

Non-heme diiron enzymes perform important functions in biology,1 including the selective oxidation of hydrocarbons by bacterial multicomponent monooxygenases (BMMs).2,3 Dioxygen activation and the conversion of C-H to C-OH bonds is accomplished at carboxylate-bridged diiron units within the α-sub-units of the hydroxylase components of these enzymes.4 The active oxidant in some of these reactions has been trapped as a kinetically competent intermediate that forms upon reaction of the diiron(II) core with O2.5 The binding of a small effector protein6 to the hydroxylase facilitates this process. The evolutionarily related2 toluene/o-xylene monooxygenase (ToMO)7 and soluble methane monooxygenase (sMMO)3 hydroxylases ToMOH and MMOH have nearly identical resting diiron(III) active-site structures,4,8-10 but ToMO cannot hydroxylate methane. Since crystallographic characterization of ToMOH has thus far been limited to a native, oxidized structure at 2.15 Å resolution,8 and in order to expand our structural characterization and mechanistic understanding of the ToMO system, we have determined X-ray crystal structures of native ToMOH to 1.85 Å resolution and of ToMOH reconstituted with Mn(II) as an analogue of the reduced, diiron(II) enzyme.11 The results, which provide insights that significantly advance our knowledge about the BMM family of enzymes, are that (i) formation of the dimetal(II) center is accompanied by a carboxylate shift and opening of a site for dioxygen binding and activation; (ii) a strategically placed, conserved asparagine residue in the dimetal(II) form of the enzyme undergoes a rotameric shift; and (iii) there exists a previously unidentified series of hydrophobic cavities analogous to those in MMOH. A technical discovery of note in this study is the use of an annealing procedure during data collection that improved the resolution from 2.55 to 1.85 Å.
Since a suitable crystal of the diiron(II) form of ToMOH was unavailable, the dimanganese(II) derivative was prepared from the apo protein and MnCl2 and structurally characterized to 2.20 Å. A 1.85 Å resolution structure of native ToMOHox was determined using crystals grown in an identical manner. Detailed information about sample preparation, Mn reconstitution, crystal annealing, data collection, and structure solution and statistics is supplied as Supporting Information.
Although the overall topology and fold of the native protein are essentially identical to those of the previously published structure of ToMOHox,8 the active site differs (Figures 1, S1, and S2). Foremost is the absence of a thioglycolate molecule bridging the two iron atoms, which was not present in the current purification buffer (Supporting Information). Instead, the iron atoms are linked either by a hydroxide anion, as observed in MMOH structures,12 or by the terminal hydroxyl group of the polyethylene glycol (PEG) molecule found in the product-binding channel (vide infra).8 Additional differences between the diiron centers of the previously reported and the present higher resolution structure of ToMOHox are minor and are summarized in Table S2.
Figure 1.
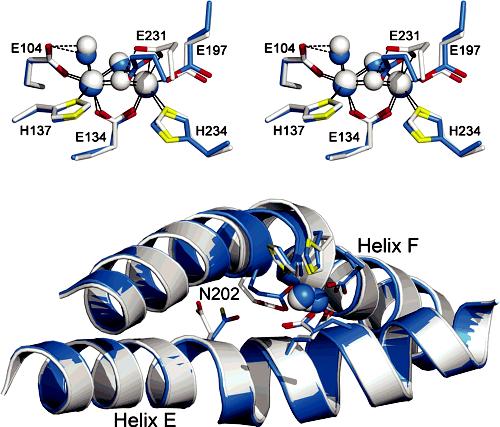
Overlaid 1.85 Å ToMOHox (white) and 2.20 Å Mn(II)-ToMOH (blue) structures. (Top) Stereoview of the active sites. (Bottom) Helices E and F, active-site ligands and metals, and N202. Fe(III) ions, Mn(II) ions, and oxygen atom species are represented as white or blue spheres. Side-chain ligands are represented as sticks in white or blue (carbon), red (oxygen), and yellow (nitrogen).
The structure of the Mn(II)-ToMOH derivative is quite revealing. The carboxylate group of E231, which is analogous to E243 in MMOH, undergoes a shift from its terminal, monodentate coordination mode in ToMOH to a bidentate-terminal, monodentate-bridging position (Figures 1 and S1). In conjunction with this carboxylate shift, the bridging hydroxide syn to the coordinated histidine ligands is displaced, while the one in the anti position becomes protonated and is semibridging, being 2.8 Å from Mn1. This structure closely resembles the core geometry of both reduced and dimanganese(II)-reconstituted MMOH (Figure S3).11,12 The results are consistent with Mn being in the divalent state and strongly suggest that ToMOHred will have the same configuration. As in the structures of MMOHox, MMOHred, and Mn(II)-MMOH, an increase in the metal-metal distance from 3.1 to 3.3 Å accompanies the carboxylate shift and oxidation state change.
The present Mn(II)-ToMOH results are nicely consistent with recently reported X-ray absorption spectroscopy (XAS) data for ToMOHred.13 This study indicated that the reduced active site is similar to that of MMOHred, both having iron coordination numbers between 5 and 6 and average Fe-Fe distances of 3.3-3.4 Å. Moreover, the XAS spectra of the diferrous hydroxylases in the presence of their respective regulatory proteins, MMOB and ToMOD, closely resemble one another, as well as those for hydroxylases in the absence of the regulatory protein. Thus, it is probable that the active-site structures of ToMOH and MMOH immediately prior to O2 activation are very similar, based on the results reported here and the XAS data. Differences in reactivity, therefore, are likely to reflect the nature of the oxygenated intermediates and/or the surrounding protein scaffolds.
In this regard, it is noteworthy that accompanying changes in the oxidation state of the dimetallic center are conformational shifts analogous to those first discovered in a comparison of the MMOHred and MMOHox structures.12 Precisely the same features can be identified by comparing the Mn(II)-ToMOH and 1.85 Å ToMOHox structures. Conformational shifts in MMOH residues N214, E240, L244, and R245 in MMOH12 are reflected in the analogous N202, Q228, S232, and R233 amino acid side chains of ToMOH (Figure 2). These residues are located on surface-exposed regions of helices E and F, adjacent to the diiron center, where the regulatory protein is postulated to bind in the MMO system10,12,14 and known to bind in the phenol hydroxylase (PH) system.15 The shift in ToMOH N202 (Figures 1 and 2) is most significant because (i) this residue is strictly conserved among BMMs; (ii) the structures reported here reveal the dependence of the side-chain orientation on oxidation state in ToMOH and MMOH; and (iii) the amide group in the analogous residue N204 in PHH is in hydrogen bond contact with the side-chain hydroxyl group of S72 (S111 in MMOB; S82 in T4MOD) of the PH regulatory component (PHM) in the PHH-PHM complex X-ray structure.15
Figure 2.
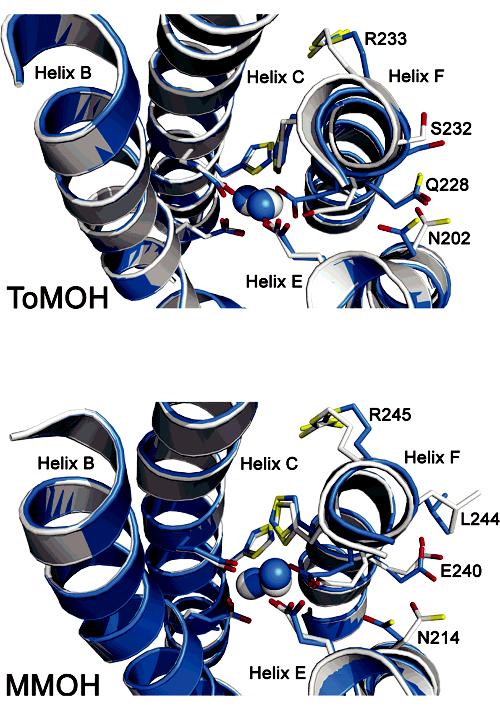
Metal center oxidation-state-dependent conformational shifts in ToMOH and MMOH. (Top) 1.85 Å ToMOHox(white) and 2.20 Å Mn(II)-ToMOH (blue). (Bottom) MMOHox(white; PDB code 1MTY) and MMOHred(blue; PDB code 1FYZ). The metal-binding four-helix bundle, active-site ligands and ions, and select side chains are shown as overlaid structures. Metal ions and helix backbones are represented as spheres and cartoons, respectively. Amino acid side chains are shown as sticks in blue/white (carbon), red (oxygen), and yellow (nitrogen).
In the ToMOHox structure, the N202 side chain points away from the diiron center and faces bulk solvent. In Mn(II)-ToMOH the side chain rotates inward, toward the active site. The resulting 80° rotameric shift in the N202 side chain occurs exclusively about the Cα-Cβ bond and is the largest among the ToMOH oxidation-state-dependent rotamer shifts thus far encountered. As with MMOH, the asparagine side-chain rotamer shift affects the local electrostatics and topology of the protein surface between helices E and F directly above the active site. In the oxidized protein, the N202 carboxamide group protrudes from the protein surface and, in this manner, can serve as a hydrogen bond donor or acceptor. Upon reduction of the metal ions, this carboxamide group is directed into the “pore” region directly above the active site and is centered between helices E and F.
Collectively, these observations reinforce prior claims that the conserved hydroxylase asparagine12 and regulatory component serine16 residues are key players in the interaction between the hydroxylase and regulatory protein components. They further suggest that regulatory component protein binding to the hydroxylase is related to the asparagine rotamer shift, which, in turn, depends on the diiron center oxidation state.
The morphology of the protein interior distinguishes MMOH and ToMOH. Xenon pressurization as well as halogenated substrate- and product-binding experiments with MMOH crystals reveal that both Xe and hydrocarbons can reside in a series of variably interconnected hydrophobic cavities in the R-subunit (Figure 3).14,17 The cavities, or pockets, trace a pathway from the active site to the protein surface and are postulated to be the means of substrate access to, or product egress from, the diiron center. Homologous pockets exist in ToMOH (Figure 3) but were not proposed as a link from the surface to the diiron center because of the occurrence of a 35-40 Å channel that connects the active site to the protein exterior via a divergent pathway. The hydrophobic pockets that traverse the interior of the ToMOH R-subunit are the active-site pocket (cavity 1), an intermediate space located between helices B and D (cavity 2), and a solvent-exposed cavity found near the interface of the α and γ subunits (cavity 3). Cavity 1 is structurally homologous to cavity 1 in MMOH, and cavities 2 and 3 are homologous to cavity 2 and a combination of cavities 3 and 4 in MMOH, respectively. It is possible that the hydrophobic pockets in ToMOH convey hydrocarbon access to the active site, but an alternative scenario is that, in ToMOH, the cavities are a consequence of its evolutionary relationship to MMOH. If the latter is the case, then the ToMOH channel may have been selected to tune its reactivity and favor hydroxylation of aromatic substrates.
Figure 3.
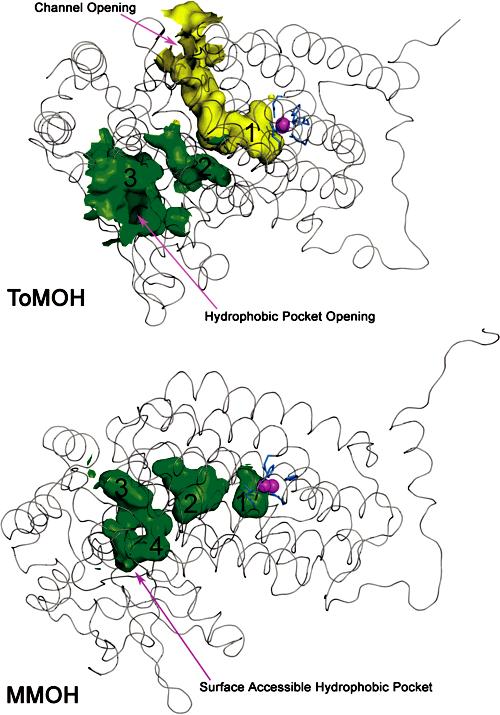
Interior surface renderings of ToMOH (top) and MMOH (bottom) R-subunits. ToMOH channel van der Waals surface is shown in yellow; hydrophobic pockets in ToMOH and MMOH are shown in green. Protein backbones (Cα trace) are represented as gray ribbons, active-site iron atoms as magenta spheres, and side-chain ligands as blue sticks.
In the ToMOHox and Mn(II)-ToMOH structures presented here, electron density in the channel has been modeled as a molecule of hexaethylene glycol (HEG), a component of PEG 400 in the crystallization buffer (Figure S4). Attempts to model water molecules into the density led to unreasonable interatomic distances. In both structures, HEG is partially disordered and exhibits high B-factors which generally increase with distance from the active site. Although our model indicates that the HEG chain ends at the channel fork, its degree of polymerization may be greater, with the remainder of the chain positionally disordered between the two channel branches. Moreover, for both structures, it was possible to model the end of the HEG molecule at the active-site metal ions either as a bridging alkoxide or simply in hydrogen bond contact with a bridging oxygen atom, presumably a hydroxide ion. These two models afforded similar refinement statistics and could not be distinguished. For presentation purposes, we have chosen the latter option, which can account for the channel electron density.
The present Mn(II)-ToMOH and ToMOHox structures contribute to the mounting structural evidence implicating BMM protein scaffolds as determinants of intersystem reaction diversity. The active sites of ToMOH and MMOH are highly homologous in both the reduced and the resting states. Control of conformational shifts by the redox state of the protein, first noted in MMOH, is conserved in ToMOH. Experimentally, we show here that ToMOH is susceptible to iron extraction and Mn reconstitution by methods established in identical experiments with MMOH.11 Crystal an-nealing by careful cryostream control at SSRL induced a dramatic resolution increase in crystalline samples of ToMOH, a procedure that should prove useful in future studies. The resulting resolution increase in the ToMOHox X-ray structure has allowed us to improve our view of the protein interior and diiron center, leading to an enhanced description of the protein scaffold, active-site ligation, and core geometry.
Supplementary Material
Acknowledgment.
This work was supported by a grant from the National Institutes of Health (GM32134 to S.J.L.). M.H.S. was supported under a NIH Biotechnology Training Grant. X-ray data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL), funded by the Department of Energy (BES, BER) and the NIH (NCRR, NIGMS). Crystallographic coordinates have been deposited in the RCSB databank, accession numbers 2INC and 2IND. All figures were generated using PyMOL.18
Footnotes
Supporting Information Available: Experimental details for sample preparation and crystallization, X-ray data collection, and structure solutions, as well as Tables S1 and S2 and Figures S1-S4 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. and references cited therein. [DOI] [PubMed] [Google Scholar]
- (2)(a).Notomista E, Lahm A, Di Donato A, Tramontano A. J. Mol. Evol. 2003;56:435–445. doi: 10.1007/s00239-002-2414-1. [DOI] [PubMed] [Google Scholar]
- (2)(b).Leahy JG, Batchelor PJ, Morcomb SM. FEMS Microbiol. Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- (3).Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew. Chem., Int. Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- (4).Sazinsky MH, Lippard SJ. Acc. Chem. Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- (5)(a).Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5)(b).Beauvais LG, Lippard SJ. J. Am. Chem. Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- (5)(c).Brazeau BJ, Lipscomb JD. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- (5)(d).Valentine AM, Stahl SS, Lippard SJ. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- (5)(e).Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- (5)(f).Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J. Am. Chem. Soc. 1994;116:7465–7466. [Google Scholar]
- (5)(g).Lee S-K, Nesheim JC, Lipscomb JD. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- (6)(a).Zhang J, Wallar BJ, Popescu CV, Renner DB, Thomas DD, Lipscomb JD. Biochemistry. 2006;45:2913–2926. doi: 10.1021/bi052256t. and references cited therein. [DOI] [PubMed] [Google Scholar]
- (6)(b).Lountos GT, Mitchell KH, Studts JM, Fox BG, Orville AM. Biochemistry. 2005;44:7131–7142. doi: 10.1021/bi047459g. [DOI] [PubMed] [Google Scholar]
- (6)(c).Scognamiglio R, Notomista E, Barbieri P, Pucci P, Dal Piaz F, Tramontano A, Di Donato A. Protein Sci. 2001;10:482–490. doi: 10.1110/ps.35701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6)(d).Walters KJ, Gassner GT, Lippard SJ, Wagner G. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7877–7882. doi: 10.1073/pnas.96.14.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6)(e).Liu Y, Nesheim JC, Lee S-K, Lipscomb JD. J. Biol. Chem. 1995;270:24662–24665. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- (7).Cafaro V, Izzo V, Scognamiglio R, Notomista E, Capasso P, Casbarra A, Pucci P, Di Donato A. Appl. Environ. Microbiol. 2004;70:2211–2219. doi: 10.1128/AEM.70.4.2211-2219.2004. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sazinsky MH, Bard J, Di Donato A, Lippard SJ. J. Biol. Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- (9)(a).Rosenzweig AC, Brandstetter H, Whittington DA, Nordlund P, Lippard SJ, Frederick CA. Proteins. 1997;29:141–152. [PubMed] [Google Scholar]
- (9)(b).Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9)(c).Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- (10).Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- (11).Sazinsky MH, Merkx M, Cadieux E, Tang S, Lippard SJ. Biochemistry. 2004;43:16263–16276. doi: 10.1021/bi048140z. [DOI] [PubMed] [Google Scholar]
- (12).Whittington DA, Lippard SJ. J. Am. Chem. Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- (13).Rudd DJ, Sazinsky MH, Lippard SJ, Hedman B, Hodgson KO. Inorg. Chem. 2005;44:4546–4554. doi: 10.1021/ic048794w. [DOI] [PubMed] [Google Scholar]
- (14).Sazinsky MH, Lippard SJ. J. Am. Chem. Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]
- (15).Sazinsky MH, Dunten PW, McCormick MS, Di Donato A, Lippard SJ. Biochemistry. 2006 doi: 10.1021/bi0618969. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng H, Lipscomb JD. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- (17).Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- (18).DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. JA064837R. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.