion accumulation and oxidative stress are also associated with changes of both soluble Aβ and deposited Aβ concentrations. Nevertheless, when Aβ reaches a concentration sufficient to produce oxidative stress, it induces its own production, so generating a vicious cycle (Bush, 2002). Therefore, if Aβ synthesis is modulated by stress conditions, Aβ production can be considered a response to an increased oxidative stress in the brain, apart from being itself a potential source of additional oxidation processes. Oxidative modifications have been observed in the cell (neuron cytoplasm) and in the extracellular lesions (amyloid plaques) of AD brains. In particular, reversible and rapidly degraded products have been detected in the cell, while stable glycation, carbonyl and lipid peroxidation products have been found in the lesions. This oxidative process is the result of an increased production of membrane permeable H2O2, at both intracellular level (mitochondria) and at extracellular level (activated microglia and Aβ-amyloid deposits) (Bush et al., 1999).
H2O2 is a reactive species, in the presence of redox-active metal ions producing hydroxyl radicals (OH•) via Fenton chemistry equation (1). Since these radicals are only able to diffuse short nanometer distances, the changes observed at nucleic acid and protein sites may identify the sites of both OH• generation and metal ion interaction. Redox active iron(II) is critical in the Fenton reaction equation (1).
Iron is localised in the endoplasmic reticulum, and also in granulis formed of lipofuscin, or age pigment, as well as in their associated vacuoles (Brunk et al., 1992). Lipofuscin is an autofluorescent pigment that accumulates progressively with age within secondary lysosomes. As a result, it has often been used as a marker and index of ageing. It is formed during a process of production of partially reduced oxygen species by mitochondria (via Fenton reaction between iron and H2O2) and following degradation via autophagocytosis within secondary lysosomes. In AD, lipofuscin may play a role in the modulation of the release of iron from damaged mitochondria, which becomes an important generator of H2O2, thus an important site of oxidative damage (Brunk & Terman, 2002).
The accumulation of metals in AD brains as well as the presence of a metal-binding site on Aβ represent promising pharmacological targets. Therefore, compounds with chelation properties, and also with the ability to block the site, so preventing the adverse generation of H2O2 catalysed by the site and the metal-induced protein aggregation, could be useful in the treatment of AD. This concept is discussed in more detail in section Therapeutic strategies.
Oxidative stress and PD: the role of iron
Post-mortem studies in PD brains indicate that a wide range of molecules undergo oxidative damage, including lipids, proteins and DNA (Dexter et al., 1989; Sanchez-Ramos et al., 1994; Alam et al., 1997). In fact, significant neurochemical, physical, histochemical and biochemical evidence confirm the hypothesis that oxidative stress generates the cascade of events, which are responsible of the preferential degeneration of melanised dopaminergic neurons in the substantia nigra pars compacta (SNc) in PD. The following phenomena have all been observed in Parkinsonian brains:
decline in the mitochondria activity (which might result from generation of ROS);
generation of H2O2 following the deamination of dopamine by monoaminooxidase and by autoxidation (dopamine oxidative metabolism);
increased activity of superoxide dismutase (which catalyses the conversion of superoxide anions to H2O2);
reduced concentration of glutathione (responsible for H2O2 clearance);
elevated level of iron in microglia, astrocytes, oligodendrocytes and dopaminergic neurons of SNc (Mizuno et al., 1989; Riederer et al., 1989; Saggu et al., 1989; Halliwell, 1992; Sofic et al., 1992; Gotz et al., 1994; Olanow & Youdim, 1996; Ye et al., 1996; Lan & Jiang, 1997; Jelliger, 1999).
Furthermore, the examination of brain material and the use of a variety of analytical techniques have demonstrated changes in the normal iron and antioxidant concentrations in SNc of PD patients (Sofic et al., 1988; Dexter et al., 1989; Hirsh et al., 1991; Olanow, 1992; Sanchez-Ramos et al., 1994; Alam et al., 1997). Interestingly, a study, in which antibodies against ferritin were used, indicated no increase in neuronal ferritin, suggesting that the elevated iron is unbound and therefore potentially reactive (Connor et al., 1995). Such iron would be able to initiate ROS-dependent oxidative stress in nigrostriatal dopamine neurons.
Synucleyns
Iron can in principle mediate the generation of hydroxyl radicals by interaction with α-synuclein, a 140-amino-acid presynaptic protein that accumulates intracellularly in the form of fibrillar aggregates in neurons and sometimes also in glia cells. The synucleins (α-, β- and γ-) are highly expressed proteins in the nervous tissue. Two missense mutations in the α-synuclein gene (A53T and A30P) are responsible for rare forms of familial PD. As a result of these mutations, the α-helical structure of the protein is disrupted and β-pleated sheet conformations are favoured, increasing the ability of the protein to self-aggregate. The accumulation of fibrillar forms of the protein is one of the decisive events occurring in the pathogenesis of PD (Ueda et al., 1993; Polymeropoulos et al., 1997; Conway et al., 1998; El-Agnaf et al., 1998; Krüger et al., 1998). The toxicity in part may be generated by oxidative damage, induced by interactions between α-synuclein fibrils and iron (Paik et al., 1999; Miranda et al., 2000). Such phenomena are supported by electron spin resonance investigations (Turnbull et al., 2001).
Neuromelanin
The vulnerability of dopaminergic neurons of SNc studies in PD has also been related to the presence of the dark-coloured polymer pigment neuromelanin (NM). Melanins are widely distributed in different body tissues and, due to their ability to bind and to concentrate transition metals, they act as protective agents against oxidative stress. The presence of NM in the human SNc suggests a similar function in the brain. The structure of NM has only been partially characterised, and its biosynthesis is still unclear (Double et al., 2003). Nevertheless, it has been hypothesised that the SNc form of NM may be the result of a complex biosynthesis starting with a reaction of autoxidation of dopamine (Figure 6). A number of different intermediates are formed such as 5,6-dihydroxyindole, 5,6-dihydroxyindole-2-carboxylic acid and cysteinyl-dopamine (Swan, 1974); coupling reactions between these substrates generate a polymer with a very high tendency to aggregate. In fact, the planar aromatic groups are able to form interconnected layers stacked together generating a domain. On the basis of UV, IR, X-ray diffraction analysis data (Cheun, 2004), both covalent bonds and hydrophobic interactions have been implicated in the formation of this multilayer three-dimensional structure. The structure of this polymeric matrix suggests high chelation ability, mainly related to the presence of phenolic groups. This metal-binding property of NM may have a role in the progression of PD (Double et al., 1999; 2000).
Figure 6.

Hypothesised pathway for the biosynthesis of neuromelanin.
Iron-binding studies have demonstrated that both natural and synthetic human NM bind iron in a saturable manner. In particular, the analysis of nonlinear binding curves reveal two classes of binding site, only one of which is a high-affinity iron site (Ben-Shachar et al., 1991; Youdim, 1994). Ferric iron is bound by NM phenol groups, forming a high spin complex with six-fold oxygen octahedral configuration in an iron oxide-hydroxide cluster form (Figure 7), a structure which as been confirmed by both X-ray absorption fine-structure spectroscopy and infrared spectroscopy (Kropf et al., 1998; Brindelli et al., 1999).
Figure 7.

Model for NM-iron coordination. Units I and II can react to form the polymer. Unit I provides a planar system and catechol functions to bind iron; unit II will be axial to this plane, hence providing crossinteractions between adjacent layers and the third coordination site.
In normal brain, NM is only 50% loaded with iron, retaining a high potential for chelation in situations where intraneuronal iron concentrations suddenly increase. Under physiological conditions, iron bound to the NM high-affinity binding sites is redox inactive and NM may thus be considered as a protective agent in the neuron. On the other hand, in pathological conditions (PD), when iron concentration increases in the brain, the high-affinity binding sites may become saturated, and low-affinity sites would then bind iron. In this case, a proportion of the bound iron would be redox active, demonstrating a toxic potential. Thus, the balance between antioxidant and pro-oxidant action of NM could be dependent on the brain iron concentration (Double et al., 2002).
Neuromelanin loss is another issue to consider. This occurs early in the pathology and the subsequent pallor of the SNc can be a first clue of PD (Kastner et al., 1992). The NM concentration in SN is very low up to the age of 2–3 years and then it continuously increases until the end of life with level approaching 3.5 μg/mg. In patients with PD, this value is reduced to 1.2–1.5 μg/mg (less than 50% of that seen in age-matched controls). Iron levels in SN are normally 20 ng/mg in the first year of life increasing to about 200 ng/mg at the forth decade of life and then remaining stable until 90 years of age (Zecca et al., 2001b; 2002). In PD, the cytoplasmic iron concentration selectively increases by 30–35% only in SNc and the normal consequential increased ferritin expression is not observed, suggesting that free iron concentration is altered in the tissue (Zecca et al., 2001a). The presence of extraneuronal NM has been observed in subjects with PD. Extracellular NM is phagocytosed by microglia-inducing astrocytic and microglial activation, chemotaxis and release of neurotoxic factors (in microglia cell cultures) (Mc Geer et al., 1998; Wilms et al., 2003). Therefore, even though NM may play a neuroprotective role at the intracellular level, once in the extracellular environment, it might cause a cytotoxic effect. In subjects with idiopathic PD, insoluble extraneuronal NM granules, released after SN neuron death, can remain in the extracellular compartment for long periods, leading to microglia activation and therefore production of neurotoxic mediators (Zecca et al., 2003).
In conclusion, it is evident that the relationship existing between metals, α-synuclein, NM and oxidative stress is crucial in the pathology of PD.
Prion disease: the role of metals (copper, manganese, iron)
The conversion of PrPC (cellular isoform of PrP) to PrPSc (scrapie isoform of PrP) is the key event in the pathogenesis of TSEs. How this conversion occurs is still unclear. Nevertheless, in the last 10 years, the role of metal ions, copper in particular, has been shown to have a critical function in the physiopathology of prion diseases, and in the process leading to the conversion of PrPSc from PrPC. The first experimental result, demonstrating that these disorders were related to metal ions, was reported in the early 1970s, when the copper chelator cuprizone was found to induce histopathological changes in mice similar to those induced by scrapie (Pattison & Jebbet, 1971).
Recently, studies have been undertaken to verify the concentrations of metals in mice affected by prion disease. Interestingly, changes of copper levels were observed in samples of brain, liver and blood. In particular, a reduction of Cu(II) was found in the brain, while significant increases were observed in the liver. Moreover, the same study showed elevated manganese levels to be widespread in the body (see below) (Thackray et al., 2002). PrPC is a surface glycosyl-phosphatidyl-inositol (GPI)-anchored monomeric protein of 253 amino acids that are organised in different domains (Figure 5). PrPC is protease sensitive and mostly expressed by neural tissue at the synaptic level. The protein is characterised at its amino terminus by five octapeptide repeats (consisting of amino residues 51–91 of the human PrP sequence). This 40 residue region and, in particular, histidine residues appear to bind up to five Cu(II) ions with a high affinity, which increases in a cooperative way from micromolar to fentomolar magnitude. A second high-affinity binding site for Cu(II) is located on the carboxy-terminal side, where two histidine residues (His96 and His111) might be specifically involved in the binding. It is likely that the copper binding induces a conformational change at the amino-terminal tail of the protein (Brown et al., 2004). There is strong evidence that the expression of PrPC is necessary for the manifestation of prion disease, as genetically modified mice lacking the PrPC gene are resistant to infection with mouse prion inoculum (Büeler et al., 1993). Moreover, these mice are also more sensitive to the toxicity of both copper and superoxide species. The physiological function of PrPC is unknown and is the object of several hypotheses. For example, it has been suggested that it possesses a superoxide dismutase (SOD)-like activity. Indeed, both purified native and recombinant PrPC have been demonstrated to possess a catalytic activity similar to that of SOD (Brown et al., 1997a, 1997b). In order to be activated, the PrPC is reported to bind copper and the C-terminal seems to be important for this activity. Accordingly, cerebellar cells from mice deficient in PrPC reveal increased sensitivity to the toxicity of copper-containing salts, a decrease in membrane copper content and a decreased activity of SOD (Brown et al., 1997a, 1997b). These data suggest a role for this protein in plasma membrane copper uptake and delivery of copper to specific target proteins. On the other hand, copper itself might regulate the binding of the protein to blood factors, such as plasminogen (Maissen et al., 2001).
The agent responsible for the transmission of the disease is thought to be PrPSc, which is the conformational variant of PrPC. The generation of PrPSc from PrPC follows a still unclear process and the only difference between the two isoforms appears to be structural. Furthermore, there is strong evidence for the existence of different PrPSc conformational variants. PrPSc characteristically has greater β-sheet content than PrPC, which is α-helical rich. It is a protease-resistant protein existing in form of extracellular aggregates. An interesting feature of PrPSc is that it does not only bind copper specifically, but also manganese. A study undertaken on a mouse scrapie model has demonstrated changes in the levels of copper and manganese in the brain of scrapie-infected mice prior to the onset of clinical symptoms (Brown et al., 1997a, 1997b). In particular, a major increase of manganese in the blood was detected in the early stages of disease. According to this study, the observed trace-metal changes could result from changes in the binding affinity of PrP when it converts from PrPC to PrPSc. Manganese binding to PrP induces β-sheet conformation and so stimulates the formation of fibrils, which generate plaques similar to those of AD. Moreover, manganese binding enhances protease resistance of PrPSc (Brown et al., 2000).
Metal ions and their chelators affect the biochemical properties of PrPSc, possibly by modifying the metal ion occupancy of the molecule; this aspect is crucial in terms of the differences between PrPC and PrPSc. Therefore, it is possible that metal imbalance triggers the conversion from PrPC to PrPSc. All these changes occur prior to the appearance of clinical symptoms, and could be involved in the onset of the pathology associated with the disease.
Oxidative stress events in the brain (lipid peroxidation, decrease in neuronal nitric oxide synthase activity) are detectable in prion disease in both infected animals and cultures (Guentchev et al., 2000; Lehmann, 2002). In particular, the concentrations of total iron, Fe(III) and the Fe(II)/Fe(III) ratio were reported to be increased in the brain of infected mice (Kim et al., 2000). Recently, the widely studied toxic peptide fragment of the PrP, PrP106-126, has been reported to be Fenton active in the presence of copper, suggesting that the cytotoxic effects of the peptide could be due to the ability to generate H2O2 as well as to aggregate (Turnbull et al., 2003).
Therapeutic strategies
The complexity of the neurodegenerative process renders the identification of a therapeutically useful approach difficult. Current treatment options are limited and are designed to alleviate the symptoms of disease. By virtue of the existence of multiple unknown factors, it is difficult to readily identify a target. Nevertheless, numerous attempts have been made to treat several of the recognised causative issues. A wide range of drugs with diverse mechanisms of action has been investigated. Although some of these approaches have demonstrated potential, when studied in cellular and animal models of acute or chronic neurodegeneration, only a few have provided convincing clinical results. Among the relevant therapeutic strategies, it is noteworthy to mention the use of antioxidants (Moosmann & Behl, 2002), excitotoxicity modulators (Bensimon et al., 1994; Hely et al., 2000; Reisberg et al., 2003) and alternative approaches such as the inhibition of the expression of amyloidogenic protein (De Felice & Ferreira, 2002), inhibition of the release of amyloidogenic peptide and inhibition of Aβ-amyloid aggregation (Conway et al., 2003; Lahiri et al., 2003; reviewed in Mason et al., 2003).
Oxidative stress, protein aggregation and redox active metal ions are all considered to be promising pharmacological targets. The apparently critical involvement of metals, particularly iron and copper, in both oxidative stress and protein aggregation processes therefore renders chelation therapy a sensible strategy. The interesting feature of a suitable chelating agent would be the ability firstly to scavenge the free redox active metal present in excess in the brain to form a nontoxic metal complex, which is then excreted, and secondly to cap the metal at its labile binding site (Aβ-amyloid, α-synuclein, PrP), preventing any mediated toxic action (Fenton activity and/or aggregation). In this second case, the newly formed stable metal complex would favour the state in which the metal is not redox active and therefore not toxic. The latter mechanism implies additional interactions between the drug and the target protein, which have to be considered in the design.
Design features of clinically useful metal chelators
One of the dominant properties of any therapeutic chelator is metal selectivity, typically a high selectivity being required, for instance in the treatment of iron overload associated with β-thalassaemia. In this latter situation, ligands with a high selectivity for iron over copper and zinc are essential, as chelation therapy is maintained for life. Unfortunately, with the proposed treatment of neurodegenerative diseases by chelation therapy, the identity of the putative toxic metal is not always firmly established. With AD, for instance, iron, copper and zinc have all been associated with the progression of the disease. In contrast, with PD iron is clearly the major target.
Although there are clear guidelines for the design of iron-selective chelating agents (Liu & Hider, 2002a), this is not the situation with copper and zinc. Furthermore, with the necessity of ready permeation of the blood–brain barrier (BBB), the size of useful chelators should probably be limited to less than 300Da, thereby excluding hexadentate ligands and seriously limiting the potential for the design of selective copper(II) and zinc(II) chelators (vide infra). Any agent that binds copper(II) tightly will also bind iron(II), zinc(II), nickel(II), cobalt(II) and manganese(II), thereby causing a potential toxic insult to most cell types (Liu & Hider, 2002b). This limitation is a major issue for the design of chelators with potential for treating neurodegeneration.
In principle therefore, there are two major classes of ligands required:
iron selective chelators for the treatment of PD;
iron/copper/zinc chelators for the investigation and eventual treatment of AD; inevitably, this latter group of chelators will be more toxic, at a given dose, than iron-selective chelators.
Iron-selective ligands
Chelating agents can be designed for either the iron(II) or the iron(III) oxidation state. Chelators that prefer iron(II) use nitrogen and sulphur atoms as ligands, for instance 2,2′-bipyridyl (2, Figure 8). Although these compounds are selective for iron(II) over iron(III), they retain an appreciable affinity for other biologically important bivalent metals such as copper(II) and zinc(II) (Table 1). Thus, the design of a low molecular weight nontoxic iron(II)-selective ligand is extremely difficult and indeed may not be possible. In contrast, iron(III)-selective chelators favour oxygen atoms as ligands, notably hydroxamates and catecholates (Table 1). Most tribasic cations, for instance aluminium(III) and gallium(III), are not essential for living cells and thus iron(III) is a practical target for ‘clinical chelator' design. An additional advantage of high-affinity iron(III) chelators is that, under aerobic conditions, they will chelate iron(II) and facilitate autoxidation to iron(III) (Harris & Aisen, 1973). Therefore, high-affinity iron(III)-selective ligands bind both iron(III) and iron(II) under most physiological conditions.
Figure 8.

General structure of iron(III) chelators. Hexadentate: desferrioxamine (DFO, 1), ethylenediaminetetraacetic acid (EDTA, 6); bidentate: 2,2′-bipyridyl (2), N,N-dimethyl-2,3,-dihydroxybenzamide (DMB) (3), acetohydroxamic acid (4), deferiprone (5), 1-hydroxypyridin-2-one (7), 3-hydroxypyridin-2-one (8), 1,10-phenanthroline (9), 8-hydroxyquinoline (10), 4-hydroxypyridopyrazine (11).
Table 1.
Metal affinity constants for selected ligands (Martell & Smith, 1974–1989)
| Ligand | Log cumulative stability constant | ||||||
|---|---|---|---|---|---|---|---|
| Fe(III) | Al(III) | Ga(III) | Cu(II) | Zn(II) | Fe(II) | pFeIIIa | |
| DFO (1) | 30.6 | 25.0 | 27.6 | 14.1 | 11.1 | 7.2 | 26 |
| 2,2′-bipyridyl (2) | 16.3 | — | 7.7 | 16.9 | 13.2 | 17.2 | — |
| N,N-dimethyl-2,3,-dihydroxy-benzamide (DMB) (3) | 40.2 | — | — | 24.9 | 13.5 | 17.5 | 15 |
| Acetohydroxamic acid (4) | 28.3 | 21.5 | — | 7.9 | 9.6 | 8.5 | 13 |
| 3-hydroxypyridin-4-one (deferiprone) (5) | 37.2 | 35.8 | 32.6 | 21.7 | 13.5 | 12.1 | 19 |
| EDTA (6) | 25.1 | 16.5 | 21.0 | 18.8 | 16.5 | 14.3 | 23.4 |
| 8-hydroxyquinoline (10) | 37.7 | — | 40.5 | 22.9 | 15.8 | — | 20.6 |
pFe3+=−log[Fe3+] when [Fe3+]total=10−6 M and [ligand]total=10−5 M at pH 7.4.
Hexadentate: DFO (1), EDTA (6); bidentate: 2,2′-bipyridyl (2), N,N-dimethyl-2,3,-dihydroxybenzamide (DMB) (3), acetohydroxamic acid (4), deferiprone (5), 8-hydroxyquinoline (10).
General requirements of iron(III) complexes with therapeutic potential
Ligands can be structurally classified according to the number of donor atoms that each molecule possesses. When a ligand contains two, three, six or more donor atoms, it is termed bidentate, tridentate, hexadentate or generally multidentate, respectively (Figure 9). Under biological conditions, the pM (pM is defined as the negative logarithm of the metal ion concentration under the following conditions: [Metal Ion]total=10−6 M, [Ligand]total=10−5 M at pH 7.4) value is a more useful parameter than the conventional stability constant to assess the ligand affinity for the metal (Raymond et al., 1984; Liu & Hider, 2002a); for clinically useful iron scavengers, a pFe3+ value ⩾20 (Table 1) is considered to be essential. Molecular size is also a critical factor, as it influences the penetration of both the wall of the gastrointestinal tract (Holander et al., 1988) and the BBB (Oldendorf, 1974). In order to achieve greater than 70% oral absorption, the chelator molecular weight should be <500 (Maxton et al., 1986). This molecular-weight limit provides a considerable restriction on the choice of chelator and may effectively exclude hexadentate ligands from consideration. Bidentate and tridentate ligands, by virtue of their much lower molecular weights, are predicted to possess higher absorption efficiencies. The fraction of the absorbed dose for a range of bidentate hydroxypyridin-4-ones (e.g. 5, Figure 8) has, for instance, been found to fall between 50 and 70%, as assessed in the rabbit (Yokel et al., 1995). This type of molecule has also been demonstrated to penetrate the BBB (Habgood et al., 1999). Hydroxypyridin-4-ones are monoprotic acids at pH 7.0 and thus form the neutral tris-iron(III) complexes. Furthermore, they possess a high affinity for iron(III) and are selective for tribasic metal cations over dibasic cations (e.g. deferiprone (5, Figure 8) in Table 1).
Figure 9.

Schematic representation of chelate ring formation in metal–ligand complexes.
Toxicity of iron(III) chelating agents
The toxicity associated with iron chelators originates from a number of factors, such as inhibition of iron-containing metalloenzymes, lack of metal selectivity, redox cycling of iron-complexes between iron(II) and iron(III) (Figure 10) and the kinetic lability of the iron complex leading to iron redistribution. General properties, which can potentially lead to toxicity, of bidentate and tridentate iron(III) chelators are summarised in Table 2. If all the coordinating ligands are charged oxygen atoms (as it is possible in the case of bidentate ligands), then the iron complex will favour the most oxidised form of iron thus preventing the redox-cycling activity. In contrast, if some of the coordinating ligands are nitrogen atoms (as in the case of tridentate ligands), then redox cycling becomes possible with the concomitant generation of free radicals. Tridentate molecules also possess the disadvantage of being able to form polymeric, high molecular weight, metal complexes, which in principle can become trapped intracellularly.
Figure 10.

Redox cycling activity of iron complexes. The reducing agent could be vitamin C or other reduced coenzymes, for instance NADPH.
Table 2.
General properties of bidentate and tridentate ligands associated with potential toxicity
| Bidentate ligands | Tridentate ligands |
|---|---|
| All the coordinating atoms can be oxygen and this renders ligands highly selective for iron(III) | It is very difficult that all the coordinating atoms can be oxygen and this may lead to poor metal selectivity |
| Labile-iron redistribution is possible | Labile-iron redistribution is possible |
| Partially coordinated 2 : 1 complexes can be formed | Partially coordinated 1 : 1 complexes can be formed |
| Polymeric complexes are not formed | Polymeric complexes, which are likely to be trapped within cells, can be formed |
In order to prevent free radical production, iron should be coordinated in such a manner as to avoid direct access of oxygen and H2O2. Most hexadentate ligands such as desferrioxamine (DFO) (1, Figure 8) are kinetically inert and reduce hydroxyl radical production to a minimum by entirely masking the surface of iron (Figure 11). However, not all hexadentate ligands are of sufficient size to completely envelope the bound iron, in which case the resulting complex may enhance the ability of iron to generate free radicals. This phenomenon is particularly marked at neutral or alkaline pH values when the solubility of noncomplexed iron(III) is severely limited. Bidentate and tridentate ligands are kinetically more labile and the iron(III) complexes tend to dissociate at low ligand concentrations (Scheme 1). Unlike hexadentate ligands, both bidentate and tridentate ligands can form partially coordinated iron complexes. The existence of such iron complexes renders the iron(III) cation surface accessible to H2O2 and thus to possible free radical production.
Figure 11.
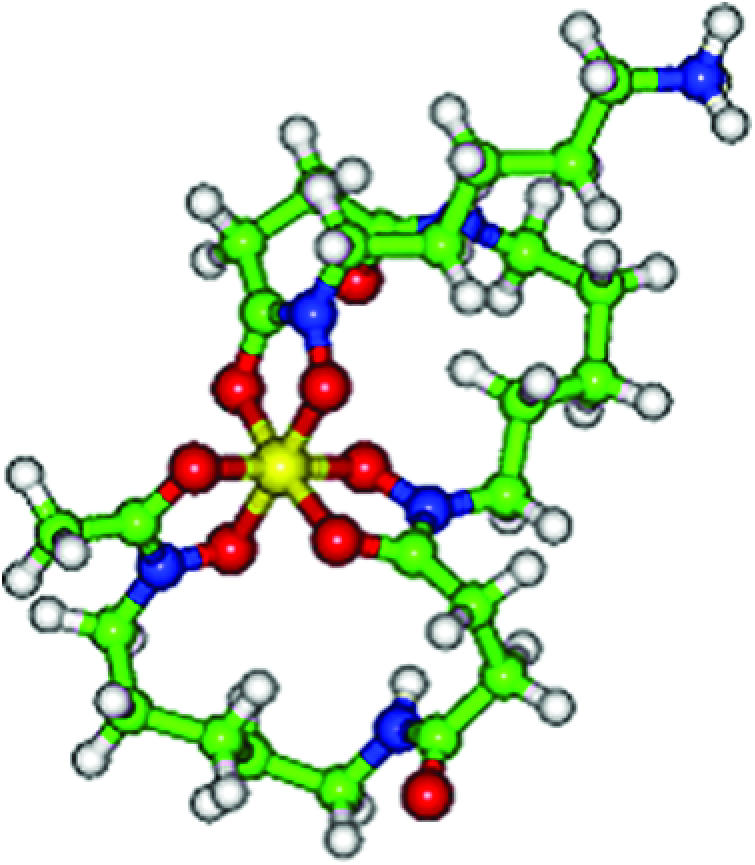
Energy minimised structure of desferrioxamine.
Scheme 1.

Dissociation of bidentate chelator-iron 3 : 1 complexes. The reaction generates the partially coordinated 2 : 1 complexes which render the iron(III) cation surface accessible to oxygen and H2O2 and thereby susceptible to possible hydroxyl radical generation.
In general, iron chelators do not directly inhibit haem iron-containing enzymes due to the inaccessibility of porphyrin-bound iron to chelating agents. In contrast, in many non-haem iron-containing enzymes such as lipoxygenase, the aromatic hydroxylase families and ribonucleotide reductase are susceptible to chelator-induced inhibition (Hider, 1995). Generally, hydrophobic chelators inhibit lipoxygenases, therefore the introduction of hydrophilic characteristics into a chelator tend to minimise such inhibitory potential (Abeysinghe et al., 1996), particularly if their introduction also induces steric interference of the chelation process at the enzyme active site (Liu et al., 2002). By careful modification of physicochemical properties, iron chelators can therefore be designed which exert minimal inhibitory influence on many metalloenzymes (Cooper et al., 1996; Liu et al., 2001).
In summary, a chelator suitable for the treatment of PD should possess a molecular weight in the region 200–400, should bind iron(III) much more tightly than iron(II), should not redox cycle or generate hydroxyl radicals under physiological conditions and should not interfere with metalloenzyme activity. Hydroxypyridin-4-ones are suitable candidates, by virtue of their high selectivity for iron and their generally favourable physical–chemical properties. Deferiprone (5, Figure 8), a hydroxypyridinone, has been used clinically for over 10 years in the treatment of transfusion-induced iron overload. It forms a stable 3 : 1 ligand iron complex, which is water soluble and readily excreted by the kidneys (Olivieri et al., 1990). Furthermore, deferiprone readily crosses the BBB (Habgood et al., 1999).
Iron/Copper/Zinc-binding ligands
Chelators with a broad selectivity for transition metals generally use nitrogen atoms as ligands, for instance 2,2′-bipyridyl (2, Figure 8) and 1,10-phenanthroline (9, Figure 8). These two general classes possess high affinities for iron, copper and zinc cations (Table 1). However, the metal complexes of these ligands are positively charged, tend to bind to membranes and, by virtue of their net charge, do not penetrate membranes readily. Thus, they are inefficient at excreting intracellularly localised transition metals. In contrast, the 8-hydroxyquinoline (10, Figure 8) chelators contain both oxygen and nitrogen atoms and hence possess intermediate properties between those of the di-nitrogen ligands and the di-oxygen ligands (Table 1) (Hider & Hall, 1991; Liu & Hider, 2002b). More importantly, because 8-hydroxyquinoline is monobasic, it forms neutral 3 : 1 complexes with iron(III) and neutral 2 : 1 complexes with both copper(II) and zinc(II). Thus, in principle, it can remove these metals from cells. However, because 8-hydroxyquinoline binds both iron(III) and iron(II) relatively tightly, iron can redox cycle between these two oxidation states with relative ease, thereby generating toxic oxygen radicals (Figure 10). Although hydroxyquinoline derivatives are used in many parts of the world for the treatment of diarrhoea (World Health Organ (WHO) Tech Rep Ser (1977); Claesen & Clements, 1989), their use has been criticised (Chetley & Gilbert, 1986) due to associated toxicity (Palm, 1932; Rose & Gawel, 1984).
At the present time, there is no obvious solution to the design of a nontoxic ligand with high affinities for iron, copper and zinc and the ability to mobilise such metals from intracellular sites.
Chelators investigated for their potential in the treatment of neurodegenerative diseases
Hexadentate chelators
Two hexadentate ligands have been investigated for the treatment of neurodegenerative disease, desferrioxamine (1, Figure 8) and a synthetic amino-carboxylate ligand, DP-109 (12, Figure 12).
Figure 12.

Chelators investigated for their potential in the treatment of neurodegenerative diseases. Hexadentate: DP-109 (12); tridentate: isonicotinoyl picolinoyl hydrazine, IPH (13); bidentate: bathocuproine (14), feralex (15), clioquinol (16), VK-28 (17).
A 2-year, single blind study was undertaken to investigate whether the progression of AD dementia could be slowed by desferrioxamine (Crapper Mclachlan et al., 1991). With this purpose, 48 patients were assigned to three different groups: DFO treated (125 mg DFO given intramuscularly twice daily, 5 days per week, for 24 months), oral placebo (lecithin) and no treatment. Activities of daily living were monitored and recorded over 24-month period at regular intervals. No differences were observed in the rate of deterioration of patients receiving either placebo or no treatment. In contrast, it was reported that DFO treatment led to a significant reduction in the rate of decline of daily living activities, leading to the conclusion that sustained administration of DFO might slow the clinical progression of dementia associated with AD. Although interesting, these findings are surprising because, by virtue of this molecule's hydrophilic nature and size, it does not penetrate the BBB and hence, access to the brain could only be achieved in the presence of a damaged BBB. In contrast, DP-109 has been designed as a pro-drug, and optimal metal chelation will only occur subsequent to the cleavage of the two long-chain ester functions (Lee et al., 2004). Indeed, this molecule has been demonstrated to possess a strong inhibition activity on plaque formation and deposition in female hAβPP-transgenic Tg 2576 mice (Hsiao et al., 1996; Lee et al., 2004). Nevertheless, under in vivo conditions, a large trans BBB flux is unlikely to be achieved due to the high molecular weight of DP-109 (>1000) and the surface activity of this strongly amphiphilic molecule.
Tridentate chelators
Aroyl hydrazone ligands are currently under investigation for their ability to penetrate the CNS. The neutral class typified by isonicotinoyl picolinoyl hydrazine (IPH) (13, Figure 12) would appear to be promising. These molecules coordinate metals via two nitrogen atoms and a single oxygen, and thus bind iron(III), copper(II) and zinc(II) tightly (Richardson, 2004). Work with animal studies is eagerly awaited, although redox cycling with iron may be a problem.
Bidentate chelators
Several ranges of bidentate ligands have been investigated for their potential to cross the BBB and to treat neurodegeneration. The hydrophobic phenanthroline analogue, bathocuproine (14, Figure 12), has been demonstrated to facilitate the solubilisation of Aβ from AD brain samples (Cherny et al., 2000). However, all metal complexes of this ligand are positively charged and are, therefore, unlikely to penetrate the BBB. In contrast, a number of 3-hydroxypyridin-4-ones (e.g. 5, Figure 8) have been demonstrated to penetrate the BBB (Habgood et al., 1999). This ability is due to the formation of neutral complexes with both iron(III) and copper(II) and, therefore, they have the potential of facilitating metal efflux from the brain.
Another interesting molecule, feralex (15, Figure 12), a closely related ligand to 3-hydroxypyridin-4-ones, has been reported to disaggregate in vitro hyperphosphorylated τ-protein, which is responsible for the formation of neurofibrillary tangle in AD (Shin et al., 2003).
To date, a range of 8-hydroxyquinoline analogues have demonstrated the greatest potential for the treatment of neurodegeneration and one compound, clioquinol (CQ) (16, Figure 12), has entered clinical trial. CQ is a small, lipophilic bioavailable metal chelator, which leads to beneficial effects in both AD and PD animal models. (Cherny et al., 2001; Finefrock et al., 2003; Kaur et al., 2003). Following oral treatment with CQ (30 mg kg−1 day−1), Aβ accumulation was markedly inhibited (49% decrease), as shown in a blinded study of APP2576 transgenic mice treated for 9 weeks. There was no evidence of neurotoxicity or increased nonamyloid pathology. General health and body weight parameters were significantly stable in the treated animals, with a conspicuous improvement after only 16 days of treatment (Cherny et al., 2001).
In the PD studies, mice previously treated with the neurotoxin MPTP were administered orally with CQ (30 mg kg−1 day−1) for 8 weeks to assess the ability of the compound to protect against MPTP-induced toxicity. Total substantia nigra (SN) iron levels were found to be reduced approximately 30% in the CQ-fed versus control animals, accordingly with the reported nontoxic range. Following CQ pretreatment, oxidative stress markers and glutathione depletion were found significantly attenuated in SN (Kaur et al., 2003). Unfortunately, many halogenated hydroxy-quinolines possess neurotoxic side effects (Tsubaki et al., 1971; Oakley, 1973). These side effects may be avoided by the use of nonhalogenated analogues, for instance the brain permeable VK-28 (Warshawsky et al., 2000) (17, Figure 12). A study centred on rats, with 6-OHDA induced striatal dopaminergic lesions (Ben-Shachar et al., 2004), has shown that, when injected either intraventricularly (1 μg in 5 ml) or intraperitoneally (1 or 5 mg kg−1 day−1 for 10 and 7 days, respectively), VK-28 is able to provide neuroprotection against 6-OHDA at very low doses. This has been confirmed by the prevention of the reduction in striatal dopamine levels and by the decrease of dopamine turnover, which are normally observed after 6-OHDA lesioning only. Moreover, this study has shown that the mechanism of action of VK-28 is more likely to be related to iron chelation properties than to any direct interference with 6-OHDA, since intranigral or intraventricular 6-OHDA initiates an increase in total iron in the substantia nigra and striatum at the sites of neurodegeneration, in monkeys, rats and mice (Ben-Shachar et al., 2004).
Conclusion
In summary, protein aggregation and oxidative stress have been demonstrated to be the major factors involved in the neurodegenerative process in AD, PD and prion disease. Metal ions play a crucial role, acting as mediators of neurotoxicity either by favouring plaque formation or redox cycling. Thus, they provide a suitable pharmacological target for the treatment of neurodegenerative diseases. In particular, bidentate chelators such as hydroxypyridinones and hydroxyquinolines would appear to possess the greatest potential for this goal. The development of an effective nontoxic therapeutic agent for such complex and comprehensive brain disorders represents an extremely challenging task. Much needs to be understood in terms of ethiopathogenesis for AD, PD and prion disease. However, chelation therapy may be considered as a valuable strategy both for the treatment and for the investigation of neurodegeneration.
Abbreviations
- AD
Alzheimer's disease
- Aβ
β-amyloid
- ALS
amyotrophic lateral sclerosis
- APP
amyloid precursor protein
- BBB
blood–brain barrier
- CNS
central nervous system
- CQ
clioquinol
- DFO
desferrioxamine
- EDTA
ethylenediaminetetraacetic acid
- 6-OHDA
6-hydroxy dopamine
- MS
multiple sclerosis
- NM
neuromelanin
- PD
Parkinson's disease
- PrP
prion protein
- PrPC
normal isoform of the prion protein
- PrPSc
scrapie isoform of the prion protein
- ROS
reactive oxygen species
- SN
substantia nigra
- SNc
substantia nigra pars compatta
- SOD
superoxide dismutase
- TSEs
transmissible spongiform encephalopathies
References
- ABEYSINGHE R.D., ROBERTS P.J., COOPER C.E., MACLEAN K.H., HIDER R.C., PORTER J.B. The environment of the lipoxygenase iron binding site explored with novel hydroxypyridinone iron chelators. J. Biol. Chem. 1996;271:7965–7972. doi: 10.1074/jbc.271.14.7965. [DOI] [PubMed] [Google Scholar]
- ADAMS J.H., CORSELLIS J.A.N., DUNCHEN L.W. Greenfield's Neuropathology 1984New York: Wiley; eds4th edn [Google Scholar]
- ALAM Z.I., DANIEL S.E., LEES A.J., MARSDEN D.C., JENNER P., HALLIWELL B. A generalised increase in protein carbonyls in the brain of Parkinson's but not incidental Lewy body disease. J. Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- ALPER G., NARAYANAN V. Friedreich's ataxia. Ped. Neurol. 2003;28:335–341. doi: 10.1016/s0887-8994(03)00004-3. [DOI] [PubMed] [Google Scholar]
- ANTZUTKIN O.N., LEAPMAN R.D., BALBACH J.J., TYCKO R. Sopramolecular structural constraints on Alzheimer's β-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- ATWOOD C.S., OBRENOVICH M.E., LIU T., CHAN H., PERRY G., SMITH M.A., MARTINS R.N. Amyloid-β: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-β. Brain Res. Rev. 2003;43:1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- BABCOCK M., DE SILVA D., OAKS R., DAVIS-KAPLAN S., JIRALERSPONG S., MONTERMINI L., PANDOLFO M., KAPLAN J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homologue of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- BELLINGER-KAWAHARA C.G., KEMPNER E., GROTH D., GABIZON R., PRUSINER S.B. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology. 1988;164:537–541. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- BENCE N.F., SAMPAT R.M., KOPITO R.R. Impairment of the ubiquitin-proteosome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- BEN-SHACHAR D., KAHANA N., KAMPEL V., WARSHAWSKY A., YOUDIM M.B.H. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lesion in rats. Neuropharmacology. 2004;46:254–263. doi: 10.1016/j.neuropharm.2003.09.005. [DOI] [PubMed] [Google Scholar]
- BEN-SHACHAR D., RIEDERER P., YOUDIM M.B. Iron melanin interaction and lipid peroxidation: implications for Parkinson's disease. J. Neurochem. 1991;57:1609–1614. doi: 10.1111/j.1471-4159.1991.tb06358.x. [DOI] [PubMed] [Google Scholar]
- BENSIMON G., LACOMBLEZ L., MEININGER V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole study group. N. Engl. J. Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- BONILLA E. Huntington disease. A review. J. Clin. Invest. 2000;41:117–141. [PubMed] [Google Scholar]
- BRADLEY J.L., BLAKE J.C., CHAMBERLAIN S., THOMAS P.K., COOPER J.M., SCHAPIRA A.H.V. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum. Mol. Gen. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- BRINDELLI M.G., TAMPELLINI D., ZECCA L. The structure of neuromelanin and its iron binding site studied by infrared spectroscopy. FEBS Lett. 1999;457:18–22. doi: 10.1016/s0014-5793(99)01001-7. [DOI] [PubMed] [Google Scholar]
- BRION S., MIROL J., PSIMARAS A.Recent findings in Pick's disease Progress in Neuropathology 1973New York: Grune and Stratton; 421–452.ed. Zimmerman, H.M., Vol. 2, pp [Google Scholar]
- BROWN D.R. Copper and prion disease. Br. Res. Bull. 2001;55:165–173. doi: 10.1016/s0361-9230(01)00453-1. [DOI] [PubMed] [Google Scholar]
- BROWN D.R., GUATIERI V., GRASSO G., IMPELLIZZERI G., PAPPALARDO G., RIZZARELLI E. Copper(II) complexes of peptide fragments of the prion protein. Conformation changes induced by copper(II) and the binding motif in C-terminal protein region. J. Inorg. Biochem. 2004;98:133–143. doi: 10.1016/j.jinorgbio.2003.09.006. [DOI] [PubMed] [Google Scholar]
- BROWN D.R., HAFIZ F., GLASSMITH L.L., WONG B.-S., JONES I.M., CLIVE C., HASWELL S.J. Consequences of manganese replacement of copper for prion protein function and proteinase resistance. EMBO J. 2000;19:1180–1186. doi: 10.1093/emboj/19.6.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN D.R., QUIN K., HERMS J.W., MADLUNG A., MANSON J., STROIME R., FRASER P.E., KRUCK T.A., VON BOHLEN A., SCHULZ-SCHAEFFER W., GIESE A., WESTAWAY D., KRETZSCHMAR H. The cellular prion protein binds copper in vivo. Nature. 1997a;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- BROWN D.R., SCHULZ-SCHAEFFER W.J., SCHMIDT B., KRETZSCHMAR H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreases SOD-1 activity. Exp. Neurol. 1997b;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- BRUNK U.T., JONES C.B., SOHAL R.S. A novel hypothesis of lipofuscinogenesis and cellular aging based on interactions between oxidative stress and autophagocytosis. Mut. Res. 1992;275:395–403. doi: 10.1016/0921-8734(92)90042-n. [DOI] [PubMed] [Google Scholar]
- BRUNK U.T., TERMAN A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Rad. Biol. Med. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- BUCCIANTINI M., GIANNONI F., CHITI F., BARONI F., FORMIGLI L., ZURDO J.S., TADDEI N., RAMPONI G., DOBSON C.M., STEFANI M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:501–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- BÜELER H., AGUZZI A., SAILER A., GREINER R.-A., AUTENRIED P., AUGET M., WEISSMAN C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- BUSH A.I. Metal complexing agents as therapies for Alzheimer's disease. Neurobiol. Ageing. 2002;23:1031–1038. doi: 10.1016/s0197-4580(02)00120-3. [DOI] [PubMed] [Google Scholar]
- BUSH A.I., HUANG X., FAIRLIE D.P. The possible origin of free radicals from amyloid β-peptides in Alzheimer's disease. Neurobiol. Ageing. 1999;20:335–337. doi: 10.1016/s0197-4580(99)00058-5. [DOI] [PubMed] [Google Scholar]
- BUTTERFIELD D.A., DRAKE J., POCERNICH C., CASTEGNA A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends Mol. Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- BUTTERFIELD D.A., KANSKI J. Brain oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 2001;122:945–962. doi: 10.1016/s0047-6374(01)00249-4. [DOI] [PubMed] [Google Scholar]
- CAMPUZANO V., MONTERMINI L., MOLTÒ M.D., PIANESE L., COSSÉE M., CAVALCANTI F., MONROS E., RODIUS F.F., DUCLOS MONTICELLI A., ZARA F., CAÑIZARES J., KOUTNIKOVA H., BIDICHANDANI S.I., GELLERA C., BRICE A., TROUILLAS P., DE MICHELE G., FILLA A., DE FRUTOS R., PALAU F., PATEL P.I., DI DONATO S., MANDEL J.-L., COCOZZA S., KOENIG M., PANDOLFO M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- CARRÌ M.T., FERRI A., COZZOLINO M., CALABRESE L., ROTILIO G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Br. Res. Bull. 2003;61:365–374. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- CHERNY R.A., ATWOOD C.S., XILINAS M.E., GRAY D.N., JONES W.D., MCLEAN C.A., BARNHAM K.J., VOLITAKIS I., FRASER F.W., KIM Y.-S. Treatment with a copper-zinc chelator markedly and rapidly inhibits b-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- CHERNY R.A., BARNHAM K.J., LYNCH T., VOLITAKIS I., LI Q.-X., MCLEAN C.A., MULTHAUP G., BEYREUTHER K., TANZI R.E., MASTERS C.L., BUSH A.I. Chelation and intercalation: complementary properties in a compound for the treatment of Alzheimer's disease. J. Struct. Biol. 2000;130:209–216. doi: 10.1006/jsbi.2000.4285. [DOI] [PubMed] [Google Scholar]
- CHETLEY A., GILBERT D. Health Action International. The Hangue: International Organisation of Consumers Unions; 1986. [Google Scholar]
- CHEUN W.L. The chemical structure of melanin. Pigm. Cell Res. 2004;17:422–424. doi: 10.1111/j.1600-0749.2004.00165_1.x. [DOI] [PubMed] [Google Scholar]
- CLAESEN M.E., CLEMENTS M.L. Ridding the world of hydroxyquinolines. Br. Med. J. 1989;299:527–528. doi: 10.1136/bmj.299.6698.527-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN A.S., SHIRAHAMA T., SKINNER M.Electron microscopy of amyloid Electron Microscopy of Proteins 1982London, UK: Academic Press; 165–205.ed. Harris, J.R., Vol. 3, pp [Google Scholar]
- COHEN F.E. Protein misfolding and prion diseases. J. Mol. Biol. 1999;293:313–320. doi: 10.1006/jmbi.1999.2990. [DOI] [PubMed] [Google Scholar]
- COMPSTON A., COLES A. Multiple sclerosis. Lancet. 2002;359:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- CONFAVREUX C., AIMARD G., DEVIC M. Course and prognosis of multiple sclerosis assessed by the computerized data processing of 349 patients. Brain. 1980;103:281–300. doi: 10.1093/brain/103.2.281. [DOI] [PubMed] [Google Scholar]
- CONNOR J.R., SNYDER B.S., AROSIO P., LOEFFLER D.A., LEWITT P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian and Alzheimer's diseased brains. J. Neurochem. 1995;65:717–724. doi: 10.1046/j.1471-4159.1995.65020717.x. [DOI] [PubMed] [Google Scholar]
- CONWAY K.A., BAXTER E.W., FELSENSTEIN K.M., REITZ A.B. Emerging β-amyloid therapies for the treatment of Alzheimer's disease. Curr. Pharm. Des. 2003;9:427–447. doi: 10.2174/1381612033391649. [DOI] [PubMed] [Google Scholar]
- CONWAY K.A., HARPER J.D., LANSBURY P.T. Accelerated in vitro fibril formation by mutant α-synuclein linked to early-onset Parkinson's disease. Nat. Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- COOPER C.E., LYNAGH G.R., HOYES K.P., HIDER R.C., CAMMACK R., PORTER J.B. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J. Biol. Chem. 1996;271:20291–20299. doi: 10.1074/jbc.271.34.20291. [DOI] [PubMed] [Google Scholar]
- CRAPPER MCLACHLAN D.R., DALTON A.J., KRUCK T.P.A., BELL M., SMITH W., KALOW W., ANDREWS D.F. Effect of desferrioxamine on the clinical progress of Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- DE FELICE F.G., FERREIRA S.T. β-Amyloid production, aggregation and clearance as targets for therapy in Alzheimer's disease. Cell. Mol. Neurobiol. 2002;22:545–563. doi: 10.1023/a:1021832302524. [DOI] [PubMed] [Google Scholar]
- DEXTER D.T., WELLS F.R., LEES A.J. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J. Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- DICKSON D.W., CRYSTAL H.A., BEVONA C., HONER W., VINCENT I., DAVIES P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging. 1995;16:285–298. doi: 10.1016/0197-4580(95)00013-5. [DOI] [PubMed] [Google Scholar]
- DOBSON M.C. Protein folding and disease: a view from the first Horizon Symposium. Nat. Drug Disc. 2003a;2:154–160. doi: 10.1038/nrd1013. [DOI] [PubMed] [Google Scholar]
- DOBSON M.C. Protein folding and misfolding. Nature. 2003b;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- DOUBLE K.L., GERLACH M., BEN-SHACHAR D., YOUDIM M.B.H., ZECCA L., RIEDERER P. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotox. Teratol. 2002;24:621–628. doi: 10.1016/s0892-0362(02)00218-0. [DOI] [PubMed] [Google Scholar]
- DOUBLE K.L., GERLACH M., SCHÜNEMANN V., TRAUTWEIN A.X., ZECCA L., GALLORINI M., YOUDIM M.B.H., RIEDERER P., BEN-SHACHAR D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003;66:489–494. doi: 10.1016/s0006-2952(03)00293-4. [DOI] [PubMed] [Google Scholar]
- DOUBLE K.L., RIEDERER P.F., GERLACH M. Significance of neuromelanin for neurodegeneration in Parkinson's disease. Drug News Dev. 1999;12:333–340. [Google Scholar]
- DOUBLE K.L., ZECCA L., COSTI P., MAUER M., GRIESINGER C., ITO S., BEN-SHACHAR D., BRINGMANN G., FARIELLO R.G., RIEDERER P., GERLACH M. Structural characteristics of human substantia nigra neuromelanin and synthetic dopamine melanins. J. Neurochem. 2000;75:2583–2589. doi: 10.1046/j.1471-4159.2000.0752583.x. [DOI] [PubMed] [Google Scholar]
- DUFFY P.E., TENNYSON V.M. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and Locus caeruleus in Parkinson's disease. J. Neuropathol. Exp. Neurol. 1965;24:398–414. [Google Scholar]
- EL-AGNAF O.M.A., JAKES R., CURRAN M.D., WALLACE A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson's disease. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- FINEFROCK A.E., BUSH A.I., DORAISWAMY P.M. Current status of metals as therapeutic targets in Alzheimer's disease. J. Am. Geriatr. Soc. 2003;51:1143–1148. doi: 10.1046/j.1532-5415.2003.51368.x. [DOI] [PubMed] [Google Scholar]
- FLOYD R.A. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- FLOYD R.A., HENSLEY K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- GHETTI B., PICCARDO P., FRANGIONE B., BUGIANI O., GIACCONE G., YOUNG K., PRELLI F., FARLOW M.R., DLOUHY S.R., TAGLIAVINI F. Prion protein amyloidosis. Brain Pathol. 1996;6:127–145. doi: 10.1111/j.1750-3639.1996.tb00796.x. [DOI] [PubMed] [Google Scholar]
- GOTZ M.E., KÜNIG G., RIEDERER P., YOUDIM M.B.H. Oxidative stress: free radical production in neural degeneration. Pharmacol. Ther. 1994;63:37–122. doi: 10.1016/0163-7258(94)90055-8. [DOI] [PubMed] [Google Scholar]
- GUENTCHEV M., VOIGTLÄNDER T., HABERLER C., GROSCHUP M.H., BUDKA H. Evidence for oxidative stress in experimental prion disease. Neurobiol. Dis. 2000;7:270–273. doi: 10.1006/nbdi.2000.0290. [DOI] [PubMed] [Google Scholar]
- HABGOOD M.D., LIU Z.D., DEHKORDI L.S., KHODR H.H., ABBOTT J., HIDER R.C. Investigation into the correlation between the structure of hydroxypyridinones and blood–brain barrier permeability. Biochem. Pharmacol. 1999;57:1305–1310. doi: 10.1016/s0006-2952(99)00031-3. [DOI] [PubMed] [Google Scholar]
- HALLIWELL B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- HARDY J., SELKOE D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- HARRIS D.A. Biosynthesis and cellular processing of the prion protein. Advan. Protein Chem. 2001;57:203–228. doi: 10.1016/s0065-3233(01)57023-0. [DOI] [PubMed] [Google Scholar]
- HARRIS D.C., AISEN P. Facilitation of Fe(II) autoxidation by Fe(III) complexing agents. Biochim. Biophys. Acta. 1973;329:156–158. doi: 10.1016/0304-4165(73)90019-6. [DOI] [PubMed] [Google Scholar]
- HARTLEY D.M., WALSH D.M., YE C.P., DIEHL T., VASQUEZ S., VASSILEV P.M., TEPLOW D.B., SELKOE D.J. Protofibrillar intermediates of amyloid b-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurones. J. Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEDGE M.L., JAGANNATHA RAO K.S. Challenges and complexities of α-synuclein toxicity: new postulates in unfolding the mystery associated with Parkinson's disease. Arch. Biochem. Biophys. 2003;418:169–178. doi: 10.1016/j.abb.2003.08.015. [DOI] [PubMed] [Google Scholar]
- HELY M.A., FUNG V.S., MORRIS J.G. Treatment of Parkinson's disease. J. Clin. Neurosci. 2000;7:484–494. doi: 10.1054/jocn.2000.0766. [DOI] [PubMed] [Google Scholar]
- HIDER R.C. Potential protection from toxicity by oral iron chelators. Toxicol. Lett. 1995;82–83:961–967. doi: 10.1016/0378-4274(95)03606-7. [DOI] [PubMed] [Google Scholar]
- HIDER R.C., HALL A.D. Clinically useful chelators of tripositive elements. Prog. Med. Chem. 1991;28:41–173. doi: 10.1016/s0079-6468(08)70363-1. [DOI] [PubMed] [Google Scholar]
- HIJAZI N., SHAKED Y., ROSENMANN H., BEN-HUR T., GABIZON R. Copper binding to PrPC may inhibit prion disease propagation. Br. Res. 2003;993:192–200. doi: 10.1016/j.brainres.2003.09.014. [DOI] [PubMed] [Google Scholar]
- HIRSH E.C., BRANDEL J.-P., GALLE P. Iron and aluminium increase in the substantia nigra of patients with Parkinson's disease: an X-ray microanalysis. J. Neurochem. 1991;56:446–451. doi: 10.1111/j.1471-4159.1991.tb08170.x. [DOI] [PubMed] [Google Scholar]
- HOLANDER D., RICKETTS D., BOYD C.A.R. Importance of probe molecular geometry in determining intestinal permeability. Can. J. Gastroenterol. 1988;2:35A–38A. [Google Scholar]
- HSIAO K., CHAPMAN P., NILSEN S., ECKMAN C., HARIGAYA Y., YOUNKIN S., YANG F., COLE G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- HUANG X., ATWOOD C.S., HARTSHORN M.A., MULTHAUP G., GOLDSTEIN L.E., SCARPA R.C., CUAJUNGCO M.P., GRAY D.N., LIM J., MOIR R.D., TANZI R.E., BUSH A.I. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- JEFFREY M., GOODSIR C.M., BRUCE M.E., MCBRIDE P.A., SCOTT J.R. Infection-specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie-infected mice. Ann. NY Acad. Sci. 1994;724:327–330. doi: 10.1111/j.1749-6632.1994.tb38923.x. [DOI] [PubMed] [Google Scholar]
- JELLIGER K.A. The role of iron in neurodegeneration: prospects for pharmacology of Parkinson's disease. Drugs Aging. 1999;14:115–140. doi: 10.2165/00002512-199914020-00004. [DOI] [PubMed] [Google Scholar]
- KASTNER A., HIRSCH E.C., LEJEUNE O. Is the vulnerability of neurons in the substantia nigra of patients with Parkinson's disease related to their neuromelanin content. J. Neurochem. 1992;59:1080–1089. doi: 10.1111/j.1471-4159.1992.tb08350.x. [DOI] [PubMed] [Google Scholar]
- KAUR D., YANTIRI F., RAJAGOPALAN S., KUMAR J., MO J.Q., BOONPLUEANG R., VISWANATH V., JACOBS R., YANG L., BEAL M.F., DI MONTE D., VOLITASKIS I., ELLERBY L., CHERNY R.A., BUSH A.I., ANDERSON J.K. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- KEEGAN B.M., NOSEWORTHY J.H. Multiple sclerosis. Ann. Rev. Med. 2002;53:285–302. doi: 10.1146/annurev.med.53.082901.103909. [DOI] [PubMed] [Google Scholar]
- KIM N., PARK S.J., JIN J.K., KWON M.S., CHOI E.K., CARP R.I., KIM Y.S. Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Br. Res. 2000;884:98–103. doi: 10.1016/s0006-8993(00)02907-3. [DOI] [PubMed] [Google Scholar]
- KLEIN W.L., KRAFFT G.A., FINCH C.E. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum. Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- KROPF A.J., BUNKER B.A., EISNER M., MOSS S.C., ZECCA L., STROPPOLO A., CRIPPA P.R. X-ray absorption fine-structure spectroscopy studies of Fe sites in natural human neuromelanin and synthetic analogues. Biophys. J. 1998;75:3135–3142. doi: 10.1016/S0006-3495(98)77755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRÜGER R., KUHN W., MÜLLER T., WOITALLA D., GRAEBER M., KÖSEL S., PRZUNTEK H., EPPLEN J.T., SCHOLS L., RIESS O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- KUKULL W.A., HIGDON R., BOWEN J.D., MCCORMICK W.C., TERI L., SCHELLENBERG G.D., VAN BELLE G., JOLLEY L., LARSON E.B. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch. Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- LAHIRI D.K., FARLOW M.R., SAMBAMURTI K., GREIG N.H., GIACOBINI E., SCHNEIDER L.S. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer's disease. Curr. Drug Targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- LAINE J., MARC M.-E., SY M.-S., AXELRAD H. Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur. J. Neurosci. 2001;14:47–56. doi: 10.1046/j.0953-816x.2001.01621.x. [DOI] [PubMed] [Google Scholar]
- LAMBERT M.P., BARLOW A.K., CHROMY B.A., EDWARDS C., FREED R., LIOSATOS M., MORGAN T.E., ROZOVSKY I., TROMMER B., VIOLA K.L., WALS P., ZHANG C., FINCH C.E., KRAFFT G.A., KLEIN W.L. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAN J., JIANG D.H. Desferrioxamine and vitamin E protect against iron and MPTP-induced neurodegeneration in mice. J. Neural Transm. 1997;104:469–481. doi: 10.1007/BF01277665. [DOI] [PubMed] [Google Scholar]
- LEE J.-Y., FRIEDMAN J.E., ANGEL L., KOZAK A., KOH J.-Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human β-amyloid precursor protein transgenic mice. Neurobiol. Aging. 2004;25:1315–1321. doi: 10.1016/j.neurobiolaging.2004.01.005. [DOI] [PubMed] [Google Scholar]
- LEHMANN S. Metal ions and prion disease. Curr. Opin. Chem. Biol. 2002;6:187–192. doi: 10.1016/s1367-5931(02)00295-8. [DOI] [PubMed] [Google Scholar]
- LIU Z.D., HIDER R.C. Design of clinically useful iron(III)-selective chelators. Med. Res. Rev. 2002a;22:26–64. doi: 10.1002/med.1027. [DOI] [PubMed] [Google Scholar]
- LIU Z.D., HIDER R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2002b;232:151–171. [Google Scholar]
- LIU Z.D., KAYYALI R., HIDER R.C., PORTER J.B., THEOBALD A.E. Design, synthesis, and evaluation of novel 2-substituted 3-hydroxypyridin-4-ones: structure–activity investigation of metalloenzyme inhibition by iron chelators. J. Med. Chem. 2002;45:631–639. doi: 10.1021/jm010817i. [DOI] [PubMed] [Google Scholar]
- LIU Z.D., LOCKWOOD M., ROSE S., THEOBALD A.E., HIDER R.C. Structure–activity investigation of the inhibition of 3-hydroxypyridin-4-ones on mammalian tyrosine hydroxylase. Biochem. Pharmacol. 2001;61:285–290. doi: 10.1016/s0006-2952(00)00551-7. [DOI] [PubMed] [Google Scholar]
- LOVELL M.A., ROBERTSON J.D., TEESDAL W.J., CAMPBELL J.L., MARKESBERY W.R. Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- LUE L.F., KUO Y.-M., ROHER A.E., BRACHOVA L., SHEN Y., SUE L., BEACH T., KURTH J.H., RYDEL R.E., ROGERS J. Soluble amyloid β-peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACCHI G., CIOFFI R.P. An in vivo and post mortem MRI study in multiple sclerosis with pathological correlation. Ital. J. Neurol. Sci. 1992;13:97–103. [PubMed] [Google Scholar]
- MAISSEN M., ROECKL C., GLATZEL M., GOLDMANN W., AGUZZI A. Plasminogen binds to disease-associated prion protein of multiple species. Lancet. 2001;357:2026–2028. doi: 10.1016/S0140-6736(00)05110-2. [DOI] [PubMed] [Google Scholar]
- MANGÉ A., MILAHAVET O., UMLAUF D., HARRIS D., LEHMANN S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–162. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- MARTELL A.E., SMITH R.M. Critical Stability Constant 1974–1989London: Plenum Press; Vols. 1–6, [Google Scholar]
- MASON J.M., KOKKONI N., STOTT K., DOIG A.J. Design strategies for anti-amyloid agents. Curr. Opin. Struc. Biol. 2003;13:526–532. doi: 10.1016/s0959-440x(03)00100-3. [DOI] [PubMed] [Google Scholar]
- MAXTON D.G., BJARNASON I., REYNOLDS A.P., CATT S.D., PETERS T.J., MENZIES I.S. Lactulose, 51Cr-labelled ethylenediaminetetra-acetate, L-rhamnose and polyethyleneglycol 400 [corrected] as probe markers for assessment in vivo of human intestinal permeability. Clin. Sci. 1986;71:71–80. doi: 10.1042/cs0710071. [DOI] [PubMed] [Google Scholar]
- MC GEER P.L., ITAGAKI S., BOYES B.E., MCGEER E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1998;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- MCLEAN C.A., CHERNY R.A., FRASER F.W., FULLER S.J., SMITH M.J., BEYREUTHER K., BUSH A.I., MASTERS C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- MERZ P.A., SOMMERVILLE R.A., WISNIEWSKY H.M., IKBAL K. Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. (Berlin) 1981;54:63–74. doi: 10.1007/BF00691333. [DOI] [PubMed] [Google Scholar]
- MIRANDA S., OPAZO C., LARRONDO L.F., MUÑOZ F.J., RUIZ F., LEIGHTON F., INESTROSA N.C. The role of oxidative stress in the toxicity induced by amyloid β-peptide in Alzheimer's disease. Prog. Neurobiol. 2000;62:633–648. doi: 10.1016/s0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- MIZUNO Y., OHTA S., TANAKA M., TAKAMIYA S., SUZUKI K., SATO T., OYA H., OZAWA T., KAGAWA Y. Deficiencies in Complex I subunits of the respiratory chain in Parkinson's disease. Biochem. Biophys. Res. Commun. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- MOOSMANN M., BEHL C. Antioxidants as treatment for neurodegenerative disorders. Exp. Opin. Invest. Drugs. 2002;11:1407–1435. doi: 10.1517/13543784.11.10.1407. [DOI] [PubMed] [Google Scholar]
- OAKLEY G.P. The neurotoxicity of the halogenated hydroxyquinolines. JAMA. 1973;225:395–397. [PubMed] [Google Scholar]
- OLANOW C.W. Magnetic resonance imaging in parkinsonism. Neurol. Clin. North Am. 1992;10:405–420. [PubMed] [Google Scholar]
- OLANOW C.W., YOUDIM M.B. Neurodegeneration and Neuroprotection in Parkinson's Disease. New York: Academic Press; 1996. pp. 55–69. [Google Scholar]
- OLDENDORF W.H. Lipid solubility and drug penetration of the blood-brain barrier. Proc. Soc. Expt. Bio. Med. 1974;147:813–816. doi: 10.3181/00379727-147-38444. [DOI] [PubMed] [Google Scholar]
- OLIVIERI N., KOREN G., HERMANN C., BENTUR Y., CHUNG D., KLEIN J., ST LOUIS P., FREEDMAN M.H., MCCLELLAND R.A., TEMPLETON D.M. Comparison of oral iron chelator L1 and desferrioxamine in iron-loaded patients. Lancet. 1990;336:1275–1279. doi: 10.1016/0140-6736(90)92962-h. [DOI] [PubMed] [Google Scholar]
- PAIK S.R., SHIN H.-J., LEE J.-H., CHANG C.-S., KIM J. Copper(II)-induced self-oligomerisation of α-synuclein. Biochem. J. 1999;340:821–828. [PMC free article] [PubMed] [Google Scholar]
- PALM A. Untersuchung in des chinolin reihe. Arch. Exp. Pathol. Pharmacol. 1932;199:176–185. [Google Scholar]
- PAN K., BALDWIN M., NGUYEN J., GASSET M., SERBAN A., GROTH D., MEHLHORN I., HUANG Z., FLETTERICK R.J., COHEN F.E., PRUSINER S.B. Conversion of α-helices β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATTISON I.H., JEBBET J.N. Histopathological similarities between scrapie and cuprizone toxicity in mice. Nature. 1971;230:115–117. doi: 10.1038/230115a0. [DOI] [PubMed] [Google Scholar]
- PEREZ G.R., WAYMIRE J.C., LIN E., LIU J.J., GUO F., ZIGMOND M.J. A role for α-synuclein in the regulation of dopamine synthesis. J. Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIEMONTE & VALLE D'AOSTA REGISTER FOR AMYOTROPHIC LATERAL SCLEROSIS (PARALS) Incidence of ALS in Italy. Evidence for a uniform frequency in Western countries. Neurology. 2001;56:239–244. doi: 10.1212/wnl.56.2.239. [DOI] [PubMed] [Google Scholar]
- POLYMEROPOULOS M.H., LAVEDAN C., LEROY E., IDE S.E., DEHEJIA A., DUTRA A., PIKE B., ROOT H., RUBENSTEIN J., BOYER R., STENROOS E.S., CHANDRASEKHARAPPA S., ATHANASSIADOU A., PAPAPETROPOULOS T., JOHNSON W.G., LAZZARINI A.M., DUVOISIN R.C., DI IORIO G., GOLBE L.I., NUSSBAUM R.L. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- PRUSINER S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- PRUSINER S.B. Molecular biology of prion disease. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- PRUSINER S.B. Prions. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRUSINER S.B. Neurodegenerative diseases and prions. N. Engl. J. Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- PRUSINER S.B., MCKINLEY M.P., BOWMAN K.A., BOLTON D.C., BENDHEIM P.E., GROTH D.F., GLENNER G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- RAYMOND K.N., MÜLLER G., MATZANKE B.F. Complexation of iron by siderophores: a review of their solution and structural chemistry and biological function. Top Curr. Chem. 1984;58:49–102. [Google Scholar]
- REISBERG B., DOODY R., STÖFFLER A., SCHMITT F., FERRIS S., MÖBIUS H.J. Memantine in moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- RICHARDSON D.R. Novel chelators for central nervous system disorders that involve alterations in the metabolism of iron and other metal ions. Ann. NY Acad. Sci. 2004;1012:326–341. doi: 10.1196/annals.1306.026. [DOI] [PubMed] [Google Scholar]
- RIEDERER P., SOFIC E., RAUSCH W.D., JELLINGER K., YOUDIM M.B.H. Transition metals, ferritin, glutathione and ascorbic acid in Parkinsonian brains. J. Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- ROCHET J.C., LANSBURY P.T. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 2000;10:60–80. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- ROHER A.E., CHANEY M.O., KUO Y.-M., WEBSTER S.D., STINE W.B., HAVERKAMP L.J., WOODS A.S., COTTER R.J., TUOHY J.M., KRAFFT G.A., BONNELL B.S., EMMERLING M.R. Morphology and toxicity of Aβ-(1-42) dimmer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J.Biol. Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- ROSE F.C., GAWEL M. Clioquinol neurotoxicity: an overview. Acta Neurol. Scand. 1984;80 (Suppl. 100):137–145. [PubMed] [Google Scholar]
- SAGGU H., COOKSEY J., DEXTER D. A selective increase in a particular superoxide dismutase activity in Parkinsonian substantia nigra. J. Neurochem. 1989;53:692–697. doi: 10.1111/j.1471-4159.1989.tb11759.x. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-RAMOS J., OVERVICK E., AMES B.N. A marker of oxyradical-mediated DNA damage (8-hydroxy-2′-deoxy-guanoxine) is increased in nigro-striatum of Parkinson's disease brain. Neurodegeneration. 1994;3:197–204. [Google Scholar]
- SERPELL L.C., BERRIMAN J., JAKES R., GOEDERT M., CROWTHER R.A. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross β-conformation. Proc. Natl. Acad. Sci. U.S.A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHASTRY B.S. Neurodegenerative disorders of protein aggregation. Neurochem. Internat. 2003;43:1–7. doi: 10.1016/s0197-0186(02)00196-1. [DOI] [PubMed] [Google Scholar]
- SHERKI Y.G., MELAMED E., OFFEN D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–975. doi: 10.1016/s0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- SHIN R.-W., KRUCK T.P.A., MURAYAMA H., KITAMOTO T. A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated τ of Alzheimer's disease. Br. Res. 2003;961:139–146. doi: 10.1016/s0006-8993(02)03893-3. [DOI] [PubMed] [Google Scholar]
- SIKORSKI P., ATKINS E.D.T., SERPELL L.C. Structure and texture of fibrous crystals formed by Alzheimer's Aβ (11–25) peptide fragment. Structure. 2003;11:915–926. doi: 10.1016/s0969-2126(03)00149-7. [DOI] [PubMed] [Google Scholar]
- SOFIC E., LANGE K.W., JELLINGER K., RIEDERER P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci. Lett. 1992;142:128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- SOFIC E., RIEDERER P., HEINSEN H. Increased iron(III) and total iron content in post mortem, substantia nigra of parkinsonian brain. J. Neural Trans. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- STAHL N., BORCHELT D.R., HSIAO K., PRUSINER S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- SWAN G.A. Structure, chemistry and biosynthesis of the melanins. Fortschr. Chem.Org. Naturst. 1974;31:521–582. doi: 10.1007/978-3-7091-7094-6_9. [DOI] [PubMed] [Google Scholar]
- TERRY R.D., MASLIAH E., SALMON D.P., BUTTERS N., DETERESA R., HILL R., HANSEN L.A., KATZMAN R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- THACKRAY A.M., KNIGHT R., HASWELL S., BUJDOSO R., BROWN D.R. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J. 2002;362:253–258. doi: 10.1042/0264-6021:3620253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOROK M., MILTON S., KAYED R., WU P., MCINTIRE T., GLABE C.G., LANGEN R. Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labelling. J. Biol. Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- TSUBAKI T., HONMA Y., HOSH M. Neurological syndrome associated with clioquinol. Lancet. 1971;1:696–697. doi: 10.1016/s0140-6736(71)92699-7. [DOI] [PubMed] [Google Scholar]
- TURNBULL S., TABNER B.J., BROWN R.D., ALLSOP D. Copper-dependent generation of hydrogen peroxide from the toxic prion protein fragment PrP106-126. Neurosci. Lett. 2003;336:159–162. doi: 10.1016/s0304-3940(02)01254-5. [DOI] [PubMed] [Google Scholar]
- TURNBULL S., TABNER B.J., EL-AGNAF O.M.A., MOORE S., DAVIES Y., ALLSOP D. α-Synuclein implicated in Parkinson's disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 2001;30:1163–1170. doi: 10.1016/s0891-5849(01)00513-5. [DOI] [PubMed] [Google Scholar]
- TWELVES D., PERKINS K.S., COUNSELL C. Systematic review of incidence studies of Parkinson's disease. Mov. Disord. 2003;18:19–31. doi: 10.1002/mds.10305. [DOI] [PubMed] [Google Scholar]
- TYCKO R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol. 2004;14:1–8. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- UEDA K., FUKUSHIMA H., MASLIAH E., XIA Y., IWAI A., YOSHIMOTO M., OTERO D.A.C., KONDO J., IHARA Y., SAITOH T. Molecular cloning of cDNA encoding an unrecognised component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UVERSKY V.N., LI J., FINK A.L. Metal-triggered structural transformations, aggregation and fibrillation of human α-synuclein. J. Biol. Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- VIGO-PELFREY C., LEE D., KEIM P., LIEBERBURG I., SCHENK D.B. Characterisation of amyloid peptide from human cerebrospinal fluid. J. Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- WALSH D.M., HARTLEY D.M., KUSUMOTO Y., FEZOUI Y., CONDRON M.M., LOMAKIN A., BENEDEK G.B., SELKOE D.J., TEPLOW D.B. Amyloid β protein fibrillogenesis: structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- WALSH D.M., KLYUBIN I., FADEEVA J.V., CULLEN W.K., ANWYL R., WOLFE M.S., ROWAN M.J., SELKOE D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- WANG S.S., BECERRA-ARTEAGA A., GOOD T.A. Development of a novel diffusion-based method to estimate the size of the aggregated Aβ species responsible for neurotoxicity. Biotechnol. Bioeng. 2002;80:50–59. doi: 10.1002/bit.10347. [DOI] [PubMed] [Google Scholar]
- WARSHAWSKY B., YOUDIM M.B.H., BEN-SHACAR D. Pharmaceutical compositions compromising iron chelators for the treatment of neurodegenerative disorders and some novel iron chelators. International Publication number WO 00/74664A2. 2000.
- WEINSHENKER B.G. The natural history of multiple sclerosis. Neurol. Clin. 1995;13:119–146. [PubMed] [Google Scholar]
- WEINSHENKER B.G., BASS B., RICE G.P., NOSEWORTHY J., CARRIERE W., BASKERVILLE J., EBERS G.C. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain. 1989;112:133–146. doi: 10.1093/brain/112.1.133. [DOI] [PubMed] [Google Scholar]
- WILMS H., ROSENSTIEL P., SIEVERS J., DEUSCHL G., ZECCA L., LUCIUS R. Activation of microglia by human neuromelanin is NF-κB-dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J. 2003;17:500–502. doi: 10.1096/fj.02-0314fje. [DOI] [PubMed] [Google Scholar]
- WISNIEWSKY H.M., COBLENTZ J.M., TERRY R.D. Pick's disease. A clinical and ultrastructural study. Arch. Neurol. 1972;26:97–108. doi: 10.1001/archneur.1972.00490080015001. [DOI] [PubMed] [Google Scholar]
- World Health Organ (WHO) Tech Rep Ser The selection of essential drugs: report of a WHO expert committee. 1977;615:1–36. [PubMed] [Google Scholar]
- XU J., KAO S.-Y., LEE F.J.S., SONG W., JIN L.-W., YANKNER B.A. Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- YE F.Q., ALLEN P.S., MARTIN W.R.W. Basal ganglia iron content in Parkinson's disease measured with magnetic resonance. Mov. Disord. 1996;11:243–249. doi: 10.1002/mds.870110305. [DOI] [PubMed] [Google Scholar]
- YOKEL R.A., FREDENBURG A.M., MEURER K.A., SKINNER T.L. Influence of lipophilicity on the bioavailability and disposition of orally active 3-hydroxypyridin-4-one metal chelators. Drug Metab. Dispos. 1995;23:1178–1180. [PubMed] [Google Scholar]
- YOUDIM M.B.H. The enigma of neuromelanin in Parkinsonian substantia nigra. J. Neural. Transm. 1994;43 (Suppl.):113–122. [PubMed] [Google Scholar]
- ZECCA L., FARIELLO R., RIEDERER P., SULZER D., GATTI A., TAMPELLINI D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson's disease. FEBS Lett. 2002;510:216–220. doi: 10.1016/s0014-5793(01)03269-0. [DOI] [PubMed] [Google Scholar]
- ZECCA L., GALLORINI M., SCHÜNEMANN V., TRAUTWEIN A.X., GERLACH M., RIEDERER P., VEZZONI P., TAMPELLINI D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001a;76:1766–1773. doi: 10.1046/j.1471-4159.2001.00186.x. [DOI] [PubMed] [Google Scholar]
- ZECCA L., TAMPELLINI D., GERLACH M., RIEDERER P., FARIELLO R.G., SULZER D. Substantia nigra neuromelanin: structure, synthesis, and molecular behaviour. J. Clin. Pathol: Mol. Pathol. 2001b;54:414–418. [PMC free article] [PubMed] [Google Scholar]
- ZECCA L., ZUCCA F.A., WILMS H., SULZER D. Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends Neurosc. 2003;26:578–580. doi: 10.1016/j.tins.2003.08.009. [DOI] [PubMed] [Google Scholar]