

The development of general methods for the synthesis of small molecule libraries is a major goal of the combinatorial chemistry research community.1–3 To address this challenge, we have developed an efficient method for library generation that is based on small molecule macroarrays.4–6 Macroarrays have the potential to address several of the drawbacks of traditional combinatorial synthesis platforms (i.e., solid-phase beads) – these focused, 20–200 compound arrays are straightforward to manipulate, inexpensive, and amenable to numerous screening applications where the array compounds are either bound to or cleaved from the planar support. In addition, we have found that the inclusion of microwave (MW)-assisted reactions during macroarray construction can accelerate library synthesis dramatically and expand the scope of accessible chemical structures.7,8
The work reported here arose from our interest in the design of synthetic ligands capable of intercepting bacterial quorum sensing.9 Cyclic dipeptides, or diketopiperazines (DKPs), have been reported to both activate and inhibit quorum sensing in certain Gram-negative bacteria.10,11 Their mode of action, however, remains unknown. The synthesis and screening of focused DKP libraries, therefore, represents a straightforward approach that couldbe used to establish structure–activity-relationships and study systematically the effects of DKPs on quorum sensing. Here, we report the efficient synthesis of DKP libraries using MW-assisted reactions and multicomponent couplings on our array platform.
Recently, we reported that small molecule macroarrays are highly compatible with Ugi four-component reactions (4CRs).5 In this work, we developed a robust, planar cellulose support system derivatized with a photolabile linker. We also demonstrated that the Ugi 4CR rate could be accelerated appreciably on this planar support by the addition of water.12 In the present study, we chose to build upon this work and examine the feasibility of DKP synthesis via Ugi 4CRs using the same planar support/linker system. DKPs have been synthesized previously on solid-phase13 and in solution14 through the use of an N-Boc-protected α-amino acid as the carboxylic acid component in the Ugi 4CR; deprotection of the amino acid after the Ugi 4CR allows for facile intramolecular attack (on an ester moiety) to give DKPs. Hulme et al. have further modified this route15 to include the “convertible isocyanide” building block (1-isocyanocyclohexene) pioneered by Keating and Armstrong (Scheme 1)16 which, upon acidic activation, allows for DKP formation to proceed via an intermediate müchnone. This route gave good to high yields of DKPs in solution, and we reasoned that it should be readily amendable to planar support-bound substrates. Furthermore, as use of the convertible isocyanide in Ugi 4CRs provides access to a broad range of structures post-condensation,16 its inclusion would expand the structural diversity possible in future macroarrays.
Scheme 1.
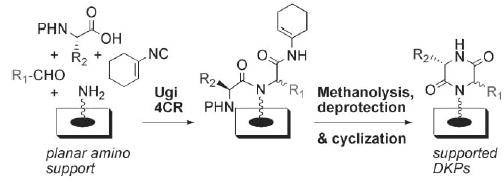
General synthetic route to diketopiperazine (DKP) macro-arrays using the convertible isocyanide. P = protecting group.
Our studies commenced with the construction of amino-derived cellulose support 1 (Scheme 2). In brief, planar cellulose (Whatman 1 Chr filter paper) was subjected to tosylation followed by reaction with a diamine spacer unit, 4,7,10-trioxa-1,13-tridecanediamine, according to our previous procedure to give support 1.4,5 By controlling the reaction times, amine loadings of ca. 8 μmol/cm2 were achieved routinely (as determined by UV Fmoc quantitation). This high functionalization level rendered the cellulose support considerably more stable to acidic solutions relative to lower loaded supports;18 such high acid stability was eventually found to be essential, as subsequent activation of the convertible isocyanide input would require relatively harsh acidic conditions (see below). Next, the photolabile linker 2, 4-{4-[1-(Fmoc-amino)ethyl]-2-methoxy-5-nitrophenoxy}butanoic acid, was coupled to support 1 in a spatially-addressed format (spot size = 0.8 cm2) under MW-assisted, carbodiimide-mediated conditions.5 N-Fmoc deprotection of the pro-linker support set the stage for the coupling of an amine input for Ugi 4CRs. We selected N-Fmoc-4-nitro-l-phenylalanine (Fmoc-Nph, 4) as the amine building block, as it provided a convenient UV chromophore for analysis of macroarray members post-cleavage.5 Coupling of Fmoc-Nph (4) to the linker-derived spots of support 3 under analogous MW-assisted conditions, followed by a second cycle of deprotection, gave amine array 5 (loading ca. 400 nmol/cm2).
Scheme 2.
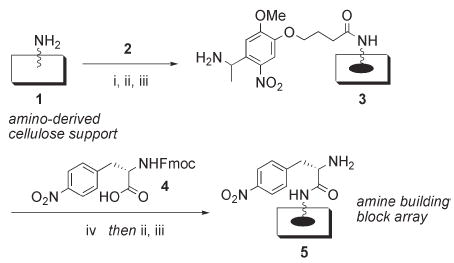
Initial planar support derivatization. Reagents and conditions: (i) N,N′-diisopropylcarbodiimide (DIC), N-hydroxysuccinimide (HOSu), diisopropylethylamine (DIPEA), NMP, MW 95 °C, 8 min. (ii) Capping step: Ac2O, DIPEA, DMF, rt, 20 min (2×).17 (iii) 20% piperidine in DMF, rt, 10 min (2×). (iv) DIC, HOSu, DIPEA, NMP, MW 100 °C, 10 min.
We next optimized the Ugi 4CR conditions on amine array 5. Isocyanocyclohexane, as opposed to the convertible isocyanide, was used in these studies, because it is commercially available and provided Ugi intermediates with heightened stabilities for routine analysis. In contrast to the work of Hulme et al.,15 we selected N-Fmoc amino acids as the carboxylic acid building blocks, as opposed to N-Boc amino acids, because an acid-stable protecting group was necessary for efficient DKP synthesis via the convertible isocyanide on planar support 5 (see below). Our previously developed water-accelerated Ugi 4CR conditions5 were found to be translatable to these alternate building blocks with only two modifications: (1) aldehyde building blocks were mixed with 2 M solutions of N-Fmoc amino acids in DMF immediately prior to use, and (2) the array spotting procedure was performed three times (3 × 10 min, 25 °C). This gave excellent conversions to a range of Ugi 4CR products (⩾95%, as determined post-cleavage).
The convertible isocyanide (synthesized according to the reported procedure16) was submitted to these optimized Ugi 4CR conditions on array 5, and we were pleased to observe high conversion (> 93%) to Ugi array 6 (Scheme 3). At this juncture, we envisioned two potential routes to generate DKP macroarrays 9 from array 6: (1) conversion of array 6 to methyl esters 7 with AcCl/MeOH according to the method of Keating and Armstrong,16 followed by basic N-Fmoc deprotection and cyclization to give DKP array 9, or (2) deprotection of the N-Fmoc group prior to subjection to acid-mediated cyclization with TFA in analogy to Hulme et al.15 The latter method was most attractive, as it required milder reaction conditions. However, only primary amide hydrolysis products (10′) and numerous unknown byproducts were generated using this route (Scheme 3).19 We therefore pursued the former methanolysis-based approach.
Scheme 3.
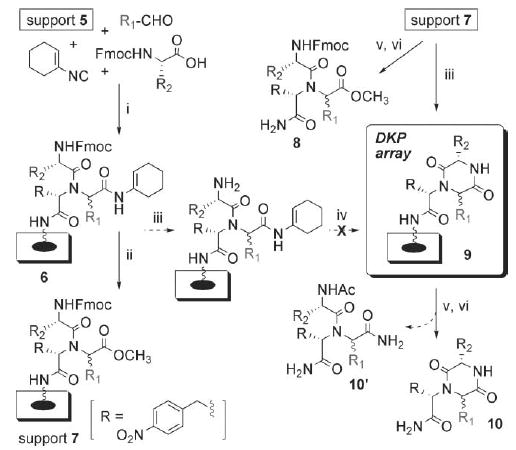
Synthesis of DKP macroarray 9 via Ugi 4CR and cyclization. Reagents and conditions: (i) H2O, rt, 10 min (3×). (ii) 10% AcCl in MeOH, MW 80 °C, 30 min. (iii) 20% piperidine in DMF, rt, 20 min (3×). (iv) 10% TFA in CH2Cl2, rt, 24 h. (v) Capping: Ac2O, DIPEA, DMF, rt, 20 min (2×).17 (vi) Photo-cleavage: hv at 366 nm in MeOH, rt, 16 h.
Support-bound Ugi product 6a was selected for a careful optimization study of the methanolysis reaction on planar support (Scheme 4). For this study, we found it most convenient to work with small, square sections of support 6a (1.2 × 1.2 cm), as opposed to larger, full array sections. Submerging squares of 6a in 10% AcCl in MeOH at room temperature for 24 h gave only 24% of the desired product 8a and 76% of the hydrolyzed product 8a′ after cleavage (Table 1, entry 1). We were able to increase the formation of 8a by warming the reaction in a heating block at 50 °C, generating 91% of 8a after 5 h (entry 3). To reduce the time required for methanolysis, we examined the reaction using MW heating in sealed vessels in a commercial MW reactor. A quick scan of MW heating conditions (entries 6–10) revealed that ramping the reaction to either 80 °C or 100 °C for 4–5 min and then holding for 10 min gave almost quantitative conversion to methyl ester 8a. We compared these results with those for conventional heating at 80 °C and found that, while 8a could be generated in 86% yield in 14 min (entry 4), complete conversion to 8a was only achieved after 38 min (entry 5). Therefore, MW heating provided a time advantage for the methanolysis reaction, and we selected MW irradiation at 80 °C for 10 min as our standard protocol. Deprotection of the N-Fmoc group after the methanolysis step with 20% piperidine in DMF led to clean cyclization to the DKP product 10a over 1 h at room temperature (ca. 98%; as determined post-cleavage; Scheme 3). These deprotection/DKP cyclization conditions proved to be general for all Ugi 4CR substrates examined in this study.
Scheme 4.
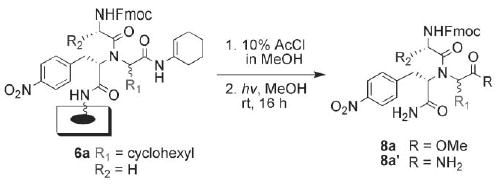
Methanolysis of Ugi 4CR product 6a. Conditions: see Table 1.
Table 1.
Optimization of the methanolysis reaction on support 6a
| Entry | Conditions | Time | Ratio 8a : 8a′a | |
|---|---|---|---|---|
| 1 | RT | 24 h | 24 : 76 | |
| Heating block | ||||
| 2 | 50 °C | 1 h | 43 : 57 | |
| 3 | 50 °C | 5 h | 91 : 9 | |
| 4 | 80 °C | 14 min | 86 : 14 | |
| 5 | 80 °C | 38 min | 100 : 0 | |
| Microwave | Ramp | Hold | ||
| 6 | 70 °C | 3 min | 10 min | 79 : 21 |
| 7 | 70 °C | 3 min | 20 min | 85 : 15 |
| 8 | 75 °C | 4 min | 10 min | 81 : 19 |
| 9 | 80 °C | 4 min | 10 min | 100 : 0 |
| 10 | 100 °C | 5 min | 10 min | 100 : 0 |
Calculated by integration of HPLC spectra with UV detection at 280 nm; Error = ± 2%. Ratio of 8a takes into account 5–15% DKP product 10a formed due to thermal cleavage of the N-Fmoc group and cyclization during methanolysis.
Three aldehydes and eight N-Fmoc l-amino acids were selected for final DKP macroarray construction (shown in Fig. 1). These largely hydrophobic building blocks were chosen in order to introduce functionality into the DKP scaffold that mimicked the functionality of the natural DKPs known to modulate quorum sensing (e.g., cyclo(l-Phe-l-Pro) and cyclo(l-Pro-l-Tyr)).10 The synthesis of initial DKP test libraries, however, revealed that our standard methanolysis conditions developed for model DKP 10a were not effective for several of the building blocks (e.g., sarcosine gave conversions of ca. 50–70% with aldehydes A–C). We therefore chose one model DKP containing Sar (10o, Table 2) for further optimization of the methanolysis reaction. Careful reevaluation of temperature and reaction times indicated that MW heating at 80 °C for 35 min (5 min ramp, 30 min hold) provided DKP 10o in good purity post-cleavage (85%). (Note, conventional heating gave purities of ca. 70% for the same reaction time.) Gratifyingly, these re-optimized conditions were found to be compatible with all of the library building blocks.
Fig. 1.
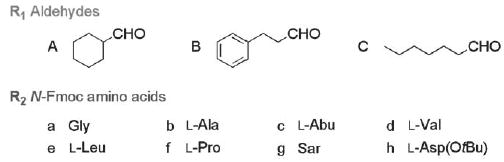
Building blocks used in the construction of DKP macroarray 9. Sar = sarcosine. l-Abu = l-aminobutyric acid.
Table 2.
Purity data for final DKP macroarray after cleavage
| Entry | R1 | R2 | Purity (%)a | Entry | R1 | R2 | Purity (%)a |
|---|---|---|---|---|---|---|---|
| 10a | A | a | 82 | 10m | B | e | 66 |
| 10b | A | b | 90 | 10n | B | f | 82 |
| 10c | A | c | 86 | 10o | B | g | 81 |
| 10d | A | d | 84 | 10pb | B | h | 82 |
| 10e | A | e | 83 | 10q | C | a | 80 |
| 10f | A | f | 90 | 10r | C | b | 80 |
| 10g | A | g | 80 | 10s | C | c | 70 |
| 10hb | A | h | 88 | 10t | C | d | 58 |
| 10i | B | a | 85 | 10u | C | e | 53 |
| 10j | B | b | 74 | 10v | C | f | 80 |
| 10k | B | c | 73 | 10w | C | g | 91 |
| 10l | B | d | 65 | 10xb | C | h | 81 |
Based on integration of HPLC spectra with UV detection at 280 nm; Error = ± 2%. For products where the diastereomeric products could be resolved by HPLC, their ratios ranged from 2 : 1 to 5 : 1.
MS data indicated that the side chain of l-Asp(OtBu) was deprotected and the methyl ester was formed during methanolysis.
The final DKP macroarray 9 was constructed on a 6 × 8 cm piece of amino support 5, with individual spots spaced 1.2 × 1.2 cm apart in a grid. The library components were applied to support 5 so as to generate 24 unique Ugi products (6). In the methanolysis step, the Ugi array (6) was immersed in 10% AcCl in MeOH and subjected to MW irradiation using the re-optimized conditions. The subsequent two steps, Fmoc-deprotection/cyclization and capping,17 were performed in a spatially addressed format to generate DKP macroarray 9 (Scheme 3). All 24 macroarray members were subjected to photo-cleavage and analyzed by LC-MS. We were pleased to observe 70% of the DKP library showed purities greater than 80% (Table 2). In addition, this library format provided ca. 25 μg of DKP per spot, which is sufficient for bacteriological assays (i.e., with reporter strains and agar overlay).9
In summary, we have demonstrated the compatibility of the small molecule macroarray platform with DKP synthesis. The use of MW-assisted reactions and Ugi 4CRs allows for libraries of DKPs to be generated in high purity in only 48 h. With further development, the macroarray synthesis platform reported here could permit the first systematic study of the effects of synthetic DKPs on bacterial quorum sensing.
Supplementary Material
Acknowledgments
We thank the NIH (AI063326-01), the Shaw Scientist Award Program, and Research Corporation (CS1309) for their generous financial support of this work. Jennifer C. O’Neill is acknowledged for her critical review of this manuscript.
Footnotes
Electronic supplementary information (ESI) available: Full experimental details for macroarray construction. See DOI: 10.1039/b604329a
Libraries of diketopiperazines have been generated in high purity using the small molecule macroarray synthesis platform.
References
- 1.Dolle RE. J Comb Chem. 2005;7:739–798. doi: 10.1021/cc050082t. [DOI] [PubMed] [Google Scholar]
- 2.Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]
- 3.Ley SV, Baxendale IR. Nat Rev Drug Discovery. 2002;1:573–586. doi: 10.1038/nrd871. [DOI] [PubMed] [Google Scholar]
- 4.Bowman MD, Jeske RC, Blackwell HE. Org Lett. 2004;6:2019–2022. doi: 10.1021/ol049313f. [DOI] [PubMed] [Google Scholar]
- 5.Lin Q, O’Neill JC, Blackwell HE. Org Lett. 2005;7:4455–4458. doi: 10.1021/ol051684o. [DOI] [PubMed] [Google Scholar]
- 6.Bowman MD, Jacobson MM, Blackwell HE. Org Lett. 2006;8:1645–1648. doi: 10.1021/ol0602708. [DOI] [PubMed] [Google Scholar]
- 7.Blackwell HE. Org Biomol Chem. 2003;1:1251–1255. doi: 10.1039/b301432k. [DOI] [PubMed] [Google Scholar]
- 8.Kappe CO, Dallinger D. Nat Rev Drug Discovery. 2006;5:51–63. doi: 10.1038/nrd1926. [DOI] [PubMed] [Google Scholar]
- 9.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J Am Chem Soc. 2005;127:12762–12763. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]
- 10.Holden MTG, Chhabra SR, de Nys R, Stead P, Bainton NJ, Hill PJ, Manefield M, Kumar N, Labatte M, England D, Rice S, Givskov M, Salmond GPC, Stewart GSAB, Bycroft BW, Kjelleberg SA, Williams P. Mol Microbiol. 1999;33:1254–1266. doi: 10.1046/j.1365-2958.1999.01577.x. [DOI] [PubMed] [Google Scholar]
- 11.Degrassi G, Aguilar C, Bosco M, Zahariev S, Pongor S, Venturi V. Curr Microbiol. 2002;45:250–254. doi: 10.1007/s00284-002-3704-y. [DOI] [PubMed] [Google Scholar]
- 12.Pirrung MC, Das Sarma K. Tetrahedron. 2005;61:11456–11472. [Google Scholar]
- 13.Szardenings AK, Burkoth TS, Lu HH, Tien DW, Campbell DA. Tetrahedron. 1997;53:6573–6593. [Google Scholar]
- 14.Wyatt PG, Allen MJ, Borthwick AD, Davies DE, Exall AM, Hatley RJD, Irving WR, Livermore DG, Miller ND, Nerozzi F, Sollis SL, Szardenings AK. Bioorg Med Chem Lett. 2005;15:2579–2582. doi: 10.1016/j.bmcl.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 15.Hulme C, Morrissette MM, Volz FA, Burns CJ. Tetrahedron Lett. 1998;39:1113–1116. [Google Scholar]
- 16.Keating TA, Armstrong RW. J Am Chem Soc. 1996;118:2574–2583. [Google Scholar]
- 17.Acetylation was performed to both block any unreacted amines on the support and provide intermediates with improved stability for analysis.
- 18.Zander N. Mol Divers. 2004;8:189–195. doi: 10.1023/b:modi.0000036253.04738.ac. [DOI] [PubMed] [Google Scholar]
- 19.We initially examined the use of N-Boc amino acids, but found that no DKP products were formed after a 24 h treatment with 10% TFA in anhydrous CH2Cl2 at 25 °C. Compounds 10′ were the predominant products observed post-cleavage (ca. 70% conversion). We speculate that hydrolysis could occur faster than the cyclization reaction and/or be promoted by residual water in the cellulose support.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.