


Abstract
Protein–RNA interactions play essential roles in a number of regulatory mechanisms for gene expression such as RNA splicing, transport, translation and post-transcriptional control. As the number of available protein–RNA complex 3D structures has increased, it is now possible to statistically examine protein–RNA interactions based on 3D structures. We performed computational analyses of 86 representative protein–RNA complexes retrieved from the Protein Data Bank. Interface residue propensity, a measure of the relative importance of different amino acid residues in the RNA interface, was calculated for each amino acid residue type (residue singlet interface propensity). In addition to the residue singlet propensity, we introduce a new residue-based propensity, which gives a measure of residue pairing preferences in the RNA interface of a protein (residue doublet interface propensity). The residue doublet interface propensity contains much more information than the sum of two singlet propensities alone. The prediction of the RNA interface using the two types of propensities plus a position-specific multiple sequence profile can achieve a specificity of about 80%. The prediction method was then applied to the 3D structure of two mRNA export factors, TAP (Mex67) and UAP56 (Sub2). The prediction enables us to point out candidate RNA interfaces, part of which are consistent with previous experimental studies and may contribute to elucidation of atomic mechanisms of mRNA export.
INTRODUCTION
The recent discovery of unconventional non-coding RNAs (ncRNAs) (1,2) suggests that RNA molecules may mediate unknown biological functions (2,3). Many RNAs form large ribonucleoprotein complexes such as the ribosome, the spliceosome and the signal recognition particle. It is expected that ncRNAs too perform some biological functions also in complex with proteins (4). Coding RNAs are modified and transported by specific proteins as occurs during mRNA splicing (5), transport (6) and translation (7). In addition, specific proteins carry out the repair of damaged RNA (8) and the editing of transcribed RNA (9). In humans, it is estimated that about 1500 proteins interact with RNA (10).
3D structures of protein–RNA complexes could provide valuable insight into the function of these complexes, however few structures of RNA-binding protein in complex with RNA were solved. In such a situation, computational statistical analysis of protein–RNA interactions may play a significant role possibly to predict complex structures.
A number of computational studies have examined interactions between proteins and nucleotides, especially DNA (11–17). These studies showed that electrostatic interactions play a major role in mediating protein–DNA associations (13,15). The protein surface mediating DNA-binding is generally characterized by a positive electrostatic potential (18). Characteristics of protein–RNA associations are thought to be similar to that of protein–DNA associations (19) and RNA-binding area on protein surface is similarly predicted by electrostatic potential calculation. However, accuracy of calculating electrostatic potential of protein surfaces is low due to the ambiguity in dielectric constant and ion strength of the environment and also due to inaccuracies in the positions of pertinent atoms (20). In addition, possible structural changes upon complex formation make predictions more difficult. Stawiski et al. (21) argued that the DNA/RNA-binding area of a protein could not be identified only by the calculated surface electrostatic potential. For example, eukaryotic release factor (eRF1) is known to interact with ribosomal RNA (rRNA) and mRNA, but the interface areas for those RNAs are still not firmly identified (22–24), even though the 3D structure of eRF1 has been known for some time (25).
Compared with a number of computational studies of protein–DNA associations, a limited number of studies have examined protein–RNA associations, likely due to the paucity of structural data of protein–RNA complexes. As the number of protein–RNA complexes deposited in the Protein Data Bank (PDB) (26) increases, some statistical analyses on interactions between amino acid residues and RNA molecules have been performed (27–30). These studies showed the importance of Arg and Lys for interactions with RNA and this is consistent with the importance of electrostatic interactions. In addition, protein–RNA interfaces are pointed out to involve more non-polar weak interactions than protein–DNA interfaces (27,30). These studies suggested possibility of applying the observed amino acid frequency of the protein–RNA interfaces to predict the RNA interfaces of other protein, but few studies have attempted this (23).
In order to better understand the nature of protein–RNA complexes based on 3D structural data, we performed statistical analyses of the amino acid residue at the RNA interfaces of 86 RNA-binding proteins. We introduced a new propensity measure for amino acid residues that focuses on a residue pair in the interface. Protein–RNA interactions are reported to involve a number of non-polar weak interactions and these weak interactions often occur in a patch (27,30). Therefore, a statistical analysis of residue pairs in the RNA interface is expected to shed light on new characteristics. We applied the determined residue propensities plus information from position-specific multiple sequence profiles based on homologous sequences to RNA interface prediction. The combination of information from these different sources enabled us to achieve reasonable accuracy in RNA interface prediction.
MATERIALS AND METHODS
Protein–RNA complexes
Protein–RNA complexes solved by X-ray crystallography were selected from the Protein Quaternary structure file Server (PQS) (31). When an entry contained multiple protein chains, each chain was treated separately. We limited this study to handle complexes containing RNA molecules with at least 3 bases and protein with at least 50 amino acid residues. Viral protein–RNA complexes were ignored, since these complexes often included only a small part of RNA and most of the RNA interfaces on proteins were not determined. The RNA-binding proteins were classified into groups based on amino acid sequence similarity. A pair of proteins was placed in the same group, when the sequence identity was >50%. A representative complex with the best resolution from each group was selected. Multiple proteins were chosen from the same group, when the interface residues differed.
Definition and identification of RNA interface and protein surface
An atom in the protein–RNA interface was defined, based on the degree of difference in the accessible surface area of the atom calculated, using the protein structure coordinates with and without RNA. When the difference was non-vanishing, the atom was considered an interface atom. The RNA interface area was defined by clustering the interface atoms based on distances between them using the single linkage clustering method. The threshold was set to 7.0 Å. In an RNA interface area, at least one pair of atoms is located within 7.0 Å and no atoms from two different RNA interface areas are within 7.0 Å. An RNA interface residue was defined as a residue with at least one RNA interface atom.
To calculate the residue propensity for the RNA interface, the frequency of residues on protein surface was required as a background distribution. Protein surface residues were defined based on the solvent accessibility of each residue. In this study, a surface residue was defined as a residue with accessibility of no <8%.
Interface residue propensity
To measure the relative importance of different amino acid types in RNA-binding interface, the residue interface propensity was calculated.
Residue singlet interface propensity
The residue singlet interface propensity (Pi) was calculated for each amino acid type [AAi (i = 1, … , 20)] as a fraction of the frequency that AAi contributes to a protein–RNA interface compared to the frequency that AAi contributes to a protein surface. The frequencies of surface AAi (fi) and interface AAi ( ) were calculated with the following equations:
| 1 |
where ni is the number of amino acid type i on the protein surface and is that in the RNA interface. The number ni was obtained from the population of non-homologous proteins in the PDB and was determined from the data for protein–RNA complexes described above. We added pseudocounts to the observed to minimize possible statistical error in caused by a paucity of data (32). These pseudocounts should reflect a priori expectations of the occurrence of the amino acid type i in the RNA interface. Since we did not have a priori expectations of the amino acid type in the interface, we set all pseudocounts to one. The residue singlet interface propensity (Pi) is
| 2 |
When Pi is more than one, amino acid type i occurs more frequently in the interface than on the protein surface. This method is similar to the one employed by Jones et al. (27).
Residue doublet interface propensity
The residue doublet interface propensity gives a measure of the pairing preference of amino acid types in protein–RNA interfaces. Amino acid type i and the adjacent amino acid type j were considered to be a doublet, if the distance between their Cβ atoms (Cα for Gly) was no more than a certain threshold. In this study, the threshold was set to 7.0 Å. This threshold value was chosen to account for neighboring residues. The residue doublet interface propensity (Pij) was calculated as follows. The frequency of doublet amino acid type ij (i, j = 1, … , 20) on the protein surface (fij) and that in the protein–RNA interface ( ) were calculated from the number of residue doublets with the following equations:
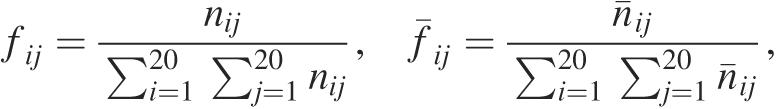 |
where nij is the number of doublet type ij on the protein surface and is that in the RNA interface. We added pseudocounts to the observed to minimize possible statistical error in caused by a paucity of data. The frequency of doublet type ij on the protein surface (fij) and that in the RNA interface ( ) can also be expressed as
| 4 |
| 5 |
where fi (or fj) and (or ) are given in Equation 1 and Cij and Dij are the surface and interface residue doublet coefficients, respectively. If amino acid types i and j had no correlation on the protein surface, then Cij = 1.0 and in the RNA interface, Dij = 1.0.
The residue doublet preference in the RNA interface was determined from Equations 2, 4 and 5 to be
| 6 |
where Pi and Pj are the residue singlet interface propensities in Equation 2. We defined
| 7 |
as the residue doublet interface propensity.
Reliability estimation of the calculated propensities
The limited dataset may reduce the statistical reliability of the calculated propensities. A bootstrap procedure was used to estimate the standard deviation of Pi and Pij. We constructed bootstrap datasets based on 1000 resamplings. Corresponding to each bootstrap dataset, we calculated the bootstrap replications of Pi and Pij and standard deviations were estimated from the replications. To assess the reliability of Pij, we tested whether the values derived from the replications followed Gaussian distribution and whether the value obtained using the entire data was within a certain deviation from the average value calculated from the replications (bootstrap percentile method). In this test, we set a 15% deviation from the average value as a reliability standard.
Position-specific multiple sequence profile
Homologous protein sequences for each group of RNA-binding proteins were extracted using BLAST (33) from a set of amino acid sequences gained from the translation of non-redundant DNA sequences stored in GenBank (34). Amino acid sequences with no <50% sequence identity to the query sequence were selected. The multiple sequence profile was then calculated from a multiple sequence alignment of those sequences. The profile Vi(x) (i = 1, … , 20) for the residue at position x in the multiple sequence alignment is the observed frequency of amino acid type i;
| 8 |
where mi(x) is the number of amino acid type i in residue position x in the multiple sequence alignment.
The profile Uij(x, y) for a pair of residue positions x and y in a multiple sequence alignment is the observed pair frequency of amino acid type i in position x and amino acid type j in position y;
| 9 |
where mij(x, y) is the number of amino acid type i in position x and amino acid type j in position y simultaneously in the multiple sequence alignment. In order to enable us to use the information from multiple sequence profile, we discarded a query sequence from our dataset, when no homologous sequence was found.
Prediction score
The prediction score, the indicator of a particular residue to bind RNA, was determined by assigning propensities and/or profiles to a residue located at the protein surface. We assumed that RNA-binding residues are located on the surface and proteins do not undergo large structural changes upon RNA-binding. We used several methods to determine the prediction scores.
Singlet score (S)
A singlet score was assigned to each surface residue. The value Sx(S) for a residue at position x was determined by amino acid type i of the residue at that position in a protein under consideration. Hence,
| 10 |
where Pi(x) is simply given by Pi from Equation 2.
Multiple sequence profile score (P)
Functional residues are generally well conserved (35). A score based on a position-specific multiple sequence profile was defined by information entropy Sx(P);
| 11 |
where Vi(x) is given by Equation 8.
Singlet plus profile score (SP)
The singlet plus profile score weights a singlet score by the probability that a specific amino acid type i is conserved within a specific protein family with an overall identity of >50%;
| 12 |
where Vi(x) and Pi are given by Equations 8 and 2, respectively.
Averaged singlet score (AS)
Binding of RNA to protein is a cooperative phenomenon involving interactions between various parts of protein and RNA. Therefore, it is likely that if one amino acid residue is involved in RNA-binding, its near neighbor residues are also involved. If this is the case, the propensity averaged over near neighbor residues should be more pertinent for the prediction of RNA-binding. We therefore examined scores averaged over residues located within a certain distance. We set a distance of 7.0 Å between Cβ (Cα for Gly) atoms. The score of residue x, when a number of residues within the distance is Nx (residue x included), is given as;
| 13 |
where y is a surface residue located within 7.0 Å from residue x and Pj(y) is simply given by Equation 2. The value of 7.0 Å was the most effective distance for prediction determined after a systematic trial of various values.
Averaged singlet plus profile score (ASP)
The singlet score is weighted by the profile and then averaged over near neighbor residues;
| 14 |
Singlet and doublet score (ASD)
The doublet propensity is defined as a correlation of amino acid types i and j in the interface as in Equations 6 and 7. Therefore, singlet and doublet score for a surface residue x of amino acid type i is defined as;
| 15 |
where Pi(x)j(y) is simply given by Pij of Equation 7.
Singlet and doublet plus profile score (ASPD)
The singlet and doublet propensities can be weighted by the probability that a specific amino acid type is conserved within a specific protein family with an overall identity of >50% (Equation 9);
Averaged singlet and doublet score (A2SD)
The singlet and doublet scores can be averaged among the residues located within a certain distance. The score of residue x, when the number of residues within the distance is Nx (residue x included), is given as;
| 17 |
where y is a surface residue located within 7.0 Å of residue x and Sy(ASD) is given by Equation 15. As the abbreviation of this score suggests, Equation 17 performs the averaging step twice. The justification for this procedure will be discussed later.
Averaged singlet and doublet plus profile score (A2SPD)
The singlet and double plus profile score can be averaged among the residues within a certain distance.
| 18 |
where y is a surface residue located within 7.0 Å of residue x and Sy(ASPD) is given by Equation 16.
Prediction evaluation
The quality of a prediction was evaluated using a jack-knife test. A set of propensities was calculated based on protein–RNA complex structures after excluding the protein to be predicted (target protein) and we examined different methods of prediction of RNA-binding residues of the target protein with the scores based on the equations in the previous section. An amino acid residue was predicted to be a protein–RNA interface residue, when its prediction score was higher than a certain threshold. The quality of the prediction was evaluated by comparing the prediction result and the real protein–RNA interfaces. We performed this procedure on each protein in the dataset. To assess the quality of various scores, the prediction was carried out for each method for a series of threshold values and the sensitivity and specificity (36) were calculated. Sensitivity was defined as the fraction of correctly predicted true RNA interface residues (the ratio of true positive to real interface residues). Specificity was defined as the fraction that a positive prediction was correct (the ratio of true positives to predicted interface residues). A prediction method was most useful when both the sensitivity and specificity were high.
RESULTS AND DISCUSSION
Dataset of protein–RNA complexes
We selected 86 RNA-binding proteins from PQS (Supplementary Table 1). The dataset contained two different threonyl-tRNA synthetase complexes, one with PDB ID 1KOG binding to mRNA and the other with PDB ID 1QF6 binding to threonyl-tRNA. The two RNA molecules bound different surfaces of the synthetase. We included three 54 kDa proteins of the signal recognition particle, each from eubacteria, archaea and eukaryote; two 19 kDa proteins of the signal recognition particle, each from archaea and eukaryote; three aspartyl-tRNA synthetases, each from eubacteria, archaea and eukaryote; and two tRNA-guanine transglycosylases, each from archaea and eubacteria. Proteins in each group were homologous, but sequence identity was <50% and the residues associated with RNA molecules in each structure were not conserved. In the 30S ribosomal proteins, we omitted the THX subunit (chain V in 1J5E) and in the 50S ribosomal proteins, we omitted the L10 (chain G in 1JJ2) and L39E (chain 1) subunits. The THX and L10 subunits were shorter than our selection criteria and L39E had no similar sequences in the sequence database, thus precluding us from building a multiple sequence profile.
The dataset contained different types of RNA including four complexes of snRNA molecules, 7 of signal recognition particle RNA (SRP RNA), 8 of mRNA, 20 tRNA, 45 ribosomal RNA (rRNA) and 2 other RNA–protein complexes.
Protein–RNA interfaces
The single linkage clustering method was used to identify clusters of RNA interface atoms on the protein surfaces with a threshold of 7.0 Å. Within 86 protein surfaces examined, 141 RNA interface patches were observed. Some of the identified patches were small. When a patch with area <100.0 Å2 was omitted, ∼1.6 patches per tRNA-binding proteins and 1.1 for other RNA-binding proteins for a total of 111 patches were identified. RNA interfaces in tRNA-binding proteins were often separated into two, one for the acceptor stem and the other for the anti-codon loop.
Of the analyzed patches, the area of the RNA interface varied from (by definition) 100 to 7000 Å2 approximately and consisted of 3–170 amino acid residues. On average, one amino acid residue occupied about 40 Å2 of the interface and these values are consistent with those observed by Nadassy et al. (13) calculated using the protein–nucleic acid complexes available at that time. The distribution of size of the area had reasonable correlation with the type of bound RNA. In our study, we noted that an interface with a size <1500 Å2 bound all types of RNA, while those >1500 Å2 bound either rRNA or tRNA and those >2900 Å2 bound only rRNA.
Residue singlet interface propensity
The residue singlet interface propensities are shown in Figure 1 in a log2 scale. The top seven high propensities were observed for Arg, Lys, Tyr, Met, His, Gly and Phe in descending order. The similar studies in the past on residue singlet interface propensity showed similar results with slight differences in the order of amino acid residues from high to low values (27–30). Our data showed that the positively charged residues (Arg, Lys) were preferred in the interface of RNA-binding proteins. The preference of Arg was pronounced. These preferences were observed not only for protein–RNA complexes, but also for protein–DNA complexes (15,16). For DNA interfaces, it was hypothesized that Arg occurred more frequently than Lys due to the length of its side-chain, its capacity to interact in different conformations and/or its ability to produce good hydrogen-bonding geometries (15).
Figure 1 .

Histogram of the calculated residue singlet interface propensity in a logarithm (log2) scale. A positive propensity indicates that a residue occurs more frequently in the interface than on the protein surface. An error bar for each propensity corresponds to standard deviations estimated from a bootstrap procedure with 1000 resamplings. The number given below the horizontal axis is the count of each amino acid type in the protein–RNA interfaces.
High propensity was observed for Tyr in RNA interfaces. Frequent stacking interactions between aromatic side chains and nucleic acid bases in a number of protein–RNA complexes may explain this observation (19,30).
Residue doublet interface propensity
The surface residue doublet coefficients Cij, the interface residue doublet coefficients Dij and the residue doublet interface propensities Pij in Equation 7 are shown in Figure 2. All values are shown in a log2 scale and are color-coded. As seen in Figure 2A, the logarithm of the surface residue doublet coefficients Cij of hydrophobic residues were weakly positive. The logarithm of the coefficients of pairs, each with positively and negatively charged residues, was also positive and those of positively charged residue pairs and those of negatively charged residue pairs were negative. These data suggest that hydrophobic residues are paired and charged residues form salt bridges on the protein surface. The interface residue doublet coefficients Dij were, as a whole, much more variable than the surface doublet coefficients (Figure 2B). The calculated coefficients for hydrophobic residue pairs were much greater than those seen for the surface residues, but the coefficients of positively and negatively charged residue pairs were negative and this was the opposite of that observed for the surface residue doublet coefficients, suggesting that salt bridge formation was not favored at RNA interfaces. Negatively charged residues were less common in the RNA interfaces and positively charged residues can mediate electrostatic interactions with RNA.
Figure 2 .

A graphical matrix of the surface residue doublet coefficients Cij (A), the interface residue doublet coefficients Dij (B) and the residue doublet interface propensities Pij (C) color-coded in a logarithm (log2) scale. Cys–Cys pair in A and Cys–Cys and Cys–Trp pairs in B are off the scale. In (C), a value with a cross mark indicates that the data are not statistically sufficient to warrant the result.
The doublet interface propensities (Figure 2C) are more reflective of Dij, because Pij was given by Dij/Cij and Cij was far less variable than Dij. In Figure 2C, a residue doublet propensity in red reflects Pij > 1.0, meaning that a particular pair was more favored in the RNA interface than on the surface in general. A residue doublet propensity in blue indicates that a particular pair was disfavored and a propensity in white indicates that the frequency of the occurrence of the two residues in a pair is independent. Boxes with a cross mark indicate that the data were not statistically sufficient to warrant significance of the result. Out of the possible 210 pairs of residue types, 173 pairs had a doublet propensity that passed the tests of significance described in Materials and Methods.
The distribution of doublet propensities displayed in a log2 scale (Figure 2C) has several interesting characteristics. The log doublet propensities of positively charged residue pairs were close to zero. Interactions with the RNA backbone may stabilize the proximity of positively charged residues, but these residues were not preferentially found as pairs in the RNA interface. The log doublet propensities of negatively charged residues were negative. The log doublet propensities of aromatic residue pairs were generally positive. Aromatic residues facilitate RNA-binding by stacking an aromatic group with RNA bases (19,30) and this stacking appears to be favored by a pair of aromatic residues. The pair of Tyr and Lys had a significant negative log doublet propensity, despite the high positive singlet propensities of these residues (Figure 1). We are not aware of a physico-chemical reason for disfavoring this pair. Doublet propensities of aliphatic residues were generally high and the top doublet propensity was observed for the Ile–Ile pair. The log singlet propensity of Ile was negative, but when Ile existed in a pair, the log doublet propensity became positive. The physico-chemical reasons causing the pair remain unclear. We have found a case that two Ile side-chains formed a planar structure possibly to provide a bed to accept an uracyl base of the RNA molecule in the spliceosomal protein. By comparing the results in Figures 1 and 2, nearly 13% (27 out of 210 cases) of the doublet propensities had signs that were the opposite of a sign given by a sum of two singlet propensities. These results emphasize the importance of the residue doublet propensity.
Ability to predict protein–RNA interfaces
Propensities and multiple sequence profiles (Equations 8 and 9) were used for predicting the RNA interfaces of the proteins listed in Supplementary Table 1. Interfaces for RNA-binding were subjected to jack-knife prediction and the quality of the prediction was evaluated in terms of sensitivity and specificity as detailed in the Materials and Methods. We tested the nine different prediction methods (Equations 10–18) and the ability of these methods to predict the correct RNA interfaces is summarized in Figure 3.
Figure 3 .

Quality of interface predictions evaluated on 86 jack-knifed datasets. The dotted lines show the evaluation of the prediction with the propensities and the solid lines show that of the prediction with the propensities plus the profile. The prediction using the profile only (P) is shown in black solid line. The predictions using the singlet propensities (S and SP) are shown in green. The predictions using the averaged singlet propensities (AS and ASP) are shown in cyan. The predictions using the doublet propensities (ASD and ASPD) are shown in red. The predictions using averaged singlet and doublet propensities (A2SD and A2SPD) are shown in orange.
Amino acid residues that are important for the function of a protein tend to be more conserved during the course of molecular evolution and the degree of conservation can be a good indicator of function (35). We quantified the degree of conservation as indicated in Equation 11 and examined whether conservation was a good predictor of RNA interfaces [the black line (P) in Figure 3]. Residues involved in forming RNA interfaces were expected to be highly conserved, but not all highly conserved surface residues were part of RNA interfaces. This may explain the rather low specificity of P. The prediction using the singlet propensity showed a similarly low specificity (Equation 10, S in Figure 3) and when this was weighted with the multiple sequence profile (Equation 12), no clear improvement in the quality of prediction was observed (SP in Figure 3). By averaging the singlet propensities over near neighbor residues (Equation 13), the quality of prediction improved (AS in Figure 3). As discussed in the Materials and Methods, RNA-binding is a cooperative phenomenon involving a number of near neighbor residues. This cooperation can be incorporated into the prediction by using the averaged singlet propensity and the improved predictive ability of this value suggests that the protein–RNA interface area has such a shape that can be covered by a set of mutually overlapping spheres with 7.0 Å radii. When the singlet score was first weighted by the multiple sequence profile and then averaged over the near neighbor residues (Equation 14), the quality of prediction was further improved (ASP in Figure 3). The multiple sequence profile provides the empirical probability of each amino acid type being found at the position under consideration. Therefore, the score weighted by the multiple sequence profile reflects the score for a homologous set of proteins and suppresses possible noise due to random amino acid occurrence. Inclusion of the doublet propensity score further improved our predictive ability (Equation 15) and the predictive ability was further enhanced by the multiple sequence profile weighting (Equation 16) (ASD and ASPD, respectively in Figure 3).
Residue doublet propensities gave information of residue pair preference shown in Figure 2. The differences between AS and ASD and between ASP and ASPD represent the contribution of the doublet propensities. Incorporation of the doublet propensities into the calculations markedly increased the specificity of our predictions and this was especially true when using a high score threshold and the resulting sensitivity was low. Averaging scores over near neighbor residues effectively increased our predictive ability (compare S to AS) and therefore, we performed an averaging procedure for the doublet information contained in ASD and ASPD. The singlet information in those scores was already averaged. Therefore, the averaging doublet information in ASD and ASPD resulted in averaging the singlet information twice (therefore, the names of scores were A2SD and A2SPD). Thus, residues as far as 14.0 Å away might influence the score. This procedure further enhanced the prediction specificity and sensitivity as seen in Figure 3 (A2SD and A2SPD).
The quality of all nine prediction methods converged to a point where sensitivity and specificity were ∼0.9 and 0.3, respectively. At this point, the threshold for determining the predicted interface was too low and most surface residues were predicted to constitute RNA interfaces in all prediction methods. When all of the surface residues were predicted as RNA interfaces, most of the interface residues were correctly predicted (sensitivity ≈ 1.0) and many non-interface residues (∼70% of the surface residues) were also positively predicted (specificity ≈ 0.3).
The prediction with the averaged singlet and doublet propensities plus the multiple sequence profile (A2SPD) achieved the best quality. Its specificity reached as high as 0.8 at the most strict threshold. We used the nine different methods (Equations 10–18) to predict the RNA-binding interface of the known RNA-binding interface for sex-lethal protein (PDB ID: 1B7F) in Figure 4. In this case, the sensitivity of the predictions was fixed close to 0.5. As shown in the figure, the specificities improved in the order of S, P, SP, AS, ASP, ASD, A2SD, ASPD and A2SPD. The improvement was manifested by the reduction in false positive residues, i.e. over-prediction (green residues in Figure 4). The difference between AS and ASD and between ASD and ASPD indicated the effects of the doublet propensity and the multiple sequence profile on the predictions, respectively.
Figure 4 .

RNA interface predictions for a known complex structure, sex-lethal protein (PDB ID: 1B7F), using nine different methods. Two figures are shown for each method, in which the structure is rotated 180° around the y (vertical) axis. Residues in yellow are true positives (correctly predicted as interface residues) and those in green are false positives (predicted as interface residues, but are not interfaces). Residues in white are true negatives (correctly predicted as non-interface residues) and those in cyan are false negatives (predicted as non-interface residues, but are interfaces). Residues in dark blue are buried residues, which were not considered in the prediction. The actual values of sensitivity (the ratio of true positive to real interface) and specificity (the ratio of true positive to predicted interface) for each case are given to the left of each figure.
Summary of our prediction method
A number of studies have presented structure-based analyses of protein–RNA interactions (27,28,30,37). Those studies showed singlet propensities similar to the present work. In our report, we were able to predict the RNA interface relatively well after incorporating the doublet propensity and the multiple sequence profile into our calculations. The specificity of prediction by A2SPD was as high as 80%. With this specificity, the prediction can determine residues that are almost certain to interact with RNA and this could advance wet-lab experiments designed to identify residues constituting the RNA interface. By mutating each of the predicted residues, there is a reasonable probability of experimentally identifying RNA interface residues with minimal cost compared with a random mutagenesis approach.
In our future work, we will try to improve the prediction of RNA interface starting with those residues predicted as RNA interface with high confidence. Our data suggests that the protein–RNA interface area can be described as a set of overlapping spheres and residues near those predicted to comprise the RNA interface are likely involved in protein–RNA interactions. Additional methods capable of delineating interface and non-interface areas around the ‘predicted core interface residue’ will allow predictions to be made with higher specificity and sensitivity. A prediction improvement of this nature was also suggested by Kloczkowski et al. (38) for improving their GOR secondary structure prediction. They suggested that by incorporating the information from a small number of residues predicted with high confidence into the next level of prediction, the overall quality of prediction should improve.
Our work was mainly based on statistical analyses of residue occurrence on the RNA interface, thus, the validity of our results strongly depends on the size of the dataset. The dataset was still small for calculating the residue doublet propensities for all pairs of residues shown in Figure 2 and some of the calculated propensities could not pass the test of significance. Our prediction method can be further improved as the number of known protein–RNA complex structures increases.
Prediction of the mRNA interface in an mRNA export system
We applied our prediction method to a nuclear mRNA export system, one of the most important protein–RNA complexes. The nuclear mRNA export system is composed of many protein subunits including TAP (Mex67) (39) and UAP56 (Sub2) (40,41). TAP shuttles between the nucleus and cytoplasm, associates with poly(A)+RNA and interacts directly with nuclear pore complexes (42–44). TAP directly binds mRNAs and promotes their export (42). The crystal structure of an RNA-binding domain (RBD) from human TAP was determined and random mutagenesis experiments and RNA-binding assays were carried out to identify residues that interact with mRNA (39). UAP56 plays important roles in the splicing reaction as well as in the nuclear export of mature mRNA (45). We applied our best prediction method (A2SPD) to the 3D structures of the RBD of TAP (PDB ID: 1KOH) and UAP56 (PDB ID: 1XTJ).
The result of prediction on TAP by A2SPD was shown in Figure 5A and B. Each residue was colored from red to blue, from high to low score. We visually located four high score patches in Figure 5A, centered at R105, R128, K218 and R249. A patch centered at R105 is an artifact because R105 was an N-terminal residue only in the crystallized 3D structure. In the wild type protein, there are an additional 104 N-terminal amino acid residues. Comparison between our prediction and previous mutagenesis experiments (39,46) showed that: (i) the residues with high propensities [R128, K129 and R249 (red letters)] affected RNA-binding; (ii) the residues with negative propensities [E164, R307, D329 and K347 (blue letters)] did not affect RNA-binding; (iii) residues K132, R233, R276, Y278 and K304 (black letters) were experimentally shown to be important for RNA-binding, but do not have high scores (false negative); and (iv) residue R186 (green letter) has a positive score, but does not affect RNA-binding (false positive). Our positive prediction for residues Y126, Y154, R158, K218 and R250 (orange letters) requires experimental verification. Of those residues, Y126, Y154 and R158 are located in a patch centered at the experimentally verified interface residue R128. R250 is located in a patch centered at R249 that was also experimentally verified as an RNA interface residue. Therefore, the residues in orange letters except for K218 are expected to be in the interface. Our prediction incorrectly identified the experimentally verified interface residues K132, R233, R276, Y278 and K304 (black letters). Except for residue K304, the remaining residues are located near interface residues predicted with high confidence and we expect that those residues can be predicted as interface residues in our future improvement.
Figure 5 .

Application of our best prediction method to 3D structures of mRNA export proteins. RNA interface prediction for the RBD domain of human TAP (A and B) and for human UAP56 (C and D). Two figures are shown for each molecule, in which the structure is rotated 180° around the x (horizontal) axis. Surface residues are colored based on the prediction scores (Equation 18). Residues in yellow to red have a high score and residues in deep green to blue have a low score. In (A and B), an amino acid residue with its number in red was predicted as a protein–RNA interface residue consistent with mutation experiments. A residue with its number in blue was not predicted as a protein–RNA interface residue consistent with mutation experiments. A residue with its number in orange was predicted as a protein–RNA interface residue, but no mutation experiments have been done to examine this. A residue with its number in black was predicted as a non-interface residue, but mutational experiments have shown a role for these residues in RNA-binding. A residue with its number in green was predicted as a protein–RNA interface residue, but mutation experiments did not suggest this residue lies at the interface.
Our prediction results for human UAP56 are shown in Figure 5C and D. UAP56 is an essential eukaryotic pre-mRNA splicing protein that also functions in mRNA export (47–49) and the specific mechanism of UAP56 in splicing and mRNA export remains unknown. The 3D structure of UAP56 is similar to proteins of the superfamily II (SF2) ATPase/helicase (40,41). UAP56 was shown to have RNA-dependent ATPase activity in vitro (41). The residues with high score, namely R48, G151, R175, R181, K295, P313, R338, R339, R380 and F381 correspond to conventional RNA interfaces found in the SF2 family of RNA helicases (conserved helicase motifs). Helicase function is required for RNA splicing (50) and these regions of UAP56 are likely involved in the RNA splicing process. The high score residues R240 and K241 do not correspond to the RNA interfaces known in SF2 family of RNA helicases. We speculate that residues near R240 and K241 are important for mRNA export, because these residues are apparently not required for helicase activity and mRNA export does not seem to require helicase activity of UAP56. Shi et al. independently suggested that the surface including these two residues could be an interface for RNA, based on their observation that the surface structure of this area changes upon ADP binding (41).
CONCLUDING REMARKS
In this work, we used existing protein–RNA 3D structures to analyzed residue propensities in protein–RNA interface with a new measure and applied the propensities to RNA interface prediction. The prediction had high specificity and can be used to predict protein–RNA interface residues from protein structures without biochemical or functional data. This method was then applied to two proteins involved in the nuclear mRNA export system. All of the prediction methods are available at http://yayoi.kansai.jaea.go.jp/qbg/kyg/.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
Acknowledgments
This work was supported by ITBL project and was carried out on ITBL computer at the Japan Atomic Energy Agency. Funding to pay the Open Access publication charges for this article was provided by CREST, JST.
Conflict of interest statement. None declared.
REFERENCES
- 1.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 2.Mattick J.S. The functional genomics of noncoding RNA. Science. 2005;309:1527–1528. doi: 10.1126/science.1117806. [DOI] [PubMed] [Google Scholar]
- 3.Ravasi T., Suzuki H., Pang K.C., Katayama S., Furuno M., Okunishi R., Fukuda S., Ru K., Frith M.C., Gongora M.M., et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–19. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 5.Jurica M.S., Moore M.J. Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 6.Moore M.J. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 7.Noller H.F. RNA structure: reading the ribosome. Science. 2005;309:1508–1514. doi: 10.1126/science.1111771. [DOI] [PubMed] [Google Scholar]
- 8.Aas P.A., Otterlei M., Falnes P.O., Vagbo C.B., Skorpen F., Akbari M., Sundheim O., Bjoras M., Slupphaug G., Seeberg E., et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 9.Bock R. Sense from nonsense: how the genetic information of chloroplasts is altered by RNA editing. Biochimie. 2000;82:549–557. doi: 10.1016/s0300-9084(00)00610-6. [DOI] [PubMed] [Google Scholar]
- 10.Keene J.D. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl Acad. Sci. USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandel-Gutfreund Y., Schueler O., Margalit H. Comprehensive analysis of hydrogen bonds in regulatory protein DNA-complexes: in search of common principles. J. Mol. Biol. 1995;253:370–382. doi: 10.1006/jmbi.1995.0559. [DOI] [PubMed] [Google Scholar]
- 12.Jones S., van Heyningen P., Berman H.M., Thornton J.M. Protein–DNA interactions: a structural analysis. J. Mol. Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 13.Nadassy K., Wodak S.J., Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 14.Pabo C.O., Nekludova L. Geometric analysis and comparison of protein–DNA interfaces: why is there no simple code for recognition? J. Mol. Biol. 2000;301:597–624. doi: 10.1006/jmbi.2000.3918. [DOI] [PubMed] [Google Scholar]
- 15.Luscombe N.M., Laskowski R.A., Thornton J.M. Amino acid-base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones S., Shanahan H.P., Berman H.M., Thornton J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003;31:7189–7198. doi: 10.1093/nar/gkg922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuchiya Y., Kinoshita K., Nakamura H. Structure-based prediction of DNA-binding sites on proteins using the empirical preference of electrostatic potential and the shape of molecular surfaces. Proteins. 2004;55:885–894. doi: 10.1002/prot.20111. [DOI] [PubMed] [Google Scholar]
- 18.Honig B., Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 19.Draper D.E. Themes in RNA–protein recognition. J. Mol. Biol. 1999;293:255–270. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- 20.Sheinerman F.B., Norel R., Honig B. Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 21.Stawiski E.W., Gregoret L.M., Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J. Mol. Biol. 2003;326:1065–1079. doi: 10.1016/s0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 22.Liang H., Wong J.Y., Bao Q., Cavalcanti A.R., Landweber L.F. Decoding the decoding region: analysis of eukaryotic release factor (eRF1) stop codon-binding residues. J. Mol. Evol. 2005;60:337–344. doi: 10.1007/s00239-004-0211-8. [DOI] [PubMed] [Google Scholar]
- 23.Kim O.T.P., Yura K., Go N., Harumoto T. Newly sequenced eRF1s from ciliates: the diversity of stop codon usage and the molecular surfaces that are important for stop codon interactions. Gene. 2005;346:277–286. doi: 10.1016/j.gene.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 24.Kolosov P., Frolova L., Seit-Nebi A., Dubovaya V., Kononenko A., Oparina N., Justesen J., Efimov A., Kisselev L. Invariant amino acids essential for decoding function of polypeptide release factor eRF1. Nucleic Acids Res. 2005;33:6418–6425. doi: 10.1093/nar/gki927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song H., Mugnier P., Das A.K., Webb H.M., Evans D.R., Tuite M.F., Hemmings B.A., Barford D. The crystal structure of human eukaryotic release factor eRF1—mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 26.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones S., Daley D.T., Luscombe N.M., Berman H.M., Thornton J.M. Protein–RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treger M., Westhof E. Statistical analysis of atomic contacts at RNA–protein interfaces. J. Mol. Recognit. 2001;14:199–214. doi: 10.1002/jmr.534. [DOI] [PubMed] [Google Scholar]
- 29.Kim H., Jeong E., Lee S.W., Han K. Computational analysis of hydrogen bonds in protein–RNA complexes for interaction patterns. FEBS Lett. 2003;552:231–239. doi: 10.1016/s0014-5793(03)00930-x. [DOI] [PubMed] [Google Scholar]
- 30.Allers J., Shamoo Y. Structure-based analysis of protein–RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 31.Henrick K., Thornton J.M. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence C.E., Altschul S.F., Boguski M.S., Liu J.S., Neuwald A.F., Wootton J.C. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- 33.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Res. 2006;34:D16–D20. doi: 10.1093/nar/gkj157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lichtarge O., Sowa M.E. Evolutionary predictions of binding surfaces and interactions. Curr. Opin. Struct. Biol. 2002;12:21–27. doi: 10.1016/s0959-440x(02)00284-1. [DOI] [PubMed] [Google Scholar]
- 36.Baldi P., Brunak S., Chauvin Y., Andersen C.A., Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 37.Jeong E., Kim H., Lee S.W., Han K. Discovering the interaction propensities of amino acids and nucleotides from protein–RNA complexes. Mol. Cells. 2003;16:161–167. [PubMed] [Google Scholar]
- 38.Kloczkowski A., Ting K.L., Jernigan R.L., Garnier J. Combining the GOR V algorithm with evolutionary information for protein secondary structure prediction from amino acid sequence. Proteins. 2002;49:154–166. doi: 10.1002/prot.10181. [DOI] [PubMed] [Google Scholar]
- 39.Ho D.N., Coburn G.A., Kang Y., Cullen B.R., Georgiadis M.M. The crystal structure and mutational analysis of a novel RNA-binding domain found in the human Tap nuclear mRNA export factor. Proc. Natl Acad. Sci. USA. 2002;99:1888–1893. doi: 10.1073/pnas.042698599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao R., Shen J., Green M.R., MacMorris M., Blumenthal T. Crystal structure of UAP56, a DExD/H-box protein involved in pre-mRNA splicing and mRNA export. Structure. 2004;12:1373–1381. doi: 10.1016/j.str.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Shi H., Cordin O., Minder C.M., Linder P., Xu R.M. Crystal structure of the human ATP-dependent splicing and export factor UAP56. Proc. Natl Acad. Sci. USA. 2004;101:17628–17633. doi: 10.1073/pnas.0408172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bear J., Tan W., Zolotukhin A.S., Tabernero C., Hudson E.A., Felber B.K. Identification of novel import and export signals of human TAP, the protein that binds to the constitutive transport element of the type D retrovirus mRNAs. Mol. Cell. Biol. 1999;19:6306–6317. doi: 10.1128/mcb.19.9.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang Y., Cullen B.R. The human Tap protein is a nuclear mRNA export factor that contains novel RNA-binding and nucleocytoplasmic transport sequences. Genes Dev. 1999;13:1126–1139. doi: 10.1101/gad.13.9.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katahira J., Strasser K., Podtelejnikov A., Mann M., Jung J.U., Hurt E. The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J. 1999;18:2593–2609. doi: 10.1093/emboj/18.9.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cullen B.R. Nuclear RNA export. J. Cell Sci. 2003;116:587–597. doi: 10.1242/jcs.00268. [DOI] [PubMed] [Google Scholar]
- 46.Coburn G.A., Wiegand H.L., Kang Y., Ho D.N., Georgiadis M.M., Cullen B.R. Using viral species specificity to define a critical protein/RNA interaction surface. Genes Dev. 2001;15:1194–1205. doi: 10.1101/gad.888201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleckner J., Zhang M., Valcarcel J., Green M.R. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 1997;11:1864–1872. doi: 10.1101/gad.11.14.1864. [DOI] [PubMed] [Google Scholar]
- 48.Luo M.L., Zhou Z., Magni K., Christoforides C., Rappsilber J., Mann M., Reed R. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature. 2001;413:644–647. doi: 10.1038/35098106. [DOI] [PubMed] [Google Scholar]
- 49.MacMorris M., Brocker C., Blumenthal T. UAP56 levels affect viability and mRNA export in Caenorhabditis elegans. RNA. 2003;9:847–857. doi: 10.1261/rna.5480803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staley J.P., Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.