

Abstract
Parasitic helminths (worms belonging to several metazoan phyla) cause considerable morbidity and mortality in humans. They are an important veterinary problem, and they result in significant economic losses in animal grazing and agriculture. Experimental studies on parasitic helminths have been limited by a lack of parasite cell lines and methods for molecular genetic analyses. We evaluated particle bombardment (biolistics) as a strategy to introduce and express nucleic acids in these multicellular parasites. By using embryos of the parasitic nematode Ascaris as a model, we developed methods to introduce and express both DNA and RNA during several stages of Ascaris embryogenesis. Biolistic transfection will facilitate experimental strategies in Ascaris embryos complementing other biochemical tools available (e.g., in vitro whole-cell embryo extracts for transcription, RNA processing, and translation). Transfection experiments with adult schistosomes further suggest that the biolistic strategy should be applicable to a variety of other parasitic helminths. The development of these methods provides molecular genetic tools to study gene expression and the biology of a variety of types and developmental stages of important helminth parasites.
Keywords: transfection, Ascaris, schistosome, spliced leader RNA, firefly luciferase
Parasitic helminths remain a significant health problem in many parts of the world leading to considerable mortality and long-term morbidity. For example, more than 1 billion people are infected with the intestinal nematode Ascaris, 120 million infected with lymphatic filariasis (roundworm/nematode), 200 million estimated infected and 20 million seriously ill with schistosomiasis (flatworm/blood fluke), 18 million infected with river blindness (nematode/filaria), 44 million pregnant women infected with hookworm (nematode), more than 20 million infected with Paragonimus spp. (flatworm), and 2.4 million infected with Fasciola (flatworm/liver fluke) (WHO, http://www.who.ch/ctd). Parasitic nematodes also pose a significant economic problem in grazing animals and agriculture (1).
Experimental studies on these helminth parasites have been hampered by a lack of molecular genetic approaches. The development of techniques for the introduction and expression of nucleic acids in parasitic helminths is critical for experimental analysis of the regulation of gene expression in these organisms. Moreover, molecular genetic methods are essential for the development of new experimental approaches for the study of parasite pathogenesis, cell biology, and other aspects of helminth biology. The multicellular nature, organization, and size of parasitic helminths poses significant problems with respect to the introduction of nucleic acids. To our knowledge, methods to introduce and express nucleic acids in parasitic helminths have not previously been developed.
Biolistics, or particle bombardment, has been used successfully to introduce nucleic acids into plant cells, animal tissues or organs, and whole organisms that typically are not amenable to more traditional methods of transfection (see reviews, refs. 2–8). Originally developed for use with plant cells, biolistics has been the most universally successful method for introduction of nucleic acids (including transformation of organelles such as mitochondria and plastids). Furthermore, biolistics provides a practical method for in vivo studies using whole organisms such as multicellular parasites for which cell lines and transgenic methods are currently not available. We initiated experiments to explore the use of biolistic transfection as a method for introducing and analyzing expression of nucleic acids in helminths. By using Ascaris embryos as a model, we systematically evaluated particle bombardment parameters for transfection and developed methods to introduce and express both DNA and RNA. Experiments suggest that this method should be applicable to adult schistosomes, multicellular parasites that are members of an evolutionarily divergent metazoan phylum. The successful introduction and expression of nucleic acids in these helminths suggests that a biolistic approach should prove useful for developing and facilitating molecular genetic analysis in these important parasites.
MATERIALS AND METHODS
Organisms.
Adult female Ascaris were purchased from Carolina Biological Supply or collected at a local slaughterhouse. Adult schistosomes were obtained from Syrian golden hamsters and incubated as described (9).
Isolation and Preparation of Ascaris Embryos.
Ascaris embryos were isolated from female uteri as described and stored at 4°C in PBS, pH 2.0 (10). Embryos were incubated in a shaking water bath at 30°C to allow embryonic development. After embryonic development, eggshells were removed with 0.4 N KOH/1.4% sodium hypochlorite for 90 min at 30°C. The embryos were collected, washed four times with PBS (pH 7.0), and resuspended at ≈9 × 106 embryos/ml in nematode blastomere media (11).
Biolistics.
Approximately 700,000 Ascaris embryos (75 μl) were evenly spread onto 35-mm Petri dishes, and the dishes were placed in a Bio-Rad Biolistic PDS-1000/HE Particle Delivery System for particle bombardment. Optimized biolistic parameters are 15-in Hg of chamber vacuum, target distance of 3 cm (stage 1), 1,350-psi particle acceleration pressure, and 1.6-μm gold microcarriers. Supplemental experimental details on schistosome biolistics are published on the PNAS web site (www.pnas.org).
Gold microcarriers were prepared, and circular plasmid DNA was precipitated onto the gold by using methods described by the manufacturer (Bio-Rad) (6) with the following modifications: 1–1.25 mg of gold particles carrying 5–7 μg of plasmid DNA were used per bombardment. For RNA biolistics, microcarriers were prepared as described by the manufacturer, but the RNA was lyophilized onto the gold particles. Briefly, in vitro-transcribed RNAs at 1 μg/μl were mixed with 1.6-μm gold microcarriers (≈5 μl of RNA/mg of gold particles/shot) and the gold/RNA suspension multiply dotted (≈30 small spots) onto macrocarriers in the macrocarrier holders. The holders were placed immediately on dry ice to freeze the dotted gold/RNA suspensions, and then lyophilized in a speed vac until dry (≈1 hr). Treatment of RNA with nuclease (RNase A or DNase I) and its purification were carried out before mixing the RNA with gold particles and lyophilization.
After bombardment, 2–4 ml of nematode blastomere media (11) was added, and the embryos were incubated in a humidified air shaker at 30°C (minimal rpm to keep the embryos in suspension) until assayed for either RNA expression or luciferase activity.
Evaluation of gold particle delivery into Ascaris embryos (or schistosomes) as a function of physical biolistic parameters was evaluated by using a plasmid DNA/4′,6-diamidino-2-phenylindole (DAPI) mix (0.6 ng DAPI/μg of DNA) precipitated onto the particles in the presence of MgCl2 instead of CaCl2. Gold particle delivery into cells was easily detected through identification of fluorescing nuclei within cells that had received a DNA/DAPI-coated gold particle.
Nucleic Acid Isolation and in Vitro Transcription.
Total Ascaris embryo RNA was purified by TRIzol Reagent (GIBCO/BRL/Life Technologies, Gaithersburg, MD). Plasmids for sequencing and bombardment were prepared by using QIAfilter midi or maxi plasmid kits (Qiagen, Chatsworth, CA).
Luciferase RNAs (1,895 nt) for biolistics were prepared from PCR templates generated by using Promega pGL-3 basic plasmid, Pfu polymerase (Stratagene), and primers that led to the addition of a T7 promoter and 30-nt poly(A) tail to the PCR product (5′ = ATCGAAATTAATACGACTCACTATAGGGATTCCGGTACTGTTG; 3′ = TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGAATGCAATTGTTGTTGTT). In vitro T7 RNA polymerase transcription of the PCR template (≈3 μg) with m7GpppG cap analog at a ratio of 5:1 cap analog/GTP was carried out by using a MegaScript kit as described by the manufacturer (Ambion, Austin, TX). Transcription reactions were DNase I-treated and extracted with TRIzol. Precipitated RNAs were washed with 70% ethanol, dissolved in water, quantitated spectrophotometrically, and examined by agarose-formaldehyde denaturing gel electrophoresis, and the translation efficiency of the RNA was determined by measurement of luciferase activity after translation in a rabbit reticulocyte extract (Promega).
RNase Mapping.
RNase mapping was carried out with ≈5–10 μg of total RNA by using a RPA II kit as described by the manufacturer (Ambion).
Luciferase Assays.
Embryos in 35-mm Petri dishes were collected in Kontes Mini Homogenizers and pelleted at 600 × g for 1 min at room temperature, media were removed, and the resulting pellet was frozen on dry ice. Embryos were thawed on ice with 150 μl of Promega 1× cell culture lysis reagent with complete, mini protease inhibitors/EDTA-free (Boehringer Mannheim), homogenized twice briefly with a motor-driven pestle, and centrifuged for 5 min at 4°C at 18,000 × g. Aliquots (20–40 μl) of the supernatant were assayed for luciferase activity by using a Promega Luciferase Assay System in a Opticomp I Luminometer (MGM Instruments, Hamden, CT). Luciferase activity was measured in the linear range of the assay as determined by comparison to serial dilutions of recombinant luciferase (Promega) and mixing experiments using Ascaris embryo extracts. Similar methods were used for adult schistosomes. In some experiments, protein concentrations of the embryo extracts were measured (Bio-Rad DC Protein Assay) and luciferase activity was normalized to protein concentration (see Fig. 4B). Experimental data were similar with or without normalization to protein concentrations.
Figure 4.

Luciferase activity in Ascaris embryos after biolistic introduction of RNA. Luciferase RNAs lyophilized onto gold particles (typically 5 μg RNA/mg gold) were introduced into 32- to 64-cell Ascaris embryos, and luciferase activity was measured in the embryos after incubation at 30°C. Relative luciferase light (RLU) units represent only ≈15–20% of the total activity in the embryos. (A) Structure of the in vitro-generated luciferase RNA. (B) Luciferase expression is RNA dependent. Embryos were assayed for luciferase activity 8 hr after RNA biolistics. Nuclease treatment of the RNA preceeded lyophilization of the RNA onto gold particle (n = 4 from two separate experiments). Maximum luciferase activity in a representative experiment was 236,423 ± 10,653 (n = 2), which represents ≈15% of the total embryo activity. No Nuclease Treatment, RNA introduced without additional treatment; DNase Treated, RNA treated with DNase I before introduction into embryos; Transcription Template, PCR generated template used for in vitro transcription of the luciferase RNA; RNA Assayed, equivalent amounts of RNA lyophilized onto beads directly assayed for luciferase activity. (C) Time course of luciferase activity in Ascaris embryos (n = 3). (D) Dose response for luciferase RNA expression in Ascaris embryos (n = 3). Varying amount of RNA (1.25–10 μg) were lyophilized onto gold particles (1 mg), and luciferase activity was measured 6 hr after biolistic introduction of the RNA.
Cloning, Sequencing, and Sequence Analysis.
Constructs were prepared by using standard cloning methods (12, 13) and verified by restriction analysis and automated fluorescent cycle sequencing. Detailed information on the constructs is available on request.
RESULTS
Expression of a Marked Spliced Leader (SL) RNA Gene in Ascaris Embryos.
Ascaris embryos at the 32- to 64-cell stage of embryogenesis (5 days development at 30°C) were chosen for development of in vivo molecular genetic methods as this stage corresponds to that previously used to prepare competent in vitro transcription, splicing, and translation extracts (14–16). Physical particle bombardment parameters (vacuum level, particle type and size, target distance, prebombardment and postbombardment incubation media, and particle acceleration pressure) were systematically evaluated to optimize introduction and expression of nucleic acid while minimizing embryo damage and maximizing viability. Conditions were developed that result in at least ≈60–80% of the embryos acquiring gold particles, half of which acquire multiple microprojectiles (see Materials and Methods for optimal biolistic conditions). After bombardment and recovery in media designed for culture of nematode blastomeres (11), most embryos remained viable and continued development during the period of time in which expression was measured.
The SL RNA gene was chosen for our initial experiments as the gene is readily transcribed in a 32- to 64-cell Ascaris in vitro transcription system (14). In addition, the SL RNA gene promoter is the best-characterized promoter in Ascaris. To distinguish between endogenous expression of the SL RNA gene and expression of an introduced gene, we used a marked SL RNA that contains an insertion of 11 nt within the SL RNA intron. This insertion has no affect on SL RNA transcription in vitro, and the construct contains the prerequisite elements for accurate transcription initiation and termination (14). We constructed an RNase mapping probe containing the 11-nt insertion that extends upstream from near the 3′ terminus of the mature SL RNA to 115 bases 5′ of its transcription initiation site. Use of this RNase mapping probe facilitated distinction between the endogenous and introduced marked SL RNA and allowed us to determine whether transcription initiation occurred accurately (see Fig. 1).
Figure 1.

Expression of a marked SL RNA gene in Ascaris embryos. (A) RNase mapping. Sixteen hours after bombardment of Ascaris embryos (2-day = ≈two cells, 3-day = ≈4–8 cells, or 5-day = ≈32–64 cells) with a marked SL RNA gene, total RNA was isolated and subjected to RNase mapping. The expected RNase mapping product for accurate initiation and expression of the marked SL RNA is 94 nt and is indicated by the upper, single arrow. Mapping of the endogenous wild-type SL RNA generates two fragments because of the mark (11-nt substitution) in the mapping probe (see B). Note that no endogenous RNAs of 94 nt are protected in untransfected or mock-transfected embryos (Embryos Alone). Embryos transfected with an SL RNA gene mutated with a block substitution from −46 to −56 of the SL RNA promoter show dramatically reduced levels of the marked RNA, similar to observations made in 32- to 64-whole-cell Ascaris embryo extracts (14). Data shown are representative of multiple experiments. (B) RNase mapping strategy and probe.
A marked SL RNA gene biolistically introduced into 32- to 64-cell Ascaris embryos was efficiently expressed as demonstrated by the protection of an RNA of the predicted size by using RNase mapping (Fig. 1A). Marked SL RNA expression in embryos depends on DNA concentration, time postbombardment, and an optimal set of physical biolistic parameters that includes microparticle type and size, target distance, vacuum, and particle accelerating pressure. As shown in Fig. 1A, the marked SL RNA gene is accurately initiated, and expression depends in part on upstream promoter elements (−46 to −56 region) previously shown to be essential in vitro (14). Introduction and expression of the SL RNA has been carried out in 2-day (two cells), 3-day (four cells), and 5-day (32–64 cells) embryos (see Fig. 1A) as well as in 10-day larvae. Expression of the SL RNA occurs as early as 1 hr postbombardment in early embryos and continues for at least 80 hr, the last time point analyzed. In sum, these data indicate that accurate and significant SL RNA expression can be obtained in Ascaris embryos after biolistic transfection of a marked SL RNA gene by using optimized physical bombardment parameters.
Expression of a Luciferase Reporter Driven by the SL RNA Promoter.
We also evaluated the use of a firefly luciferase reporter gene construct driven by the SL RNA promoter previously shown to be functional after biolistic transfection. This construct contains 115 bases upstream of the SL RNA transcription initiation site, the SL, and 8 bases of the SL RNA intron, the pGL-3 luciferase ORF, and a 287-bp Ascaris 3′ untranslated region (UTR) and polyadenylation signal. The Ascaris 3′ UTR and polyadenylation signal were derived from a gene encoding a protein with similarity to the 16-kDa proteolipid subunit of vacuolar ATP synthase (gene 12 = ATP synthase, GenBank accession no. M33757; nucleotides 3056 to 3342) (SL RNA gene, GenBank accession no. M27961). After biolistic transformation with this construct, high levels of luciferase activity in Ascaris embryos were readily detected and dependent on expression time, SL RNA promoter sequences and their orientation, and physical biolistic parameters (see Fig. 2). Levels of luciferase activity were typically ≈50- to 200-fold above background, depending somewhat on the lot of 1.6-μm gold microcarriers. Ascaris embryos have no endogenous luciferase activity and embryo extracts do not interfere with luciferase activity or its assay. Luciferase activity was not present in the actual plasmid DNA introduced nor derived from bacterial contamination of the media. Luciferase expression derived from the SL RNA promoter was measurable as early as ≈ 8 hr after transfection into two-cell embryos. In 32- to 64-cell embryos, luciferase expression continued to increase for at least 80 hr postbombardment (Fig. 2B).
Figure 2.

Luciferase expression from an SL RNA promoter in 32- to 64-cell Ascaris embryos. Relative luciferase light units represent ≈25% of the total activity in the embryos. Data shown illustrate a representative experiment. (A) Organization of SL RNA promoter luciferase constructs. Constructs are based on the Promega pGL-3 basic plasmid substituting the Ascaris vacuolar ATP synthase 3′ UTR and polyadenylation signal for the analogous simian virus 40 region in pGL-3 = pGL-3/AscUTR (part 1). Minimal and extended upstream SL RNA gene promoter regions (part 2), an inverted minimal SL RNA promoter (= flip) (part 3), and mutations (block or a splice donor nucleotide substitution) in the promoter (part 4) were fused upstream of the firefly luciferase ORF. (B) Time course of luciferase expression. Luciferase activity in transfected embryos from a minimal SL RNA gene promoter was measured as a function of time after bombardment (n = 3). (C) SL RNA promoter analysis. Luciferase activity in transfected embryos was measured 16 hr after bombardment. Maximal luciferase activity in this representative experiment was 122,112 ± 11,365 relative light units (SE; n = 4).
Previous analysis of SL RNA transcription in vitro indicated that promoter elements required for maximal expression of the SL RNA were limited to ≈100 bases upstream of the transcription initiation site and the SL sequence itself (14). Other transcriptional elements such as enhancers typically are not readily measured by using in vitro transcription systems. Therefore, to explore the potential role of additional upstream sequences on SL RNA promoter activity in vivo, we generated constructs extending varying distances upstream from the SL RNA gene with the largest construct (−1038) extending through the entire Ascaris SL RNA/5S tandemly repeated genomic unit (17). Promoter regions extending upstream varied in length as follows: −115, −341, −526, and −1038. After biolistic introduction, Ascaris SL RNA gene sequences upstream from −115 increased the level of in vivo transcription as measured by luciferase expression. Furthermore, in initially exploring the use of the SL RNA promoter to develop a luciferase reporter assay, we were concerned that the presence of a splice donor site downstream from the SL might result in aberrant internal cis-splicing within the luciferase ORF that would decrease luciferase activity. Consequently, we also generated and examined a construct in which the GT in the splice donor site after the SL was mutated to AT. Luciferase activity was readily detectable in constructs with the GT present as shown in Fig. 2. Notably, however, the levels of luciferase activity were ≈ 5-fold less as a result of the splice donor mutation. One likely interpretation of this observation is that in addition to the SL sequence, the donor sequence contributes to and constitutes part of the promoter for the SL RNA gene.
Expression of Other Luciferase Reporter DNA Constructs.
To further evaluate the use of firefly luciferase as a reporter for other Ascaris promoters or RNA processing elements (e.g., trans-splicing), we generated constructs with DNA insertions upstream of a pGL-3 plasmid containing the previously described Ascaris 3′ UTR and polyadenylation signal inserted downstream of the luciferase ORF (pGL-3/AscUTR) (Figs. 2A and 3A). Biolistic transfection of constructs with putative promoter regions from two mRNA encoding Ascaris genes, a translation elongation factor (eF-1-gamma) (T. Nilsen, personal communication) and fert-1 (GenBank accession no. U07350) (18) led to significant levels of luciferase activity compared with levels generated with the promoterless plasmid (pGL-3/AscUTR) or a construct containing the ribosomal RNA promoter (Fig. 3B).
Figure 3.

Luciferase activity in Ascaris embryos: mRNA gene promoter and trans-splice acceptor DNA constructs. Reporter constructs (see A) were introduced into 32- to 64-cell Ascaris embryos. Error bars in B-D represent SEM (n = 4) for these representative experiments. C and D are independent experiments. Relative luciferase light units represent ≈ 15–25% of the total activity in the embryos. (A) Organization of luciferase constructs containing putative Ascaris promoters and a trans-splice acceptor. (B) Luciferase expression derived from eF-1-gamma (translation elongation factor 1-gamma), fert-1, and rRNA promoter sequences 48 hr after transfection. (C) Luciferase expression 20 hr after transfection with DNA constructs containing the Ascaris vacuolar ATP synthase subunit gene trans-splice acceptor. (D) Luciferase expression 48 hr after transfection with DNA constructs containing the Ascaris vacuolar ATP synthase subunit gene trans-splice acceptor.
In trans-splicing trypanosomes (parasitic protozoa), expression of a protein ORF can be driven by constructs containing a ribosomal RNA promoter followed by a trans-splice acceptor site upstream of the ORF (19–21). High levels of luciferase activity were observed in Ascaris embryos transfected with a pGL-3/AscUTR construct containing the Ascaris ribosomal RNA promoter (22–24) followed 3′ with a 105-bp trans-splice acceptor from the vacuolar ATP synthase subunit gene (Fig. 3C) (15, 25–29). Transfection of a series of additional constructs containing variations of the ATP synthase trans-splice acceptor resulted in the following observations (see Fig. 3 C and D): (i) maximal luciferase activity was observed with only the 105-bp trans-acceptor present; (ii) luciferase activity decreases ≈60% when an additional ≈900-bp sequence upstream of the trans-acceptor was present (1-kb acceptor); (iii) a minimal ATP synthase trans-splice acceptor of 11 bp exhibited some, although much lower, levels of luciferase activity (8%); and (iv) addition of ≈1.6 kb of rRNA promoter sequence upstream of the trans-splice acceptor also led to a ≈60% decrease in luciferase activity. In sum, these data suggest that embryo luciferase activity derived from transfection with these constructs appears attributable primarily to the presence of the 105-bp trans-splice acceptor fragment from the vacuolar ATP synthase gene (Fig. 3 C and D). Insertion of additional sequences upstream of the trans-splice acceptor may decrease the amount of read-through transcription derived from promoter activity within the upstream pGL-3 sequences. This pGL-3 promoter activity, however, apparently must not result in significant levels of mature, translatable luciferase mRNA in the absence of efficient trans-splicing. There may be some inherent promoter activity within the vacuolar ATP synthase gene sequences, but additional experiments will be required to define such activity.
Overall, the data obtained by using DNA constructs indicate that different promoters and RNA processing elements can be readily analyzed by measuring firefly luciferase activity after biolistic introduction of constructs into Ascaris embryos.
Expression of RNA in Ascaris Embryos.
Microinjection or electroporation of RNA previously has been used to examine the role of cap structure and poly(A)-tail length on translation and stability in vivo, questions often not easily addressed by using DNA constructs (30, 31). Biolistic transfection of luciferase RNAs lyophilized onto gold particles led to significant levels of firefly luciferase activity in Ascaris embryos (Fig. 4). Luciferase activity derived from biolistic introduction of the RNA, although not affected by DNase I, was eliminated by pretreatment of the RNA with RNase before lyophilization onto the gold particles. Luciferase activity was absent in the luciferase RNA preparations and the RNA transcription template. Biolistic transfection of luciferase RNA exhibited a dose-and time-dependent response.
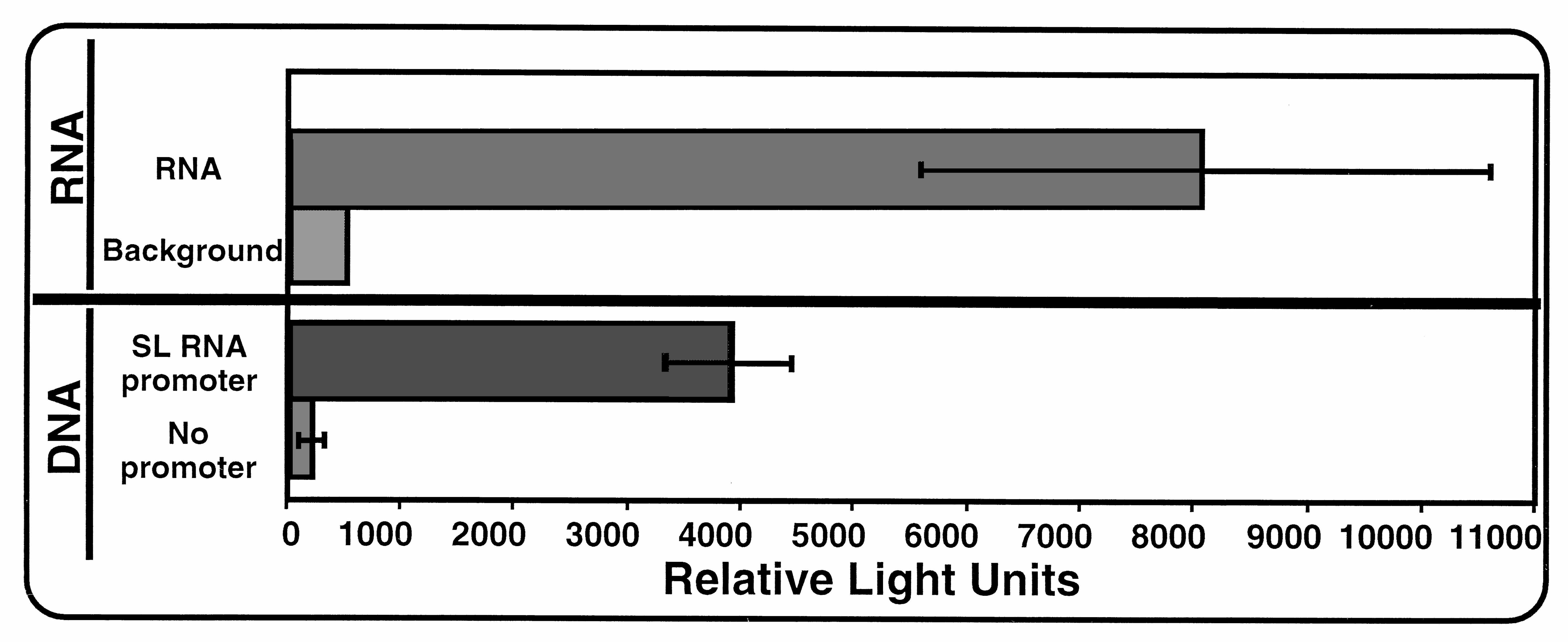
Schistosome Biolistics.
We also explored the utility of biolistics for introduction and expression of DNA and RNA into another parasitic helminth, Schistosoma mansoni. Schistosomes belong to an evolutionarily divergent metazoan phylum (Platyhelminthes), exhibit a distinctly different body plan from that of nematodes, and as adults, are quite large (≈1 mm in width and 1 cm in length) in comparison to the Ascaris embryos. Approximately 10–15 adult schistosome worm pairs in media were placed in the center of 35-mm Petri dishes, media were removed, and physical biolistic parameters were carried out as described for Ascaris embryos. Bombarded worms were incubated for 8–24 hr, collected, homogenized, and assayed for luciferase activity. Transfection of a DNA construct led to luciferase expression in adult schistosomes that was ≈20-fold above an identical control construct without a promoter. Luciferase activity derived from introduction of RNA was as high as ≈20-fold above background. Schistosome transfection data are published as supplemental data on the PNAS web site, www.pnas.org. The DNA construct contained the S. mansoni SL RNA gene (−450 to +38; SL RNA gene, GenBank accession no. M34074) fused upstream of the pGL-3 luciferase ORF followed by an S. mansoni enolase 3′ UTR and polyadenylation signal (225 bp; Enolase gene, GenBank accession no. U33177; nucleotides 1006–1230). For RNA biolistics, the luciferase RNA described for Ascaris RNA biolistics (Fig. 4A) was used.
DISCUSSION
By using biolistics we have demonstrated that DNA can be successfully introduced and expressed in Ascaris embryos as early as the two-cell stage (2 days development at 30°C) and through L1 larvae (10 days, 30°C). Expression of the transfected DNA can be measured at the RNA level via RNase mapping or at the protein level through the use of a luciferase reporter gene. Expression depends on optimal biophysical paramaters, DNA concentration, expression time, and the requirement for either upstream Ascaris promoter elements or a trans-splice acceptor. We also have demonstrated that in addition to DNA, RNA can be effectively introduced and expressed in Ascaris embryos. Expression of luciferase activity derived from introduction of RNA is RNase sensitive, DNase I resistant, and dependent on both RNA concentration and time after bombardment. RNA expression can be measured as early as 1 hr after transfection whereas protein expression requires ≈ 6–8 hr.
Development of biolistic DNA transfection in Ascaris embryos will facilitate the use of a variety of experimental paradigms to investigate gene expression and the biology of these organisms. The development of RNA transfection by using biolistics should facilitate analyses of mRNA metabolism in vivo. For example, the role of SL sequences, their trimethylguanosine caps, and poly(A)-tail lengths on mRNA translation and stability in vivo remain important and outstanding questions with respect to the molecular and functional significance of trans-splicing in helminths. These questions now can be addressed. In addition, RNA biolistics should allow analysis of mRNA translational regulation or stability in synchronously developing Ascaris embryos from one-cell stage through L1 larvae.
We also have demonstrated that the biolistic method can be used to introduce and express DNA and RNA in adult schistosomes. Although expression levels in schistosomes are currently not high, optimization of biophysical, genetic, and other transfection parameters are likely to improve expression as we observed in developing the method for Ascaris embryos. Additionally, our initial experiments in Ascaris also included biolistic transfection with Caenorhabditis elegans β-galactosidase reporter vectors (32). As a positive control for the Ascaris experiments, the reporter was introduced into mixed stage C. elegans on agar in Petri dishes, and β-galactosidase expression was observed in several C. elegans stages (data not shown). This finding indicates that this methodology can be used with other nematode species and life cycle stages. Preliminary experiments further indicate that this method also works in the parasitic, filarial nematode Brugia malayi (Thomas Unnasch, personal communication).
Our results indicate that particle bombardment is a practical and relatively efficient method for introducing and expressing nucleic acids in several multicellular helminths and should be applicable to other helminths of various sizes, stages, and types. The use of biolistics in transfecting both multicellular organisms and embryos: (i) is a rapid and simple method for transfection of a large number of organisms, tissues, and cells; (ii) produces reasonably high transformation efficiencies and allows introduction of nucleic acids into virtually any cell type; (iii) is amenable to measurement of low levels of expression and quantitative analysis in transient transfection experiments with a luciferase reporter; (iv) is flexible in that biophysical parameters can be adjusted so that particles carrying nucleic acid will reach only cells near the surface or also penetrate into cells deep within a multicellular organism; and (v) previously has been shown to also be an effective method for transformation of organelles (4, 7, 8). The potential for transfection of any cell in an organism should facilitate both general analyses in a variety of cell types and organs as well as tissue-specific expression by using constructs with cell- or tissue-specific promoters. Biolistic transfection also should facilitate molecular genetic analyses in a variety of other parasitic helminths including: (i) other parasitic nematodes such as other filaria, plant parasitic nematodes, and hookworms; (ii) parasitic flatworms including tapeworms, monogenes, and other trematodes; and (iii) various developmental stages of parasitic helminths such as the larval stages of schistosomes (miracidia, sporocysts, cercariae, and shistosomula). Space limitations preclude a discussion of the range of experimental applications that transient transfection of helminths may facilitate. One new and powerful experimental strategy that can now be explored in parasitic helminths is RNA interference (33). It is also conceivable that biolistics subsequently could be used to develop transgenic parasites for experimental purposes.
Supplementary Material
Acknowledgments
We thank Timothy Nilsen and his laboratory for generously supplying constructs and clones of Ascaris genes (SL RNA, translation elongation factor eF-1-gamma, and vacuolar ATP synthase subunit). We also thank Fritz Muller for clones of the Ascaris fert-1 gene and the ribosomal RNA repeat. We thank Scott Emmons for providing C. elegans cultures and Andrew Fire for kindly providing us with C. elegans vector kits. This research was supported in part by grants from the National Institutes of Health (AI 39714 to R.E.D. and AI 27219 to P.T.L.).
ABBREVIATIONS
- SL
spliced leader
- UTR
untranslated region
References
- 1.Sasser J N, Freckman D. In: Vistas in Nematology. Dickson D W, Veech J A, editors. Hyattsville, MD: Society of Nematologists; 1987. pp. 7–14. [Google Scholar]
- 2.Christou P. Methods Cell Biol. 1995;50:375–382. [PubMed] [Google Scholar]
- 3.Johnston S A. Nature (London) 1990;346:776–777. doi: 10.1038/346776a0. [DOI] [PubMed] [Google Scholar]
- 4.Klein T M, Fitzpatrick-McElligott S. Curr Opin Biotechnol. 1993;4:583–590. doi: 10.1016/0958-1669(93)90081-7. [DOI] [PubMed] [Google Scholar]
- 5.Maden M. Curr Biol. 1994;4:281–284. doi: 10.1016/s0960-9822(00)00066-x. [DOI] [PubMed] [Google Scholar]
- 6.Sanford J C, Smith F D, Russell J A. Methods Enzymol. 1993;217:483–509. doi: 10.1016/0076-6879(93)17086-k. [DOI] [PubMed] [Google Scholar]
- 7.Butow R A, Henke R M, Moran J V, Belcher S M, Perlman P S. Methods Enzymol. 1996;264:265–278. doi: 10.1016/s0076-6879(96)64026-9. [DOI] [PubMed] [Google Scholar]
- 8.Daniell H. Methods Enzymol. 1993;217:536–556. doi: 10.1016/0076-6879(93)17088-m. [DOI] [PubMed] [Google Scholar]
- 9.Gregoire R J, Shi M H, Rekosh D M, LoVerde P T. J Immunol. 1987;139:3792–3801. [PubMed] [Google Scholar]
- 10.Maroney P A, Hannon G J, Nilsen T W. Mol Biochem Parasitol. 1989;35:277–283. doi: 10.1016/0166-6851(89)90214-4. [DOI] [PubMed] [Google Scholar]
- 11.Shelton C A, Bowerman B. Development (Cambridge, UK) 1996;122:2043–2050. doi: 10.1242/dev.122.7.2043. [DOI] [PubMed] [Google Scholar]
- 12.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 13.Ausubel F M, Brent R, Kingston R E, Moore D D, Smith J A, Seidman J G, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 14.Hannon G J, Maroney P A, Ayers D G, Shambaugh J D, Nilsen T W. EMBO J. 1990;9:1915–1921. doi: 10.1002/j.1460-2075.1990.tb08318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannon G J, Maroney P A, Denker J A, Nilsen T W. Cell. 1990;61:1247–1255. doi: 10.1016/0092-8674(90)90689-c. [DOI] [PubMed] [Google Scholar]
- 16.Maroney P A, Denker J A, Darzynkiewicz E, Laneve R, Nilsen T W. RNA. 1995;1:714–723. [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsen T W, Shambaugh J, Denker J, Chubb G, Faser C, Putnam L, Bennett K. Mol Cell Biol. 1989;9:3543–3547. doi: 10.1128/mcb.9.8.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spicher A, Etter A, Bernard V, Tobler H, Muller F. Dev Biol. 1994;164:72–86. doi: 10.1006/dbio.1994.1181. [DOI] [PubMed] [Google Scholar]
- 19.Lee M G, Van der Ploeg L H. Annu Rev Microbiol. 1997;51:463–489. doi: 10.1146/annurev.micro.51.1.463. [DOI] [PubMed] [Google Scholar]
- 20.Rudenko G, Chung H M, Pham V P, Van der Ploeg L H. EMBO J. 1991;10:3387–3397. doi: 10.1002/j.1460-2075.1991.tb04903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zomerdijk J C, Kieft R, Borst P. Nature (London) 1991;353:772–775. doi: 10.1038/353772a0. [DOI] [PubMed] [Google Scholar]
- 22.Muller E, Neuhaus H, Tobler H, Muller F. EMBO J. 1990;9:2849–2856. doi: 10.1002/j.1460-2075.1990.tb07474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briner G, Muller E, Neuhaus H, Back E, Muller F, Tobler H. Nucleic Acids Res. 1987;15:6515–6538. doi: 10.1093/nar/15.16.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Back E, Muller F, Tobler H. Nucleic Acids Res. 1984;12:1313–1332. doi: 10.1093/nar/12.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hannon G J, Maroney P A, Yu Y T, Hannon G E, Nilsen T W. Science. 1992;258:1775–1780. doi: 10.1126/science.1465612. [DOI] [PubMed] [Google Scholar]
- 26.Hannon G J, Maroney P A, Nilsen T W. J Biol Chem. 1991;266:22792–22795. [PubMed] [Google Scholar]
- 27.Maroney P A, Hannon G J, Shambaugh J D, Nilsen T W. EMBO J. 1991;10:3869–3875. doi: 10.1002/j.1460-2075.1991.tb04956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maroney P A, Yu Y T, Jankowska M, Nilsen T W. RNA. 1996;2:735–745. [PMC free article] [PubMed] [Google Scholar]
- 29.Denker J A, Maroney P A, Yu Y T, Kanost R A, Nilsen T W. RNA. 1996;2:746–755. [PMC free article] [PubMed] [Google Scholar]
- 30.Gallie D R, Lucas W J, Walbot V. Plant Cell. 1989;1:301–311. doi: 10.1105/tpc.1.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallie D R. Genes Dev. 1991;5:2108–2116. doi: 10.1101/gad.5.11.2108. [DOI] [PubMed] [Google Scholar]
- 32.Mello C, Fire A. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- 33.Sharp P A. Genes Dev. 1999;13:139–141. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}