

Abstract
Although extensive effort has been applied toward understanding the mechanism by which enediynes cleave DNA, a continuous assay for this phenomenon is still lacking. In fact, with the exception of assays for DNase, continuous assays for most DNA cleavage events are unavailable. This article describes the application of “molecular break lights” (a single-stranded oligonucleotide that adopts a stem-and-loop structure and carries a 5′-fluorescent moiety, a 3′-nonfluorescent quenching moiety, and an appropriate cleavage site within the stem) to develop the first continuous assay for cleavage of DNA by enediynes. Furthermore, the generality of this approach is demonstrated by using the described assay to directly compare the DNA cleavage by naturally occurring enediynes [calicheamicin and esperamicin), non-enediyne small molecule agents (bleomycin, methidiumpropyl–EDTA–Fe(II), and EDTA–Fe(II]), as well as the restriction endonuclease BamHI. Given the simplicity, speed, and sensitivity of this approach, the described methodology could easily be extended to a high throughput format and become a new method of choice in modern drug discovery to screen for novel protein-based or small molecule-derived DNA cleavage agents.
Keywords: calicheamicin, esperamicin, assay, molecular beacon, bleomycin
Calicheamicin (Fig. 1), 1, from Micromonospora echinospora spp. calichensis, is over 1,000 times more potent than doxorubicin, clinically one of the most useful antitumor agents available. A prominent member of the enediyne family, 1 is a premiere example of nature's ingenuity (1–6). Of the two distinct structural regions within 1 (7, 8), the aryltetrasaccharide is composed of a unique set of carbohydrate and aromatic units that site-specifically deliver the metabolite into the minor groove of DNA (9), whereas the aglycone, or “warhead,” consists of a highly functionalized bicyclo[7.3.1]tridecadiynene core structure with an allylic trisulfide serving as the triggering mechanism. Aromatization of the bicyclo[7.3.1]tridecadiynene core structure, via a 1,4-dehydrobenzene-diradical, results in the site-specific oxidative double-strand scission of the targeted DNA, and this extraordinary reactivity has sparked considerable interest in the pharmaceutical industry (10, 11). Whereas extensive effort has been applied to understanding the mechanism by which enediynes cleave DNA, a continuous assay for this phenomenon is still lacking. In fact, with the exception of assays for DNase, continuous assays for most enzymatic and small molecule-catalyzed DNA cleavage events are unavailable.
Figure 1.

Nonenzymatic DNA-cleaving agents: calicheamicin γ1I from M. echinospora (1), esperamicin A1 from A. verrucosospora (2), bleomycin from S. verticillus (3), MPE–Fe(II) (4), and EDTA–Fe(II) (5).
In our effort to understand calicheamicin biosynthesis, self-resistance, and mode of action (1, 2, 12, 13), we now report the design and application of a modified hairpin-forming oligonucleotide to continuously assess DNA cleavage by enediynes. The substrate oligonucleotide for these assays is based on “molecular beacon” design, a single-stranded oligonucleotide that adopts a stem-and-loop structure and carries a 5′-fluorescent moiety and a 3′-nonfluorescent quenching moiety (14, 15; Fig. 2a). The stem design keeps these two moieties in close proximity to each other to provide fluorescence quenching by fluorescence resonance energy transfer (FRET) and, in our design, also includes a DNA-binding recognition sequence for the corresponding cleavage agent (Fig. 2b). Scission of this stem by the agent leads to immediate separation of the fluorophore–quencher pair and results in a spontaneous fluorescent signal that directly correlates to the extent of DNA cleavage. To delineate the function of these oligonucleotides from molecular beacons, they have been designated “molecular break lights” (as in DNA strand “break”). The general utility of the break light assay is further expanded to provide a direct comparison of the cleavage efficiencies by naturally occurring enediynes (Fig. 1; 1 and esperamicin, 2), non-enediyne small molecule agents (bleomycin, 3, methidiumpropyl–EDTA–Fe(II), 4, and EDTA–Fe(II), 5), as well as the restriction endonuclease BamHI. The molecular break light assay is advantageous over previous FRET-based DNA cleavage assays in that one can achieve a significantly higher signal to noise ratio (≈40) in comparison with assays based on oligonucleotide pairs (≈2) with a single oligonucleotide substrate (14, 15). Furthermore, the molecular break light assay exceeds the sensitivity of assays based upon fluorescence correlation spectroscopy >10-fold, a very sensitive technique that also requires extremely specialized instrumentation (16). The sensitivity of this assay also rivals the typical discontinuous assay for detection of DNA-damaging agents known as the biochemical induction assay (BIA). Given the simplicity, speed, and sensitivity of this approach, the described methodology could easily be extended to a high throughput format and become a new method of choice in modern drug discovery to screen for novel protein-based or small molecule-derived DNA cleavage agents.
Figure 2.

A schematic diagram of molecular beacons, molecular break lights, and the specific break lights used in this study. The solid lines represent covalent bonds, dashed lines represent hydrogen bonding, letters represent arbitrary bases, the gray shaded ball represents the fluorophore (FAM), the black ball represents the corresponding quencher (DABCYL), and the dashed wedges represent fluorescence. (a) Principle of operation of molecular beacons. Target hybridization leads to a separation of the fluorophore–quencher pair and a corresponding fluorescent signal. (b) Principle of operation of molecular break lights. Cleavage of the stem by an enzymatic or nonenzymatic nuclease activity results in the separation of the fluorophore–quencher pair and a corresponding fluorescent signal. (c) Molecular break lights used in this study. The stem of break light A contains a preferred calicheamicin recognition site (in bold), and the stem of break light B carries the BamHI recognition site (in bold). The predicted cleavage sites are illustrated by arrows.
Materials and Methods
Materials.
All oligonucleotides used for the described studies were purchased from GIBCO/BRL. The BamHI endonuclease used in the kinetic studies (10 units/μl) was obtained from Promega. Esperamicin was a generous gift of Kin Sing (Ray) Lam (Bristol-Myers Squibb), and bleomycin sulfate (Blenoxane) was kindly provided by Ben Shen (University of California, Davis). All other reagents described were obtained from commercial sources. Blenoxane (a mixture containing approximately 70% bleomycin A2 and 30% bleomycin B2) was dissolved in water and optically standardized (ɛ291 = 1.7 × 104 M−1⋅cm−1; ref. 17). All Fe containing solutions were prepared fresh from (NH4)2Fe(SO4)2 daily with 1 mM H2SO4 to prevent hydrolysis and oxidation (18).
Spectrofluorometry.
Samples were analyzed with a FluoroMax-2 spectrofluorometer equipped with DataMax for Windows (Instruments SA, Edison, NJ) and the temperature controlled (30°C, unless otherwise noted) by a Haake Circulator DC10. All samples were filtered before analysis and analyzed via a time base scan (λex = 485 nm, λem = 517 nm) in a Suprasil quartz cuvette (10-mm path) fitted with a magnetic stirring bar in a total volume of 2 ml. Reactions were equilibrated to the incubation temperature before initiation of DNA cleavage as was evident by a steady background emission over 10 min. Total cleavage of the labeled oligonucleotide, confirmed by PAGE, was defined as the maximum fluorescence emission possible under saturated cleaving conditions. Emission units were converted to the amount of labeled oligonucleotide used within a procedure, thereby equating labeled oligonucleotide degradation as a function of the emission of fluorescence.
BamHI Digestion.
Determination of BamHI-specific cleavage was performed in 10 mM Tris⋅HCl (pH 7.9)/50 mM NaCl/10 mM MgCl2/1 mM DTT with 3.4 nM of break light B and varying amounts of break light B lacking the fluorophore and quenching moieties (0, 3.8, 7.7, 38.5, 77.0, 192.5, and 385 nM) at 37°C. The reaction was initiated with 10 units of BamHI enzyme and monitored over a time course of 15 min. The initial rate of DNA cleavage was determined from data within the first 100 s of initiation, which was then adjusted according to Eq. 1. These adjusted values were used for the reciprocal plot from which the Michaelis–Menten kinetic parameters were determined.
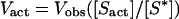 |
1 |
Enediyne-Induced Cleavage.
Enediyne antibiotics calicheamicin and esperamicin at varying concentrations (0.31, 0.78, 1.6, 3.17, 15.9, and 31.7 nM) were incubated in 40 mM Tris⋅HCl (pH 7.5) with 3.2 nM of break light A. DNA cleavage was initiated with the addition of 1 μl 100 mM DTT (50 μM final concentration) and monitored over 10 min. Pseudofirst order kinetic parameters were used to determine the initial velocities at each given enediyne concentration. Specifically, graphical representation of the data were based upon Eq. 2, where [A]t is the concentration of cleaved oligonucleotide at a given time (t) and [A]0 is the initial concentration of oligonucleotide in the assay. Least squares analysis gave the slope (k), or rate, which was converted to V by the relationship in Eq. 3. The maximum velocity achieved (Vmax) was then selected from the range of concentrations examined.
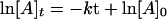 |
2 |
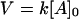 |
3 |
Bleomycin-Induced Cleavage.
Bleomycin-mediated cleavage was adapted from procedures outlined by Giloni et al. (19). Concentrations of bleomycin (9.5, 19, 47.5, 95, 142.5, and 190 nM) were incubated in 40 mM potassium phosphate buffer (pH 7.5) with 3.2 nM of break light A. The reaction was initiated by the addition of 65 mM Fe(II) and monitored over 5 min. This protocol was repeated with the addition of 5 mM sodium ascorbate to the above conditions. Pseudofirst order kinetic parameters were used to determine the initial velocities at each given bleomycin concentration as previously described.
Iron (II)-Chelator-Induced Cleavage.
EDTA–Fe(II)-mediated oligonucleotide degradation was adapted from procedures outlined by Tullius and coworkers (20). In this assay, 33.8 nM break light B was incubated in 40 mM Tris (pH 7.5)/2.5 mM sodium ascorbate/0.0075% H2O2. Cleavage was initiated by addition of EDTA/Fe(II) in a 2:1 molar ratio (final iron concentrations: 1.3, 3.1, 6.3, 12.5, 31.3, and 125 μM). Methidiumpropyl–Fe-EDTA (MPE)–Fe(II)-mediated degradation was adapted from procedures outlined by Van Dyke and Dervan (21). In this assay, 33.8 nM break light B was incubated in 40 mM Tris (pH 7.5)/2.5 mM sodium ascorbate/0.75 ppm H2O2. Cleavage was initiated by addition of MPE/Fe(II) in a 1.2:1 molar ratio (final iron concentrations: 0.13, 0.25, 0.50, 1.5, 2.5, 5.0, 10.0 μM). This protocol was repeated for 3.17 nM calicheamicin-specific oligonucleotide with the same MPE/Fe(II) molar ratios (final iron concentrations: 0.13, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0 μM). Pseudofirst order kinetic parameters were used to determine the initial velocities at each given agent concentration as previously described.
Results and Discussion
Design and Construction of Molecular Break Lights.
We prepared two scission beacons for the experiments described (Fig. 2c). Break light A was composed of a 10-base-pair stem that contained the known calicheamicin recognition sequence 5′-TCCT-3′ (7), whereas break light B carried the BamHI endonuclease recognition sequence 5′-GGATCC-3′ (21, 22). The length of break light B also considered the requirement of a 3-base-pair overhang required for BamHI recognition, and the stem of break light A was adjusted to a comparable length and melting temperature (22, 23). The loop of both probes consisted of a T4 loop to ensure nonhybridizing interactions. The 5′-fluorophore of both probes was fluorescein (FAM, absorbancemax = 485 nm, emissionmax = 517 nm), whereas the corresponding 3′-quencher was 4-(4′-dimethylaminophenylazo)benzoic acid (DABCYL). Previous studies have shown DABCYL to serve as a universal quencher in molecular beacons, and there is significant spectral overlap (1.02 × 10−15M−1·cm3) between the emission spectrum of FAM and the absorption spectrum of DABCYL. In a typical molecular beacon, the quenching efficiency of this pair via FRET has been shown to be essentially complete (99.9%), providing a significant enhancement of the signal to noise ratio as compared with typical complementary oligonucleotide pair FRET-based assays (14, 15).
Enzymatic Cleavage as Proof of Principle.
The first test was to demonstrate the specificity of the designed scission beacons via enzymatic cleavage. Specifically, only break light B should cleave in the presence of the restriction endonuclease BamHI, whereas both A and B should be digested by the nonspecific nuclease DNaseI. As anticipated, Fig. 3a reveals a time-dependent and [BamHI]-dependent increase of fluorescence only with B, whereas A shows no change at 37°C. Fig. 3b illustrates an increase of fluorescence over time with either break light A or B when digested with DNaseI, which is also [DNaseI]-dependent. In comparison, control samples containing break lights alone or break lights in the presence of BSA gave no change in fluorescence over >2 h at 37°C. Given the lack of fluorescence in the absence of enzyme, the designed break lights show no appreciable melting at the designated assay temperature. Furthermore, these experiments clearly demonstrate the specificity of cleavage by BamHI for B and, for the first time, illustrate the principle application of molecular break lights to assess DNA cleavage.
Figure 3.

The demonstration of molecular break light specificity and general proof of principle. The observed change in fluorescence intensity over time of an assay containing 3.2 nM break light at 37°C. (a) Break light A with 100 units of BamHI (□), break light B with 100 units of BamHI (○), and break light B without enzyme (●) (10 mM Tris⋅HCl/50 mM NaCl/10 mM MgCl2/1 mM DTT, pH 7.9; λEx = 485 nm, λEm = 517 nM). (b) Break light A with and 10 units of DNaseI (□), break light B with 10 units of DNaseI (○), and break light A without enzyme (●) (40 mM Tris⋅HCl/10 mM MgSO4/1 mM CaCl2, pH 8.0; λEx = 485 nm, λEm = 517 nM).
Interestingly, the fluorescence maximum intensity obtained upon complete BamHI cleavage was only 75% that observed in the presence of DNaseI at the same concentration of molecular break light. Furthermore, after the BamHI reaction was complete, the addition of BamHI showed no change, whereas the addition of DNaseI resulted in additional cleavage to give the expected 100% fluorescence maximum. This observation suggests the polyguanidine tail left attached to FAM upon BamHI digestion quenches the fluorescent signal by ≈25%. Consistent with this finding, PAGE analysis of the reaction products confirmed the presence of a 3-base overhang after excess treatment with BamHI, which is completely degraded upon DNaseI digestion. As a result, the fluorescence maximum observed with excess BamHI was designated 100% cleavage for the BamHI kinetic studies described below.
BamHI Steady-State Kinetic Determination and Sensitivity Limits.
Although continuous assays for nonspecific nucleases have been based upon ΔA260 as a function of cleavage of generic chromosomal DNA (e.g., sonicated herring sperm DNA) (24), only a few examples of continuous restriction endonuclease assays have been reported (25–29). Thus, most restriction endonuclease steady-state kinetic determinations have relied upon discontinuous assays using radioactive DNA probes, electrophoresis, and subsequent phosphoimager analysis (22, 23, 30–32). To demonstrate the utility of molecular break lights for this application, we determined the steady-state kinetic parameters for a commercially available BamHI. In our assay, the dependence of BamHI hydrolysis on substrate concentration was investigated using mixtures of a fixed amount of break light B and varying amounts of an analogous nonlabeled oligonucleotide (lacking both FAM and DABCYL) over a wide substrate concentration range. The apparent competitive inhibition observed because of the phenomenon of “carrier dilution” was corrected to give the appropriate kinetic parameters as previously described (32).
As illustrated in Fig. 4a, the velocity curves decrease with an increase in initial substrate concentration, although the true velocity has actually increased because of the carrier dilution by the nonlabeled oligonucleotide. The observed velocity (Vapp) is related to the actual velocity (Vact) by Eq. 1, where [Sact] and [S*] are the total substrate concentration and break light B concentration, respectively. The reciprocal plot after correction for this phenomenon is illustrated in Fig. 4b.
Figure 4.

The determination of BamHI steady-state kinetic parameters using break light B. (a) The observed change in fluorescence intensity over time of an assay containing a constant 3.2 nM break light B at 37°C (6 mM Tris⋅HCl/100 mM NaCl/6 mM MgCl2/1 mM DTT, pH 7.5; λEx = 485 nm, λEm = 517 nM), BamHI (10 units), and varying nonlabeled substrate oligonucleotide. Total substrate concentrations (including break light): 389 nM (○), 196 nM (□), 81 nM (⋄), 42 nM (▵), 11 nM (●), 7.5 nM (■), and 3.4 nM (⧫). (b) Lineweaver–Burke plot from a after correction for the carrier dilution effect.
From Fig. 4b, the determined Km = 8.9 ± 0.5 nM and Vmax = 0.024 ± 0.001 nM s−1. Although these values differ slightly from previously reported values for BamHI of Km = 0.4 nM and Vmax = 0.009 nM s−1 (21), kinetic parameters of restriction endonucleases vary significantly depending upon the oligonucleotide substrate (32). It should be acknowledged that our examination of three different commercial sources of BamHI (Promega, New England Biolabs, and GIBCO/BRL) gave markedly distinct specific activities (ranging roughly an order of magnitude). Thus, the differences in our reported kinetic parameters could also simply reflect distinctions in the enzyme preparation and/or commercial assay buffers. Most important, the utility of molecular break lights to assess the kinetic parameters of enzymatic DNA cleavage has been demonstrated. Furthermore, it is expected this approach could be directed toward any endonuclease by simply changing the recognition sequence found within the molecular break light stem.
A recent fluorescence correlation spectroscopy assay for the restriction endonuclease EcoRI using 0.8 nM of dual fluorophoric-labeled dsDNA and a highly specialized fluorescence correlation spectrometer reported a detection limit of 1.6 pM EcoRI (16). Under the conditions containing even slightly less oligonucleotide (0.68 nM molecular break light), cleavage was easily detectable to 3.7 pM BamHI. Furthermore, because of significantly low signal to noise of this assay, increasing the molecular break light concentration (34 nM) lowered the detection limit into the fM range (0.12 pM BamHI).
Enediyne-Catalyzed Cleavage.
Previous assays for enediyne cleavage of DNA relied upon discontinuous assays using radioactive DNA probes, electrophoresis, and subsequent phosphoimager analysis (3). In contrast, by using break lights one can directly follow the extent of DNA cleavage by a specific enediyne in real time with high sensitivity. To demonstrate, Fig. 5 illustrates enediyne concentration-dependent cleavage of break light A with either 1 or 2 in the presence of excess reductive activator DTT. Under the conditions described, this assay allows the detection of 1 in the pM range. This sensitivity compares to that of the BIA, the method of choice in detecting DNA-damaging agents (33). Also, the sensitivity can be significantly enhanced by simply increasing the concentration of the molecular break light in the assay as demonstrated with the iron-dependent agents. The observed maximum fluorescence obtained upon cleavage of 3.2 nM break light A with either 1 or 2 was identical to that observed with DNaseI, consistent with complete degradation of the oligonucleotide. As controls, incubation of molecular break light A with either DTT or enediyne alone revealed no change in fluorescence. Furthermore, although there is some debate regarding the “specificity” of 1, break light B was cleaved by 1 at an identical rate. This supports the view that the specificity of 1 is more dependent upon context and perhaps less so on DNA sequence (1–7). It should also be noted that 1 leads to predominately double-stranded cleavage, whereas 2 provides single-stranded nicks, and the current molecular break-light assay cannot distinguish these two phenomena.
Figure 5.

Cleavage of break light A by calicheamicin and esperamicin. The observed DNA cleavage over time of an assay containing 3.2 nM break light A at 37°C (40 mM Tris⋅HCl, pH 7.5; λEx = 485 nm, λEm = 517 nM), DTT (50 μM) and varied enediyne. (a) Calicheamicin concentrations: 31.7 nM (○), 15.9 nM (□), 3.2 nM (⋄), 1.6 nM (▵), 0.78 nM (●), and 0.31 nM (■). (b) Esperamicin concentrations: 31.7 nM (○), 15.9 nM (□), 3.2 nM (⋄), 1.6 nM (▵), 0.78 nM (●), 0.31 nM (■), and 0.15 nM (♦).
Interestingly, two distinct rates were observed in the enediyne molecular break-light assay. The first (0–50 s) is a lag time most likely attributed to the enediyne activation (34, 35), whereas the second (50–200 s) is indicative to the initial velocity of DNA cleavage. To confirm this, assays were also established in which DTT and enediyne were first preincubated for 1–5 min followed by initiation via the addition of the substrate oligonucleotide. In these preincubation experiments, the previously observed “lag time” attributed to activation was no longer evident, whereas the initial velocity of DNA cleavage was identical to that determined in the standard assay. Preincubation for longer periods (>30 min) revealed the same phenomenon, suggesting “activated” enediynes are perhaps more stable in an aqueous aerobic environment than previously estimated (1–7).
Cleavage Catalyzed by Fe(II)-Dependent Agents.
To further demonstrate the utility of molecular break lights, we investigated the ability to assess DNA cleavage catalyzed by Fe(II)-dependent agents. The agents selected include the natural metabolite from Streptomyces verticillus, bleomycin (Fig. 1, 3), and two DNA-footprinting reagents, MPE–Fe(II) (Fig. 1, 4) and EDTA–Fe(II) (Fig. 1, 5). Although the precise mechanism of DNA cleavage by 3 is still controversial (36), 4 and 5 cleave DNA via the generation of diffusable hydroxy radicals, which ultimately contribute to oxidative DNA cleavage. Of these three, 3 also contains a strong minor groove-binding constituent, whereas 4 carries a DNA intercalator. As with the previous enediyne assays, reported assays for cleavage by these agents have all relied upon discontinuous systems; thus, molecular break lights should present an obvious advantage. Fig. 6 illustrates agent concentration-dependent cleavage of break light A. Under the conditions described, this assay allows the detection of 3 in the nM range, which represents a slight increase in sensitivity over the BIA (32) and reiterates the power of this assay to detect the production of naturally produced DNA-damaging agents. To increase the sensitivity for the less efficient reagent 5, oligo concentration was increased 10-fold (32 nM; Fig. 6D). As a comparison, 4 was also examined at this higher molecular break light concentration (Fig. 6C). Finally, although ascorbate is critical for efficient DNA cleavage by 4 and 5, the addition of ascorbate did not affect DNA cleavage by 3.
Figure 6.

Cleavage of break light A by Fe(II)-dependent agents. (A) The observed DNA cleavage over time of an assay containing a constant 3.2 nM break light A at 37°C (50 mM sodium phosphate/2.5 mM ascorbate, pH 7.5; λEx = 485 nm, λEm = 517 nM) and varied bleomycin. Bleomycin concentrations: 200 nM (○), 100 nM (□), 50 nM (⋄), 25 nM (▵), 12.5 nM (●), 5 nM (■), and 2.5 nM (▴). (B) The observed DNA cleavage over time of an assay containing a constant 3.2 nM break light A at 37°C (40 mM Tris⋅HCl/2.5 mM ascorbate, pH 7.5; λEx = 485 nm, λEm = 517 nM) and varied MPE. Fe(II) concentrations: 8 μM (○), 4 μM (□), 2 μM (⋄), 1 μM (▵), 500 nM (●), 250 nM (■), and 125 nM (▴). (C) The observed DNA cleavage over time of an assay containing a constant 32 nM break light A at 37°C (40 mM Tris⋅HCl/2.5 mM ascorbate, pH 7.5; λEx = 485 nm, λEm = 517 nM) and varied MPE. Fe(II) concentrations: 50 nM (○), 125 nM (□), 250 nM (⋄), 500 nM (▵), 1 μM (●), and 2 μM (■). (D) The observed DNA cleavage over time of an assay containing a constant 32 nM break light A at 37°C (40 mM Tris⋅HCl/2.5 mM ascorbate, pH 7.5; λEx = 485 nm, λEm = 517 nM) and varied Fe(II)–EDTA. Fe(II) concentrations: 12.5 μM (○), 6.3 μM (□), 3.1 μM (⋄), and 1.3 μM (▵).
A Comparison of Efficiencies.
The nonenzymatic cleavage agents presented are essentially involved in single turnover events; thus, their direct comparison to an enzyme-catalyzed event is difficult. In fact, significant controversy exists regarding the more simplistic comparison of synthetic and biological catalysts in general (37). A direct correlation of the turnover (Vapp/[cleavage agent]) for 1, 2, 3, 4, and 5 indicates the maximum turnover when [beacon A] = 3.2 nM (representing at least 76.8 nM cleavage sites) occurs in the range of 0.78–1.6 nM for the enediynes, 2.5 nM for 3, and 125 nM 4. At the higher molecular break light concentration, [A] = 32 nM, and maximum turnover occurs in the range of 50 nM 4 and 1.3 μM 5. These maximum turnover values are summarized in Table 1 in a somewhat unconventional attempt to correlate the cleavage efficiencies of this highly diverse group of DNA cleavage agents, where 4, assayed at both concentrations of oligonucleotide, serves as the common agent in both sets. Table 1 suggests the addition of an intercalator (4) to the Fe(II)-chelation domain enhances the cleavage efficiency almost 103-fold in comparison to Fe(II)–EDTA (5), and the addition of a specific minor groove binder (3) increases this efficiency an additional 10-fold. Although the cleavage efficiencies of 1 and 2 are nearly identical, the near 10-fold enhancement over 3 may be attributed to direct hydrogen abstraction (versus diffusable active radical species formed from iron-dependent agents) in the formation of the DNA backbone radicals, which ultimately lead to oxidative cleavage. Most important, Table 1 illustrates that these spectacular enediynes are as efficient as an enzyme as the kcat of BamHI is identical to the observed maximum turnover of 1.
Table 1.
A comparison of cleavage efficiencies
| Agent | Vmax, nM sec−1 | Turnover,* sec−1 | Comparison to EDTA† |
|---|---|---|---|
| Enzymatic | |||
| BamHI | 0.024 ± 0.001 | 0.007‡ | 4.8 × 105 |
| Small molecule catalyzed | |||
| Esperamicin A1 | 0.007 ± 0.001§ | 0.009 | 6.1 × 105 |
| Calicheamicin γ′1 | 0.011 ± 0.002§ | 0.007 | 4.8 × 105 |
| Bleomycin | 0.009 ± 0.001§ | 0.001 | 6.8 × 104 |
| Methidiumpropyl-EDTA | 0.003 ± 0.001§ | 2.4 × 10−5 | 1.6 × 103 |
| Methidiumpropyl-EDTA | 0.118 ± 0.004¶ | 0.002 | 1.6 × 103 |
| EDTA | 0.002 ± 0.001¶ | 1.5 × 10−6 | 1.0 |
Defined as Vmax/[Agent].
Fold enhancement over EDTA turnover.
Also known as kcat.
[DNA]total = 3.2 nM.
[DNA]total = 32 nM.
Conclusions
There are a few reports of the application of FRET to assay enzymatic cleavage using a fluorescent-modified oligonucleotide/unlabeled oligonucleotide complement pair (25–29). One limitation of these early efforts, however, was often the significant background fluorescence as a result of poor fluorescence quenching by the hybridizing strand. Molecular break lights present some obvious advantages to these previous applications. First, quenching is intramolecular and complete, which eliminates the need for a second oligonucleotide and completely removes undesired background fluorescence. The presented studies also clearly reveal that the molecular break light assay has very broad utility. Furthermore, the sensitivity of this unique assay rivals the typical discontinuous BIA assay for detection of DNA-damaging agents and exceeds the sensitivity of assays based upon fluorescence correlation spectroscopy >10-fold, a very sensitive technique that also requires extremely specialized instrumentation. Given the simplicity, speed, and sensitivity of this approach, the described methodology could easily be extended to a high throughput format and become a new method of choice in all applications that require an assay for DNA cleavage.
Acknowledgments
We gratefully acknowledge the Wyeth–Ayerst Research Division of American Home Products for the generous supply of calicheamicin, Dr. Kin Sing (Ray) Lam (Bristol-Myers Squibb) for the kind gift of esperamicin, and Professor Ben Shen (Department of Chemistry, Univ. of California, Davis) for Blenoxane. J.S.T. is an Alfred P. Sloan Research Fellow and Rita Allen Foundation Scholar. This work was supported in part by National Institutes of Health Grants GM58196 and CA84374, the Mizutani Foundation for Glycoscience, Cancer Center Support Grant CA-08748, and by a grant from the Special Projects Committee of the Society of Memorial Sloan–Kettering Cancer Center.
Abbreviations
- MPE
methidiumpropyl–EDTA–Fe(II)
- BIA
biochemical induction assay
- FAM
fluorescein
- FRET
fluorescence resonance energy transfer
- DABCYL
4-(4′-dimethylaminophenylazo)benzoic acid
Note Added in Proof.
FRET-based assays have also recently been reported for ribozyme-catalyzed RNA cleavage (38) and DNaseI-catalyzed single-stranded DNA cleavage (39).
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.240460997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.240460997
References
- 1.Thorson, J. S., Sievers, E. L., Ahlert, J., Shepard, E., Onwueme, K. C. & Ruppen, M. (2000) Curr. Pharm. Des., in press. [DOI] [PubMed]
- 2.Thorson J S, Shen B, Whitwam R E, Liu W, Li Y, Ahlert J. Bioorgan Chem. 1999;27:172–188. [Google Scholar]
- 3.Borders D B, Doyle T W. Enediyne Antibiotics as Antitumor Agents. New York: Dekker; 1995. [Google Scholar]
- 4.Smith A L, Nicolaou K C. J Med Chem. 1996;39:2103–2117. doi: 10.1021/jm9600398. [DOI] [PubMed] [Google Scholar]
- 5.Nicolaou K C, Smith A L, Yue E W. Proc Natl Acad Sci USA. 1993;90:5881–5888. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolaou K C, Dai W-M. Angew Chem Int Ed Engl. 1991;30:1387–1416. [Google Scholar]
- 7.Zein N, Poncin M, Nilakantan R, Ellestad G A. Science. 1989;244:697–699. doi: 10.1126/science.2717946. [DOI] [PubMed] [Google Scholar]
- 8.Zein N, Sinha A M, McGaharen W J, Ellestad G A. Science. 1988;240:1198–1201. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 9.Kumar R A, Ikemoto N, Patel D J. J Mol Biol. 1997;265:187–201. doi: 10.1006/jmbi.1996.0718. [DOI] [PubMed] [Google Scholar]
- 10.Sievers E L, Appelbaum F R, Spielberger R T, Forman S J, Flowers D, Smith F O, Shannon-Dorcy K, Berger M S, Bernstein I D. Blood. 1999;93:3678–3684. [PubMed] [Google Scholar]
- 11.Bernstein I D. Leukemia. 2000;14:474–475. doi: 10.1038/sj.leu.2401663. [DOI] [PubMed] [Google Scholar]
- 12.Whitwam R E, Ahlert J, Holman T R, Ruppen M, Thorson J S. J Am Chem Soc. 2000;122:1556–1557. [Google Scholar]
- 13.Jiang J, Biggins J B, Thorson J S. J Am Chem Soc. 2000;122:6803–6804. [Google Scholar]
- 14.Tyagi S, Kramer F R. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi S, Bratu D P, Kramer F R. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 16.Kettling U, Koltermann A, Schwille P, Eigen M. Proc Natl Acad Sci USA. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger R M, Horwitz S B, Peisach J. Biochemistry. 1985;24:3623–3629. doi: 10.1021/bi00335a034. [DOI] [PubMed] [Google Scholar]
- 18.Burger R M, Projan S J, Horwitz S B, Peisach J. J Biol Chem. 1986;261:15955–15959. [PubMed] [Google Scholar]
- 19.Giloni L, Takeshita M, Johnson F, Iden C, Grollman A P. J Biol Chem. 1981;256:8608–8615. [PubMed] [Google Scholar]
- 20.Dixon W J, Hayes J J, Levin J R, Weidner M F, Dombroski B A, Tullius T D. Methods Enzymol. 1991;208:380–413. doi: 10.1016/0076-6879(91)08021-9. [DOI] [PubMed] [Google Scholar]
- 21.Van Dyke M W, Dervan P B. Nucleic Acids Res. 1983;11:5555–5567. doi: 10.1093/nar/11.16.5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinsch B, Mayer H, Kula M-R. Nucleic Acids Res. 1980;8:2547–2559. doi: 10.1093/nar/8.11.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinsch B, Kula M-R. Nucleic Acids Res. 1980;8:623–633. doi: 10.1093/nar/8.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorson J S, Chapman E, Schultz P G. J Am Chem Soc. 1995;117:9361–9362. [Google Scholar]
- 25.Lee S P, Porter D, Chirikjian J G, Knutson J R, Han M K. Anal Biochem. 1994;220:377–383. doi: 10.1006/abio.1994.1353. [DOI] [PubMed] [Google Scholar]
- 26.Erskine S G, Halford S E. Gene. 1995;157:153–156. doi: 10.1016/0378-1119(94)00616-z. [DOI] [PubMed] [Google Scholar]
- 27.Lee S P, Han M K. Methods Enzymol. 1997;278:343–363. doi: 10.1016/s0076-6879(97)78019-4. [DOI] [PubMed] [Google Scholar]
- 28.Koltermann A, Kettling U, Bieschke J, Winkler T, Eigen M. Proc Natl Acad Sci USA. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh S S, Eis P S, Blumeyer K, Fearon K, Millar D P. Nucleic Acids Res. 1994;22:3155–3159. doi: 10.1093/nar/22.15.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinsch B, Kula M-R. Nucleic Acids Res. 1981;9:3159–3174. doi: 10.1093/nar/9.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nardone G, Chirikjian J G. Gene Amplif Anal. 1987;5:147–184. [PubMed] [Google Scholar]
- 32.Roy K B, Vrushank D, Jayaram B. Anal Biochem. 1994;220:160–164. doi: 10.1006/abio.1994.1313. [DOI] [PubMed] [Google Scholar]
- 33.Greenstein M, Wildey M J, Maiese W M. In: Endiyne Antibiotics as Antitumor Agents. Borders D B, Doyle T W, editors. New York: Dekker; 1995. pp. 17–28. [Google Scholar]
- 34.Myers A G, Cohen S B, Kwon B M. J Am Chem Soc. 1993;166:1255–1271. [Google Scholar]
- 35.Chatterjee M, Smith P J, Townsend C A. J Am Chem Soc. 1996;118:1938–1948. [Google Scholar]
- 36.Hecht S M. J Nat Prod. 2000;63:158–168. doi: 10.1021/np990549f. [DOI] [PubMed] [Google Scholar]
- 37.Jacobsen E N, Finney N S. Chem Biol. 1994;1:85–90. doi: 10.1016/1074-5521(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 38.Vitiello D, Pecchia D B, Burke JM. RNA. 2000;6:628–637. doi: 10.1017/s1355838200990964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J J, Geyer R, Tan W. Nucleic Acids Res. 2000;28:e52. doi: 10.1093/nar/28.11.e52. [DOI] [PMC free article] [PubMed] [Google Scholar]