

Abstract
Mass spectrometric characterization of the surfactant protein A (SP-A) receptor 210 (SP-R210) led to the identification of myosin (Myo) XVIIIA and nonmuscle myosin IIA. Antibodies generated against the unique C-terminal tail of MyoXVIIIA revealed that MyoXVIIIA, MyoIIA, and SP-R210 have overlapping tissue distribution, all being highly expressed in myeloid cells, bone marrow, spleen, lymph nodes, and lung. Western blot analysis of COS-1 cells stably transfected with either MyoXVIIIA or MyoIIA indicated that SP-R210 antibodies recognize MyoXVIIIA. Furthermore, MyoXVIIIA but not MyoIIA localized to the surface of COS-1 cells, and most importantly, expression of MyoXVIIIA in COS-1 cells conferred SP-A binding. Western analysis of recombinant MyoXVIIIA domains expressed in bacteria mapped the epitopes of previously derived SP-R210 antibodies to the neck region of MyoXVIIIA. Antibodies raised against the neck domain of MyoXVIIIA blocked the binding of SP-A to macrophages. Together, these findings indicate that MyoXVIIIA constitutes a novel receptor for SP-A.
Surfactant protein A (SP-A)2 is a lipid-associated lung collectin playing an important role in binding and clearance of pathogens and the regulation of inflammatory responses in the lung (1, 2). Studies in vitro have demonstrated that SP-A is involved in both innate and adaptive host defense through its ability to regulate both pro- and anti-inflammatory activities of macrophages (3-12), induce phagocytosis of microbes (13-21) and apoptotic cells (22, 23), stimulate chemotaxis (24-26), inhibit lymphocyte proliferation (27-29), and inhibit dendritic cell differentiation (30). Studies in SP-A null mutant mice support roles of SP-A in microbial clearance (31-35), inflammation (5, 8, 32, 33, 35-40), and adaptive immunity (41).
The mechanisms that mediate a plethora of SP-A functions in mucosal host defense of the lung are not characterized in detail. A number of cell-surface molecules have been implicated to mediate SP-A function. For example, SP-A alters the binding of lipopolysaccharide to CD14, while modulating peptidoglycan and zymosan-induced inflammation in macrophages through TLR-2 (3, 6, 42). Guillot et al. (4) showed that SP-A can stimulate macrophages through TLR-4. More recently, it was shown that macropinocytosis of apoptotic cells by SP-A is mediated by the binding of SP-A to a complex of calreticulin and CD91 (43) on macrophages. In addition, binding of SP-A to SIRPα suppressed macrophage inflammation in alveolar macrophages (23, 43). Most interestingly, Kuroki and co-workers (13) and Schlesinger and co-workers (19) have shown that SP-A binding can induce phagocytosis indirectly through the up-regulation of the scavenger and mannose receptors, respectively. Earlier, it was demonstrated that the binding of SP-A to the high affinity SP-A receptor SP-R210 (44) coordinates macrophage activation and phagocytosis of mycobacteria (17, 45), suppresses the proliferation of T lymphocytes (46), and alters the metabolic activity of alveolar type II epithelial cells (44). Therefore, it was crucial to determine the molecular identity of the SP-R210 receptor.
In the present study, we used a variety of biochemical, molecular, and cellular experiments to determine that cell-surface MyoXVIIIA is the high affinity SP-R210 receptor. Together our findings establish the first physical link between SP-A-mediated functions and the unconventional cell-surface MyoXVIIIA.
EXPERIMENTAL PROCEDURES
Materials
Chemicals were from Sigma, unless noted otherwise. United States Department of Agriculture tested fetal bovine serum was purchased from either Sigma or Hyclone (Logan, UT) and heat-inactivated at 56 °C. CHAPS detergent was from Calbiochem. Whatman 0.2-μ PES filters, Millipore Centricon-Plus filtration units, Greiner tissue culture dishes, and Cell-Gro DMEM and RPMI tissue culture media were purchased from Fisher. Sulfo-NHS-biotin, streptavidin-agarose, and protein G-Sepharose were from Pierce. HiTrap protein G-Sepharose, PD-10-Sepharose pre-packed columns, and NHS-activated Sepharose were from Amersham Biosciences. Na125 I and ECL chemiluminescence kit were from PerkinElmer Life Sciences. Polyclonal antihuman platelet MyoIIA was from BTI Biomedical Technologies (Stoughton, MA). PE-conjugated goat anti-rabbit IgG and PE-conjugated streptavidin were from BD Biosciences. Most electrophoresis supplies and molecular weight standards were from Bio-Rad. NOVEX 3-8% Tris acetate gels, cell dissociation medium, ExpressHyb buffer, Trizol, RadPrime random primer labeling kit, and pcDNA3.1 expression vector were from Invitrogen. The Improm-II reverse transcription system, Wizard SV miniprep DNA purification system, and TAQ polymerase were from Promega (Madison, WI). The GeneJuice transfection reagent was from Novagen (San Diego, CA). Sequence grade trypsin was from Roche Applied Science. Restriction enzymes, T4 ligase, and Vent®-polymerase were from New England Biolabs (Beverly, MA). The COS-1, U937, and THP-1 cell lines were from the American Tissue Culture Collection (Manassas, VA). The MyoXVIIIA cDNA KIAA0216 clone ha04661 was obtained from the Kazusa DNA Research Institute (Chiba, Japan). The human MyoIIA cDNA cloned into the pCMV-XL6 expression vector was purchased from Origene (Rockville, MD). The mouse and human cDNA for LRP-5 and LRP-6 were obtained from Dr. J. F. Hess, Merck (47, 48). Therapeutic lung lavage obtained from alveolar proteinosis patients was a gift of Dr. Francis McCormack, University of Cincinnati College of Medicine (Cincinnati, OH).
Animals
Wild type C57BL/6 pathogen-free mice, 4-6 weeks of age, were obtained from The Jackson Laboratories (Bar Harbor, ME). Pathogen-free Sprague-Dawley rats of 200-250 g weight were obtained from Charles River Breeding Laboratories (Worcester, MA). Animals were used in accordance with institutional animal care and use committee protocols.
Purification of SP-A
Human SP-A was purified from frozen alveolar proteinosis lavage by modification of a procedure described previously (49). Surfactant aggregate was concentrated by centrifugation at 5,000 × g and suspended in 50 ml of PBS, and 5-ml aliquots were stored frozen at -20 °C. Surfactant lipid was extracted by a dropwise addition of concentrated lavage to isobutyl alcohol (1:5 volume ratio). Delipidated protein was centrifuged at 20,000 × g, partially dried under nitrogen gas, completely dried in a lyophilizer, and then rehydrated in extraction buffer overnight (EB: 25 mm Tris, pH 7.5, 0.15 m NaCl, and 20 mm octyl-β-D-glucoside). Rehydrated surfactant was extracted three times by vigorous vortexing and centrifugation at 20,000 × g for 20 min in EB. Insoluble SP-A was then dialyzed for 48 h against four changes of solubilization buffer (SB: 5 mm HEPES, pH 7.5, 0.02% azide). Insoluble protein was removed by centrifugation at 50,000 × g. The supernatant was adjusted to 20 mm in CaCl2 and 1 m in NaCl by addition of 0.5 m CaCl2 and NaCl crystals to re-precipitate SP-A. SP-A was then collected at 20,000 × g for 20 min and washed three times in 5 mm HEPES, pH 7.5, 20 mm CaCl2, 1 m NaCl. The precipitated SP-A was solubilized in 5 mm HEPES, 5 mm EDTA, pH 7.5, at a concentration of 1 mg/ml, and dialyzed extensively against SB to remove EDTA. The purified protein was sterilized over a Whatman 0.2-μ PES filter, and 1-mg aliquots were lyophilized and stored frozen in an anhydrous environment at -20 °C. This procedure removed contaminating IgG and albumin.
Noncovalent Immobilization of SP-A
Purified SP-A (1 mg/ml) was dialyzed against MES, pH 6.5, and incubated for 2h on ice with 0.1 volume of fresh 1 mg/ml sulfo-NHS-biotin to label SP-A at its N terminus. Biotinylated SP-A was purified on PD-10-Sepharose pre-equilibrated in 5 mm HEPES, pH 7.5. Biotinylated SP-A (0.5 mg/ml) was then added to 1.5 ml of packed streptavidin-agarose beads equilibrated in a modified detergent extraction buffer containing CHAPS (50) (DEB: 20 mm Tris, pH 7.5, 1% CHAPS, 5 mm MgCl2, 5mm CaCl2). Biotinylated SP-A and streptavidin beads were rotated for 2h at 4°C and then washed with DEB to remove unbound SP-A. Approximately 75% of biotinylated SP-A bound to the streptavidin-agarose beads under these conditions.
Cell Culture
All cell lines were cultured in DMEM or RPMI supplemented with 10% fetal bovine serum and maintained in a humidified tissue culture incubator under an atmosphere of 95% air, 10% CO2 for DMEM grown cells and 5% CO2 for RPMI grown cells.
Isolation of SP-R210
Rat SP-R210 was obtained by detergent and salt extraction of rat lung membranes as described previously (44). Mouse SP-R210 was isolated from murine alveolar monocytes (mAM) (51). Frozen stocks of mAM cells were seeded in 10 150-mm2 tissue culture flasks and cultured to confluence. Confluent adherent cells were lifted in DMEM containing 0.05% trypsin and 5 mm EDTA, and cells were reseeded into 10 roller bottles containing 200 ml of DMEM. The cells were cultured 4 days in a humidified roller bottle incubator to a density of 300 million cells per flask. The cells in each flask were washed twice in ice-cold PBS and then lysed in 20 ml of DEB buffer containing a protease inhibitor mix (44). Post-nuclear supernatants were obtained at 600 × g and allowed to stand overnight. Insoluble protein aggregates were then removed by centrifugation at 100,000 × g for 1 h. Clarified supernatants were concentrated 10-fold over 30,000 Mr cut-off Centricon-Plus filtration units. The concentrated extracts were pooled and pre-adsorbed with 10 ml of streptavidin-agarose beads. Pre-adsorbed extracts were obtained by centrifugation at 25,000 × g and rotated overnight at 4 °C with biotinylated SP-A·streptavidin-agarose beads. Bound proteins were washed in a 1 × 5-cm glass column with 25 ml each of DEB buffer and then with detergent free-DEB buffer. SP-A-bound proteins were eluted in 0.15-ml fractions using DEB containing 10 mm EDTA and 1 m NaCl. Eluted proteins were visualized on silver-stained SDS-polyacrylamide gels, and SP-R210 was monitored on Western blots using SP-R210 antibodies (44). Appropriate fractions were pooled and concentrated by centrifugation to a final volume of 20 μl over a 100,000 Mr cut-off Microcon-100 filtration unit.
Mass Spectrometry
The rat membrane SP-R210 was separated by SDS-PAGE as described previously and visualized using colloidal blue (Bio-Rad). The SP-R210 band was excised and in-gel digested with trypsin, and iodoacetamide-treated peptides were extracted according to a standard procedure (52). Matrix-assisted laser desorption ionization (MALDI) mass spectra were obtained on a MALDI-TOF SPEC SE mass spectrometer in reflectron mode (Waters, Milford, MA). Peptides were searched against Swiss-Prot and TREMBL data bases using the Peptident tool (53). The search was restricted to mammalian or rodent species for proteins between 160 and 240 kDa and pI 5-7. The pI restriction was based on two-dimensional gel electrophoresis results indicating a heterogeneous SP-R210 band within the 5-7 pI range. The mass tolerance was set at ± 0.1-0.5 Da. For peptide sequence analysis, mAM SP-R210 was isolated as described above, fractionated by SDS-PAGE on a Tris acetate gel (3-8%), and visualized by silver staining according to the procedure of Sanchez et al. (54). The silver-stained SP-R210 band was excised and subjected to in-gel digestion with trypsin. The resulting peptides were analyzed by nanoflow HPLC interfaced to electro spray ionization on a quadrupole ion mass spectrometer (LCQ) (Thermo Finnigan, San Jose, CA) (55). MS/MS spectra were searched against a nonredundant protein data base using SEQUEST (56). Sequence assignments were verified by manual interpretation of the corresponding MS/MS spectra.
Primary Structure Analysis
Domain identification was accomplished using Pfam (57). Transmembrane segments were evaluated using TMbase (58). Short sequence motifs were determined using the eukaryotic linear motif resource (59). Sequence alignment was performed using MacVector software.
Expression of MyoXVIIIA and MyoIIA in COS-1 Cells
A blunt-end BsaAI cDNA fragment of human MyoXVIIIA from KIAA0216 cDNA clone hs04661 was inserted into the EcoRV site of the pcDNA3.1 expression vector. This cDNA fragment encompasses the predicted open reading frame with flanking 68 and 250 bp of 5′- and 3′-UTR. The protein encoded by this cDNA is designated as MyoXVIIIAβ/SP-R210S (see “Results”). Expression plasmids containing MyoXVIIIAβ/SP-R210S in the sense or antisense orientations were produced in Escherichia coli, purified by standard procedures, and transfected into COS-1 cells using GeneJuice. Stable transfectants with plasmid in the sense orientation (MyoXVIIIAβ/SP-R210S-COS-1 cells) or antisense orientation (control-COS-1 cells) were selected in the presence of 300 μg/ml neomycin. The entire 7.5-kb MyoIIA cDNA was subcloned into the NotI restriction site of the pcDNA3.1 vector.
Polyclonal Antibodies
Polyclonal antibodies against gel-purified rat SP-R210 were described previously (44). The C-terminal domains (MyoXVIIIAct) and the neck (MyoXVIIIAn) domains of MyoXVIIIA were expressed in bacteria (60), and polyclonal antibodies were generated commercially in rabbits (CoCalico Biological Laboratories, Reamstown, PA) by a standard protocol using TiterMax (Sigma) as primary adjuvant. Antigen boost was in incomplete Freund’s adjuvant. The MyoXVIIIAct antigen was supplied as a lyophilized powder. Immunoaffinity-purified MyoXVIIIAct antibody was obtained by affinity chromatography using recombinant MyoXVIIIAct covalently attached to NHS-activated Sepharose. The MyoXVIIIAn antigen was rendered insoluble by dialysis in PBS and supplied as a lyophilized powder. Total polyclonal IgG was purified by affinity chromatography on a HiTrap protein G-Sepharose column.
Northern Blot Hybridization
Total RNA from indicated tissues and cells was isolated using Trizol reagent according to the manufacturer’s directions. For Northern hybridization, 10 μg of total RNA was separated using 1.2% agarose denaturing gels. The membranes were blocked for 1h in Express Hyb buffer and hybridized in the same buffer with indicated 32 P-labeled cDNA probes for 1h at 65°C. Membranes were washed three times for 30 min each in 3, 2, and 1× SSC in the presence of 0.1% SDS at 65 °C.
Western Blot Analysis
Total protein was isolated from the indicated tissues using Trizol reagent after the isolation of total RNA according to the manufacturer’s directions. Protein concentration was measured using the BCA assay and BSA as standard. Protein was used immediately after isolation. Proteins, 20-40 μg, were separated on SDS-polyacrylamide gels, transferred to nitrocellulose, and probed first with indicated primary and then horseradish peroxidase-conjugated secondary antibodies. Bound antibodies were visualized by enhanced chemiluminescence.
Immunoprecipitation
mAM cells, seeded at 4 × 106 cells, were grown for 48 h to 90% confluency in 150-mm2 tissue culture flasks (Greiner). Next, the cells were washed in PBS, pH 7.4, lifted from tissue culture flasks in enzyme-free cell dissociation medium (Invitrogen), and washed again in PBS. The cells were then lysed in a modified immuno-precipitation buffer (50) (IP: 50 mm HEPES, pH 7.5, 1% CHAPS, 0.1 m NaCl, 5 mm CaCl2, 5mm MgCl2, supplemented with DEB protease inhibitor mixture (44)) at a concentration of 1 × 107 cells per ml at 4 °C for 30 min with constant rotation. Post-nuclear lysates obtained at 14,000 × g were rotated at 4 °C for 1h in the presence of 0.1 mg/ml control IgG. 50 μl of a 30% protein G-Sepharose suspension in IP buffer was then added, and the extracts were rotated for an additional hour at 4 °C. Pre-adsorbed extracts were then incubated 2h to overnight with the indicated antibodies. To capture immune complexes, lysates were incubated with 50 μl of protein G-Sepharose beads for 2h at 4°C.The beads were then washed three times in IP buffer, two times in detergent free IP buffer containing 0.5 m NaCl, and an additional two times in IP buffer. Protein G-bound proteins were eluted in 2× Laemmli buffer. Denatured proteins were then separated by 7% SDS-PAGE and analyzed on Western blots as indicated.
SP-A Binding
Human SP-A was radio labeled with 125 I and used in radioligand binding assays according to a procedure described previously (44). Binding assays were also performed using N-terminally biotinylated SP-A. Bound SP-A was visualized by flow cytometry using phycoerythrin-conjugated streptavidin.
Flow Cytometry
Cells were washed with PBS, lifted from the tissue culture dishes in cell dissociation medium, and placed in FACS blocking buffer (PBS supplemented with 5% heat-inactivated goat serum, 0.5% BSA) at a concentration of 5-10 million cells per ml. Next, the cells were incubated with 1 μg of rabbit control or immune antibody for 30 min, washed twice in binding buffer, and then incubated for 30 min with 0.5 μg of PE-conjugated goat anti-rabbit IgG. To measure the binding of SP-A, mAM cells at 2 × 106 cells/ml were incubated with 25 μg/ml biotinylated SP-A on ice for 2h in 25 mm HEPES, pH 7.5, 1.5 mm CaCl2, 0.2 mm MgCl2, 1% BSA (44) and then washed twice in binding buffer and incubated for 30 min with 0.5 μg of PE-conjugated streptavidin. Control or MyoXVIIIAn antibodies at 50 μg/ml were added together with SP-A. Labeled samples were analyzed by flow cytometry using a Coulter Epics Elite instrument and Expo32 software.
RESULTS
Peptide Sequencing of SP-R210
The identity of SP-R210 was confirmed by two distinct approaches. Rat SP-R210 was obtained after extensive extraction of detergent-insoluble membranes with potassium chloride to deplete MyoIIA (44). Then peptide fingerprints were obtained by MALDI-assisted mass spectrometry following in-gel trypsin digestion of rat SP-R210. This analysis resulted in peptide fingerprints that matched the unconventional mouse and human myosin MyoXVIIIA (61-63) (TABLE ONE). Only fragments of the rat homo-log are currently reported in the NCBI data base. The SP-R210 band had a more complex composition with an additional fingerprint matching mouse, rat, and human rho-dependent citron kinase (64) and the lipoprotein receptor-related protein LRP-5 (47, 48) in both mouse and human. We expressed mouse and human LRP-5 and its close homolog LRP-6 in COS-1 cells, but these receptors were not recognized by SP-R210 antibodies and did not confer SP-A binding, suggesting that they may have an indirect role in SP-A function (not shown). Because a single receptor could not be identified in the SP-R210 band, we next sought to purify SP-R210 in soluble form by affinity chromatography in order to sequence its components by electrospray ionization mass spectrometry. By timing the length in culture, we arrived at conditions where most immunoreactive SP-R210 could be released from insoluble cytoskeletal proteins of mAM cells (51) by using a modified extraction buffer (50) containing CHAPS detergent instead of Triton X-100 (see “Experimental Procedures”). Solubilized SP-R210 was captured by affinity chromatography using N-terminally immobilized SP-A. Fig. 1A illustrates that most immunoreactive SP-R210 in mAM extracts bound to noncovalently immobilized SP-A as less than 5% of immunoreactive SP-R210 was found in the flow-through (Fig. 1A), indicating that solubilized mAM SP-R210 represents the same protein recovered in insoluble form in rat lung membranes. SP-R210 was eluted slowly over several fractions in the presence of 1 m NaCl and 10 mm EDTA (Fig. 1B, lanes 10-17). Most interestingly, a significant fraction of SP-R210 remained bound to the SP-A affinity beads (Fig. 1B, lane R), suggesting a tight association between immobilized SP-A and SP-R210. The salt/EDTA-resistant SP-A·SP-R210 complex was completely eluted in the presence of 10 mm dithiothreitol, but in this case the SP-R210 band was contaminated with SP-A as judged by Western blot analysis (data not shown). To obtain sequence information, the silver-stained pooled SP-R210 (see “Experimental Procedures”) (Fig. 1, B and C, lanes 11-16) was excised and digested with trypsin, and the resulting digest was then analyzed by nanoflow HPLC interfaced to electrospray ionization on an LCQ mass spectrometer. Sequences of 26 peptides were obtained in this experiment. As shown in TABLE TWO, XVIII of the sequenced peptides derived from two members of the myosin family of proteins. Thus, 16 peptides were identified as cellular MyoIIA, and two peptides were identified as MyoXVIIIA. The identification of MyoXVIIIA is consistent with the results of TABLE ONE. The sequence of peptides 1 and 2 is conserved in both mouse and human MyoXVIIIA (TABLE TWO). The sequence of 10 MyoIIA peptides is identical in both mouse and human homologs of MyoIIA (TABLE TWO). Peptides 6 and 14 are identical to mouse MyoIIA, differing by a single amino acid residue from the human homolog. Both peptides 7 and 9 are homologous to residues 342-355 of the human MyoIIA peptide, but the alanine at position 353 indicates that these peptides are derived from mouse MyoIIA. However, peptide 7 lacks leucine at position 349 and contains an alanine insertion at position 343. This novel peptide has not been reported in the current annotation of the C57BL/6 mouse genome. Peptide 9 contains valine at position 343 instead of isoleucine. Peptide 9 is identical to a MyoIIA variant in Xenopus laevis (GenBank™ accession A59282), suggesting the presence of an additional variant of MyoIIA in mAM cells or polymorphism in this area of the molecule. Similarly, peptide 15 contains methionine at position 869 instead of threonine in both reported mouse and human MyoIIA sequences, but the leucine at position 874 matches human MyoIIA. Of the eight additional peptides that were sequenced (not shown on TABLE TWO), four were identical to human SP-A, indicating that some SP-A leaked from the column during the high salt wash. In addition, there were three peptides identical to ferritin light chain, and one identical to ferritin heavy chain. The finding of 22-kDa ferritin subunits in the SP-R210 band suggests a stable association between ferritin and SP-A and a potential role of SP-A in local iron availability or transport (65). Because MyoXVIIIA was identified by both fingerprint analysis and direct peptide sequencing, we next hypothesized that MyoXVIIIA is a valid candidate SP-A receptor molecule. To further focus on characterization of MyoXVIIIA as the SP-R210 receptor, we first had to gain better understanding of the MyoXVIIIA structure.
TABLE ONE.
Identification of mouse Myosin XVIIIA in rat lung SP-R210 Following detergent and salt extraction of rat lung membranes, gel-purified SP-R210 was excised and in-gel trypsin-digested, and peptides were fractionated by MALDI-assisted mass spectrometry and searched against mammalian proteins in Swiss-Prot and TREMBL data bases using the PeptIdent tool. The search was restricted for iodoacetamide-modified peptides against proteins in the 5-7 pI and 160-240-kDa mass range and a mass tolerance of a 0.1-0.5 Da. A total of 31 peptides ranging in mass between 666.56 and 3499.25 was measured. One or two missed cleavages were allowed.
| Peptide | Measured mass | Calculated mass | ΔMass | Peptide location in mouse MyoXVIIIA |
|---|---|---|---|---|
| 1 | 910.490 | 910.427 | 0.064 | 1643DFESEKR1649 |
| 2 | 1168.550 | 1168.597 | -0.046 | 1097LLDAMRMYR1105 |
| 3 | 1500.980 | 1500.766 | -0.215 | 1971NKGPSKAPSDDGSLK1985 |
| 4 | 1608.930 | 1608.933 | -0.001 | 305LKVQPIPELSELSR318 |
| 5 | 1861.820 | 1861.938 | 0.119 | 1512VVSLEAELQDISSQESK1528 |
| 6 | 1972.090 | 1972.009 | -0.093 | 698AAYLLGCSLEELSSAIFK715 |
| 7 | 2159.980 | 2159.984 | 0.003 | 1351AAEINGEVDDDDAGGEWRLK1370 |
| 8 | 2426.470 | 2426.446 | -0.024 | 1014KKSLCIQIKLQVDALIDTIKR1034 |
| 9 | 2466.060 | 2466.288 | 0.228 | 1428CQRLTAELQDTKLHLEGQQVR1448 |
FIGURE 1.
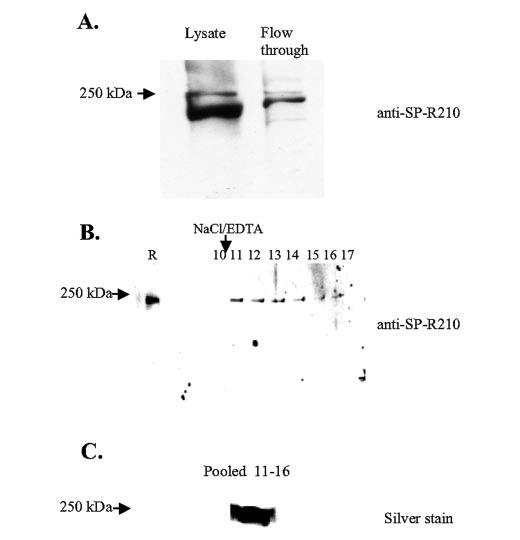
Purification of mouse SP-R210. Mouse SP-R210 was affinity-purified from mAM cells using immobilized biotinylated SP-A as described under “Experimental Procedures.” A, detection of SP-R210 by Western analysis in unprocessed cell lysate and the flow-through eluent after affinity chromatography. B, Western analysis of SP-R210 fractions eluted by 1m NaCl, 10 mmEDTA (lanes 10-17). Lane R indicates SP-R210 retained on the column. C, silver staining of concentrated SP-R210 from the size-fractionated pooled fractions. For Western analysis proteins were fractionated on 8-10% SDS-polyacrylamide gels. Purified SP-R210 was electrophoresed on a 3-8% Tris acetate gel.
TABLE TWO.
Identification of myosin XVIIIA and cellular myosin IIA in mouse SP-R210 Mouse SP-R210 was purified from mAM cells by affinity chromatography and size exclusion as described under “Experimental Procedures.” The excised silver-stained SP-R210 band was in-gel trypsin-digested, and the resulting peptides were analyzed by nanoflow HPLC interfaced to electrospray ionization on a quadrupole ion trap mass spectrometer (52). Sequenced SP-R210 peptides are aligned to the corresponding mouse (Ms) and human (Hm) MyoXVIIIA and MyoIIA peptides. Underlined boldface letters are single amino acid substitutions in the corresponding HmMyoIIA sequence.
| A) Myo XVIIIA | ||
| Peptide 1 AGSATVLSGSIAGLEGGSQLALR | Peptide 2 | ELQTQYDALKK |
| MsMyoXVIIIA 973RAGSATVLSGSIAGLEGGSQLALR996 | MsMyoXVIIIA | 1323KELQTQYDALKK1334 |
| HuMyoXVIIIA977RAGSATVLSGSIAGLEGGSQLALR1000 | HuMyoXVIIIA | 1327KELQTQYDALKK1338 |
| B) Nonmuscle myosin IIA | ||
| Peptide 3 EDQSILCTGESGAGK | Peptide 4 | QTLENERGELANEVK |
| MsMYOIIA 165REDQSILCTGESGAGK180 | MsMYOIIA | 1219KQTLENERGELANEVK1234 |
| HuMYOIIA 165REDQSILCTGESGAGK180 | HuMYOIIA | 1219KQTLENERGELANEVK1234 |
| Peptide 5 QLLQANPILEAFGNAK | Peptide 6 | LQVELDSVTGLLSQSDSK |
| MsMYOIIA 209RQLLQANPILEAFGNAK225 | MsMYOIIA | 1277KLQVELDSVTGLLSQSDSK1295 |
| HuMYOIIA 209RQLLQANPILEAFGNAK225 | HuMYOIIA | 1277KLQVELDNVTGLLSQSDSK1295 |
| Peptide 7 VIASGVLQGNIAFK | Peptide 8 | DFSALESQLQDTQELLQEENR |
| MsMYOIIA 341RVISGVLQLGNIAFK355 | MsMYOIIA | 1301KDFSALESQLQDTQELLQEENR1322 |
| HuMYOIIA 341RVI SGVLQ LGNIVFK355 | HuMYOIIA | 1301KDFSALESQLQDTQELLQEENR1322 |
| Peptide 9 VVSAVLQLGNIAFK | Peptide 10 | ALEQQVEEMK |
| MsMYOIIA 341RVISGVLQLGNIAFK355 | MsMYOIIA | 1529RALEQQVEEMK1539 |
| HuMYOIIA 341RVISGVLQLGNIVFK355 | HuMYOIIA | 1529RALEQQVEEMK1539 |
| Peptide 11 VVFQEFR | Peptide 12 | LEVNLQAMK |
| MsMYOIIA 711RVVFQEFR718 | MsMYOIIA | 1557RLEVNLQAMKA1566 |
| HuMYOIIA 711RVVFQEFR718 | HuMYOIIA | 1557RLEVNLQAMKA1566 |
| Peptide 13 ALELDSNLYR | Peptide 14 | DLEAHIDTANK |
| MsMYOIIA 745KALELDSNLYR755 | MsMYOIIA | 1620KDLEAHIDTANK1631 |
| HuMYOIIA 745KALELDSNLYR755 | HuMYOIIA | 1620KDLEAHIDSANK1631 |
| Peptide 15 LMEMET LQSQLMAEK | Peptide 16 | QLEEAEEEAQR |
| MsMYOIIA 867RLTEMETMQSQLMAEK882 | MsMYOIIA | 1877RQLEEAEEEAQR1888 |
| HuMYOIIA 867RLTEMETLQSQLMAEK882 | HuMYOIIA | 1877RQLEEAEEEAQR1888 |
| Peptide 17 ELEDATETADAMNR | Peptide 18 | IIGLDQVAGMSETALPGAFK |
| MsMYOIIA 1898RELEDATETADAMNR1912 | MsMYOIIA | 617RIIGLDQVAGMSETALPGAFK637 |
| HuMYOIIA 1898RELEDATETADAMNR1912 | HuMYOIIA | 617RIIGLDQVAGMSETALPGAFK637 |
Analysis of MyoXVIIIA Structure
Fig. 2A illustrates the domain organization of the two major isoforms of MyoXVIIIA. The classification of this myosin is based on the sequence of the myosin motor domain (63). In addition, this myosin has a typical IQ motif and a dimeric coiled-coil domain as shown in Fig. 2A. The C-terminal 126-140 amino acids is unique to MyoXVIIIA (Fig. 2A, MyoXVIIIAct). The amino acid sequence of the long and short variants of MyoXVIIIA is shown in Fig. 2B, Underlined in lowercase letters are the locations of identified peptides 1 and 2 (TABLE TWO). The long variants of this myosin isoform are distinguished by the presence of an N-terminal PDZ protein interaction domain and are shown on Fig. 2A as MyoXVIIIAα/MysPDZα/SP-R210L. The short variants of MyoXVIIIA lacking the PDZ and KE sequence are shown on Fig. 2A as MyoXVIIIAβ/MysPDZβ/SP-R210S. Obinata and co-workers (61) identified the MysPDZα variant of MyoXVIIIA in stromal fibroblasts as a novel myosin having the PDZ protein interaction domain and a KE-rich sequence at the amino terminus, and subsequently, Mori et al. (62) reported the characterization of the MysPDZβ in spleen. These MyoXVIIIA variants are generated by alternative RNA splicing. Northern analysis using a cDNA probe representing the C-terminal coding sequence indicates the expression of multiple tissue and cell-specific MyoXVIIIA mRNA species (Fig. 3A). Thus, spleen, liver, and kidney express a 7.5-kb mRNA as reported by Furusawa et al. (61) for MysPDZα. However, the message expressed by mAM and bone marrow cells is slightly smaller at 7.0 kb in length, hence the designation of SP-R210L on Fig. 2A. On this note, RT-PCR analysis of MyoXVIIIA mRNA supports the expression of more N-terminal variants of MyoXVIIIA in mAM cells.3 Muscle and heart express larger messages of 8.0-9.0 kb. In addition to the 7.0-7.5-kb MyoXVIIIA messages, spleen and bone marrow also express shorter 6.0-6.5-kb variants (Fig. 3A), reflecting the expression of MyoXVIIIAβ/MysPDZβ/SP-R210S (Fig. 4). These shorter mRNAs do not hybridize with probes representing the PDZ domain (data not shown) consistent with previous studies (62). The short message is strongly expressed in the monocytic THP-1 and promonocytic U937 cell lines (Fig. 4A) and was found in lung and mAM cells on long exposure of the Northern blots (not shown). Two short variants of MyoXVIIIA have been identified. The sequence alignment in Fig. 3B compares the 5′-UTR mRNA sequence of the MysPDZβ expressed in mouse spleen cells (62) to the mRNA of MyoXVIIIAβ produced by human KG-1 myelocytes (62). The latter mRNA is studied further in the present report and is termed as MyoXVIIIAβ/SP-R210S to reflect the role of this isoform in SP-A binding (see below). The protein sequence of MyoXVIIIAβ/SP-R210S is shown in boldface letters in Fig. 2B. The predicted start codon in MyoXVIIIAβ/SP-R210S is at position 485 bp downstream from the start codon of MysPDZβ (arrows in Fig. 3B). A 69-nucleotide insertion in the human MyoXVIIIAβ/SP-R210S splice variant introduces two in-frame stop codons (underlined in Fig. 3B). Although an equivalent MyoXVIIIAβ/SP-R210S splice variant in mouse remains to be established, it is also possible that MysPDZβ may express more than one variant by alternative start codon usage.
FIGURE 2.

Primary structure analysis of MyoXVIIIA. A, N-terminal 485 amino acids of the longest MyoXVIIIA contain a KE motif, a PDZ domain, clusters of phosphorylation (PO3) sites for casein kinases, protein kinases A and B, and a proline-rich SH3-binding site. An N-terminal KE-rich sequence is present in MysPDZ. Additional phosphorylation sites were also detected in the motor domains and the unique C terminus. The myosin heavy chain motor domain is found between amino acids 485 and 1186. The neck region between amino acids 1186 and 1246 contains an IQ motif and a C-mannosylation site. The dimeric coiled-coil domain spans amino acids 1246 and 1938, and the N-terminal sequence from 1938 to 2035/2054 is unique to MyoXVIIIA. The square box in the C terminus indicates an alternatively spliced coiled-coil insertion. The vertical shaded box in the motor domain outlines a putative transmembrane helix (TM). The short isoform of MyoXVIIIA lacks the N-terminal 485 amino acids. The long and short isoforms are designated as MyoXVIIIAα/MysPDZα/SP-R210L and the short as MyoXVIIIAβ/MysPDZβ/SP-R210S, respectively; their boundaries indicated by solid arrowheads. Antibodies were made against recombinant neck MyoXVIIIAn and C-terminal MyoXVIIIAct domains; the boundary of each domain is indicated by opposite arrows. B, the amino acid sequence of the MyoXVIIIAβ/SP-R210S isoform (see Fig. 3B) is shown in boldface letters. The underlined peptides in lowercase letters were identified by MS/MS (TABLE TWO). The boxed sequence is a putative transmembrane helix. The putative signal peptidase cleavage site MyoXVIIIAβ/SP-R210S is underlined at position 48 of the boldface sequence.
FIGURE 3.
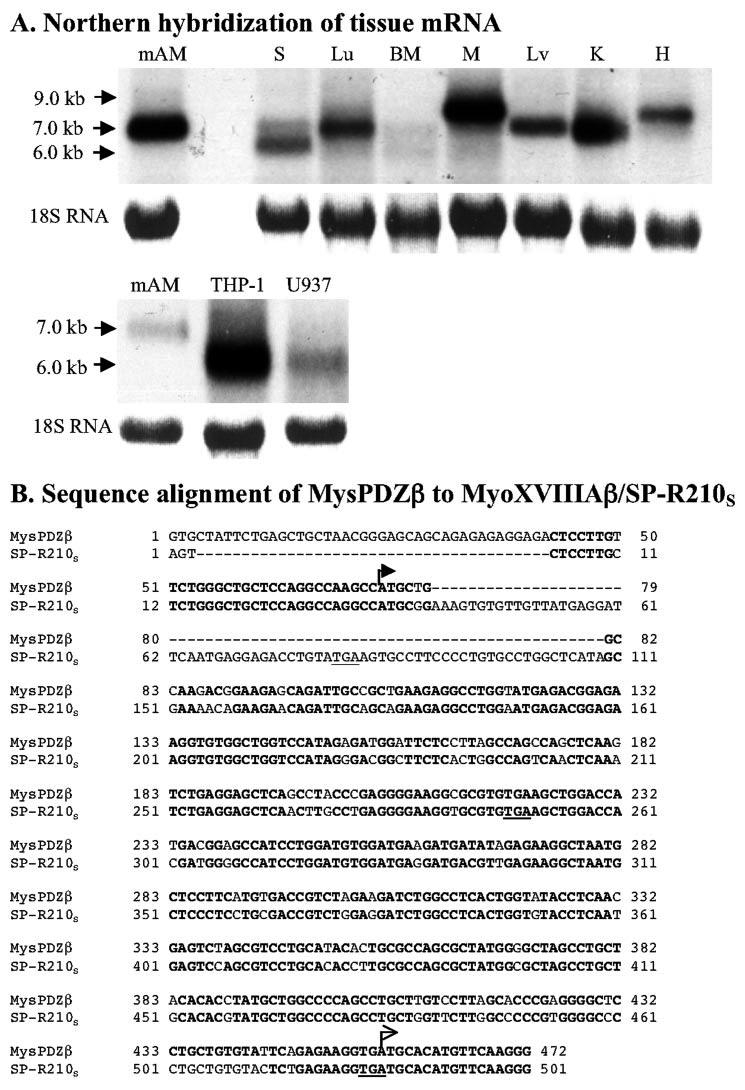
Expression of MyoXVIIIA mRNA variants. A, Northern hybridization using a human 300-bp 32 P-labeled C-terminal cDNA was carried out as described under “Experimental Procedures.” Each lane was loaded with 10 μg of total RNA. The C-terminal probe hybridized to tissue- and cell-specific mRNAs ranging between 6.0 and 9.0 kb. S, spleen; Lu, lung; BM, bone marrow; M, muscle; Lv, liver; K, kidney; H, heart. B, comparison of 5′-UTR DNA sequence of mouse MysPDZβ and human SP-R210S. Two arrows indicate the start codons of MysPDZβ and SP-R210S, respectively.
FIGURE 4.

Tissue expression of MyoXVIIIA, SP-R210, and MyoIIA. Western analysis was carried out using anti-MyoXVIIIAct (A), anti-SP-R210 (B), or anti-MyoIIA (C) antibodies. Proteins were separated on 8 (A) or 7% SDS-polyacrylamide gels (B and C). Each lane was loaded with 20 μg of protein except lung that was loaded with 40 μg of protein. S, spleen; Lu, lung; BM, bone marrow; M, muscle; Lv, liver; K, kidney; H, heart; LN, lymph node.
Identification of SP-R210 as MyoXVIIIA
To determine the tissue distribution of MyoXVIIIA variants, we generated polyclonal antibodies against the common C terminus of MyoXVIIIA/MysPDZ/SP-R210 (MyoXVIIIAct on Fig. 2A) isoforms. Given the identification of MyoIIA shown in TABLE TWO, we compared the tissue distribution of MyoXVIIIA, SP-R210, and MyoIIA by Western blot analysis. The results of Fig. 4 demonstrate that MyoXVIIIAct antibodies recognize short 210-220-kDa (SP-R210S) and long 230-250-kDa protein species reflecting the expression of short and long variants of MyoXVIIIA mRNA species in lung, bone marrow, and the immunopoietic organs spleen and lymph (Fig. 4A). Smaller species closer to 150 kDa were also detected. Previously we showed that SP-R210 isolated from U937 is highly labile to proteolysis (44). More recently, Hamilton and co-workers (66) identified a 110-kDa form of MyoXVIIIA in U937 cells. Most interestingly, the MyoXVIIIAct antibodies did not detect MyoXVIIIA protein in heart, liver or muscle extracts (Fig. 4A) suggesting low MyoXVIIIA protein levels despite the strong mRNA expression of MyoXVIIIA (Fig. 3A) or additional C-terminal splicing of the MyoXVIIIA RNA in these tissues. Kidney has low but detectable expression of MyoXVIIIA and SP-R210 (Fig. 4, A and B). Western analysis using antibodies to the originally described rat SP-R210 (44) and commercial antibodies to MyoIIA indicate overlapping tissue distribution of MyoXVIIIA (Fig. 4A), SP-R210 (Fig. 4B), and MyoIIA (Fig. 4C), all being highly expressed in immunopoietic organs. However, the proteins detected by both MyoIIA and SP-R210 antibodies in the lung were significantly more intense compared with the MyoXVIIIAct antibodies, suggesting that these antibodies detect only a fraction of MyoXVIIIA isoforms having the cognate C-terminal domain. In this regard, during the course of this work, we identified two additional splice variants of the C-terminal domain differing by the insertion of a 15-amino acid coiled-coil domain (gray box, Fig. 2A), the isoform with the longer C-terminal domain being highly expressed in lung (60). However, the overlapping tissue distribution may also be related to cross-reactivity of SP-R210 antibodies with both MyoXVIIIA and MyoIIA. To establish the specificity of these antibodies, we generated stable COS-1 cells transfected with either the MyoXVIIIAβ/SP-R210S (67) isoform or MyoIIA. COS-1 cells do not express MyoIIA (68). Fig. 5 demonstrates that control COS cells express an endogenous 250-kDa protein cross-reacting only with MyoXVIIIAct antibodies, suggesting that this endogenous MyoXVIIIA does not display the epitopes recognized by SP-R210 antibodies (top and bottom panels of Fig. 5, A1 and A2). Both SP-R210 and MyoXVIIIAct antibodies readily recognize MyoXVIIIAβ/SP-R210S (top and bottom panels of Fig. 5, A1 and A2) but not MyoIIA, whereas antibodies to MyoIIA are specific to MyoIIA-expressing cells only (middle panels of Fig. 5, A1 and A2). Together, these results indicate that SP-R210 antibodies recognize the MyoXVIIIAβ isoform. Given the identification of MyoIIA shown on TABLE TWO, we also wanted to determine whether MyoXVIIIA and MyoIIA coisolate as a complex in the absence of SP-A. Immunoprecipitation experiments using mAM cells and MyoXVIIIAct antibodies were carried out, and immunoprecipitated proteins were probed with SP-R210, MyoXVIIIAct, and MyoIIA antibodies. The results on Fig. 5B demonstrate that both short and long MyoXVIIIA/SP-R210 variants were readily precipitated by MyoXVIIIAct antibodies that were recognized by both SP-R210 and MyoXVIIIAct antibodies, but only a single MyoIIA protein was recognized by the MyoIIA antibodies. The presence of a single species recognized by anti-MyoIIA indicates that MyoXVIIIA and MyoIIA coprecipitate as a complex that is not because of cross-reactivity of the SP-R210 antibody with MyoIIA.
FIGURE 5.

Identification of SP-R210 as MyoXVIIIA and coimmunoprecipitation with MyoIIA. A, control, MyoXVIIIAβ/SP-R210S (A1), and MyoIIA-expressing COS-1 (A2) cells were obtained as described under “Experimental Procedures” and were probed with MyoXVIIIAct, MyoIIA, or SP-R210 antibodies on Western blots. Control cells were transfected with plasmids having either MyoXVIIIAβ/SP-R210S or MyoIIA cDNA cloned in the antisense orientation. Each lane was loaded with Laemmli lysates from 70,000 cells. B, cell lysates from mAM cells were incubated with MyoXVIIIAct antibodies, and immunoprecipitated (i.p.) proteins were analyzed on Western blots (w.b.) using SP-R210, MyoXVIIIAct, or MyoIIA antibodies as indicated. Proteins were separated on 7% SDS-polyacrylamide gels; the gels were allowed to run off until the 37-kDa marker reached the bottom of the gel to allow separation of long and short isoforms of MyoXVIIIA.
Expression of MyoXVIIIAβ/SP-R210S Confers SP-A Binding
We next hypothesized that if indeed MyoXVIIIAβ/SP-R210S confers SP-A binding, it is likely to be localized on the surface of COS-1 cells. The flow cytometry results of Fig. 6, A and B, demonstrate that MyoXVIIIAβ/SP-R210S is readily localized to the cell-surface of stably transfected COS-1 cells by both MyoXVIIIAct (Fig. 6A) and original SP-R210 antibodies (Fig. 6B). A low level of the endogenous MyoXVIIIA is also present on the surface of control COS-1 cells. In contrast, stable transfection of MyoIIA (Fig. 6C) did not render this protein to the cell surface. This finding indicates that the targeting of MyoXVIIIAβ/SP-R210S to the cell surface is specific to this protein and not likely an artifact of exogenous protein over expression. Moreover, Fig. 7 demonstrates that expression of MyoXVIIIAβ/SP-R210S induced saturable 125 I-SP-A binding, conferring over 2.5-fold increase in SP-A binding from 500,000 ± 70,000 sites/cell in control cells to 1,200,000 ± 120,000 sites/cell in MyoXVIIIAβ/SP-R210S-COS-1 cells. The dissociation constants are similar with a Kd of 15.8 ± 3.8 nm in control and 13.1 ± 2.5 nm in MyoXVIIIAβ/SP-R210S-expressing COS-1 cells, suggesting that endogenous MyoXVIIIA also confers some SP-A binding.
FIGURE 6.

Cell-surface localization of MyoXVIIIAβ/SP-R210S. Control-, MyoXVIIIAβ/SP-R210S-, and MyoIIA-COS-1 cells were obtained as described under “Experimental Procedures.” Flow cytometric analysis on intact cells using either MyoXVIIIAct (A) or original SP-R210 (B) (44) antibodies demonstrated cell-surface expression of MyoXVIIIAβ/SP-R210S. In contrast, MyoIIA antibodies did not detect MyoIIA on the surface of MyoIIA-COS-1 cells (C). The open histograms show staining with nonimmune polyclonal rabbit IgG (A-C). Gray histograms show staining with rabbit anti-MyoXVIIIAct (A), SP-R210 (B), or anti-MyoIIA (C) antibodies.
FIGURE 7.

Expression of MyoXVIIIAβ/SP-R210S in COS-1 cells confers SP-A binding. Binding of SP-A to control and MyoXVIIIAβ/SP-R210S-expressing cells was determined using 125 I-labeled SP-A. The saturation isotherm and Scatchard analysis of binding data (inset) show 2.5-fold increase in SP-A binding to MyoXVIIIAβ/SP-R210S-expressing cells.
Mapping of Inhibitory Antibodies
In order to generate inhibitory antibodies to block SP-A binding, we produced recombinant neck (MyoXVIIIAn) and C-terminal (MyoXVIIIAct) domains (Fig. 2A) with His tags at their C terminus (60). The 47-kDa MyoXVIIIAn and 16-kDa MyoXVIIIAct domains are shown on the colloidal blue-stained gel on Fig. 8A, lanes 1 and 2, respectively. The MyoXVIIIAn domain is heterogeneous with smaller fragments that are also His-tagged at the C terminus (60). The Western blot analysis on Fig. 8A, lanes 3 and 4, indicates that the SP-R210 antibodies described earlier (17, 44-46) recognize MyoXVIIIAn but not MyoXVIIIAct. Because the SP-R210 antibodies recognized mainly the full-length recombinant protein, these results indicate that some SP-R210 epitopes are located within a 7-10-kDa region of MyoXVIIIA between the IQ motif and part of the coiled-coil domain. To determine whether the MyoXVIIIAn domain mediates SP-A binding, we generated new polyclonal antibodies against MyoXVIIIAn (Fig. 8A, lane 5). Next, we evaluated the effect of MyoXVIIIAn antibodies on SP-A binding. Fig. 8B shows that the MyoXVIIIAn antibodies reduced SP-A binding 70% compared with control level of SP-A binding in the presence of preimmune IgG. Antibodies to the C-terminal MyoXVIIIAct domain also did not block SP-A binding (not shown). Furthermore, the anti-MyoXVIIIAn antibodies blocked SP-A binding in a concentration-dependent manner to a maximum 60% of control (Fig. 8C). Higher concentrations of MyoXVIIIAn antibodies did not lead to complete inhibition, suggesting that either mAM cells express additional SP-A-binding sites or that the coverage of SP-A-binding epitopes by the MyoXVIIIAn antibodies is incomplete. Together, these results indicate that the SP-A receptor SP-R210 is identical to cell-surface isoforms of MyoXVIIIA.
FIGURE 8.
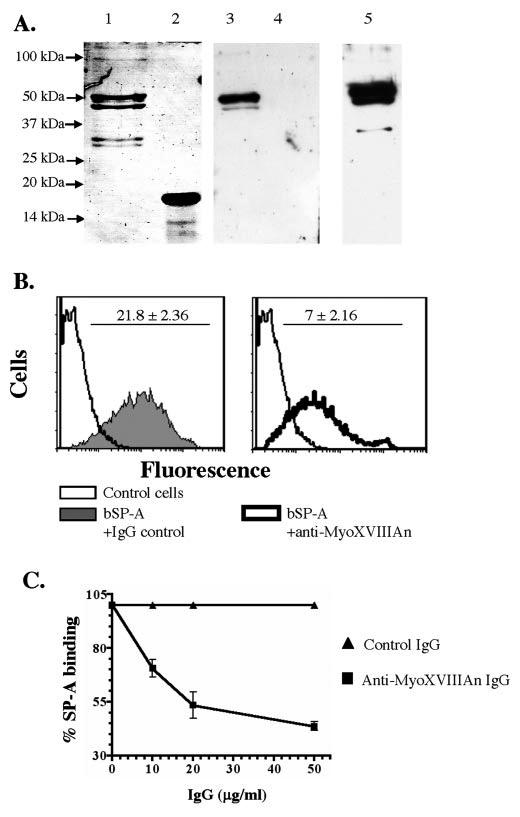
The neck domain of MyoXVIIIA/SP-R210 mediates SP-A binding to macrophages. A, purified recombinant MyoXVIIIAn (lane 1) and MyoXVIIIAct (lane 2) proteins are shown on a colloidal blue-stained 10% SDS-PAGE. Western blot analysis indicated that SP-R210 antibodies (44) recognized only the MyoXVIIIAn domain (lane 3) but not MyoXVIIIAct (lane 4). Specific antibodies against MyoXVIIIAn were generated in rabbits (lane 5). Each lane was loaded with 100 ng of protein. For Western blotting antibodies were used at 0.1 μg/ml. Antibodies were pre-adsorbed against E. coli extracts. B, the effect of MyoXVIIIAn antibodies on the binding of biotinylated SP-A (bSP-A) was determined by flow cytometry. Bound bSP-A was measured using PE-conjugated streptavidin. Anti-MyoXVIIIAn antibodies (right panel) decreased the mean fluorescence intensity of bound SP-A by 70% compared with preimmune IgG (left panel). Antibodies were used at 50 μg/ml. C, concentration-dependent inhibition of SP-A binding by MyoXVIIIAn antibodies. Data are means ± S.E., n = 6.
DISCUSSION
Previous studies utilizing inhibitory antibodies ascribed functional roles for SP-R210 on macrophage phagocytosis and activation (17, 44, 45), T lymphocyte proliferation (46), and lipid secretion in alveolar type II epithelial cells (44), suggesting an important physiological role of SP-R210 in regulating SP-A functions in the lung. Here we have determined by several lines of evidence, including mass spectrometry, cell-surface localization by flow cytometry on live cells, heterologous expression, and development of blocking antibodies, that SP-R210 is a cell-surface form of MyoXVIIIA acting as a high affinity SP-A receptor. Previous studies identified two major isoforms of this novel myosin in stromal and hematopoietic cells and tissues (61, 62). Here we also demonstrate multiple mRNA variants being expressed in a tissue- and cellspecific manner, but, in addition, we demonstrate the distinct and dominant distribution of MyoXVIIIAβ/SP-R210S in immune cells and organs implying that MyoXVIIIA isoforms have important roles in immune modulation. The present work is the first to assign a functional role for the MyoXVIIIA myosin class as cell-surface proteins. In parallel with the finding that MyoXVIIIA and MyoIIA coimmunoprecipitate, our findings also support a model where SP-A functions are mediated through exofacial MyoXVIIIA and submembrane MyoIIA.
The MyoXVIIIA gene encodes diverse mRNA and protein species, the latter having the ability to assume different subcellular localizations. Mori et al. (69) and Isogawa et al. (70) generated MysPDZ fusion proteins having fluorescent protein at the N or C terminus of mouse and human MyoXVIIIA, respectively, to visualize their subcellular localization. These studies showed that MyoXVIIIA localized to the actin cytoskeleton by a mechanism that required the KE motif at the N terminus (69, 70) but without the need for ATP hydrolysis unlike other myosins (70). Mori et al. (69) also determined that the PDZ domain is sufficient for the localization of MysPDZα to the inner surface of the plasma membrane. In addition, MysPDZα was found to colocalize with ER/Golgi membranes, suggesting a potential role in membrane traffic. In contrast, MysPDZβ, lacking both KE and PDZ sequences, had a diffuse cytoplasmic localization. The exofacial targeting of MyoXVIIIA shown in the present work is based on cell-surface localization with specific antibodies, and functional expression analyses demonstrating the presence of heterologous and endogenous protein on the cell surface. Based on these results, it is possible that some of the MyoXVIIIΑ proteins fused to N- or C-terminal fluorescent proteins are also expressed on the cell surface (69, 70). The mechanisms that mediate the exit of MyoXVIIIA isoforms to the cell surface as shown in the present report are not presently known. Computational analysis of the primary structure of MyoXVIIIAβ/SP-R210S (Fig. 2A) suggests a signal peptidase site preceded by a short hydrophobic stretch having the potential to serve as an N-terminal signal sequence. A putative transmembrane helix is present within the motor domain (Fig. 2A). Most interestingly, there are several proximal methionine residues that may potentially be utilized as start codons at the N terminus of both long and short variants of MyoXVIIIA to express multiple MyoXVIIIA isoforms with different subcellular localizations. For instance, utilization of alternative initiation codons in the plectin mRNA has been shown to generate plectin variants with different subcellular sites (71). In addition, the MysPDZβ and MyoXVIIIAβ/SP-R210S isoforms of MyoXVIIIA have different 5′-UTR sequences (Fig. 3B), indicating that alternative splicing of small exons may contribute further to the diversity and function of MyoX-VIIIA isoforms. On this note, there are alternative small exon variants at the C terminus of MyoXVIIIA (Fig. 2A) (60), and several mRNA species from different tissues having internal deletions or insertions of small exons have already been published in NCBI (GenBank™ accession numbers AY692137, AY692138, and AY692139, NM_078471, BC039612, and NM_203318). The generation of functional diversity by alternative splicing of small exons occurs frequently in the myosin family genes (72). In addition, it is also possible that internal motifs targeting MyoXVIIIA to ER/Golgi membranes may facilitate its secretion to the cell surface. For instance, internal targeting motifs are thought to be responsible for ER transport of the intermediate filament vimentin to the cell surface of a subset of microvascular endothelial cells and activated macrophages (73, 74). Computational analysis of the MyoXVIIIA primary structure also indicates a C-mannosylation motif (75), convertase-type dibasic motifs (76), and more ubiquitous casein kinase and protein kinase phosphorylation sites, each having a potential role in post-translational processing, cellular signaling, and localization. Posttranslational cleavage of MyoXVIIIA is thought to be responsible for the generation of a 110-kDa isoform in U937 cells (66). It has also been shown that PDZ domains play a role in polarized protein secretion (77). In the present case, it is notable that both long and short isoforms of MyoXVIIIA can be expressed in the same cell, possibly having a role in targeting of MyoXVIIIA isoforms to the cell surface.
Based on our findings we postulate that surface MyoXVIIIA/SP-R210 isoforms link a variety of SP-A functions via intracellular MyoIIA. Previously, it was demonstrated that the binding of SP-A to macrophages signals the reorganization of the actin cytoskeleton (78). Downstream, a number of critical cell functions have been ascribed to MyoIIA in live cells such as receptor capping (79), cell shape (80), cytokinesis (81), and vesicle transport (68). In macrophages, multiple myosin motor proteins are involved in phagosome formation (82, 83), although the role of MyoIIA in phagocytosis is less clear. In addition, MyoIIA associates with the uropod during T cell motility, tethering the formation of immune synapses during antigen recognition (12, 84). In the same context, MyoIIA interacts directly with the chemokine receptor CXCR4 in T lymphocytes (85). The results of TABLE TWO suggest the presence of an additional cellular MyoII isoform coisolating with MyoXVIIIA/SP-R210. Adelstein and co-workers (86) have already identified a third nonmuscle myosin II family member, MyoIIC. Mechanistically, the aforementioned intracellular functions of MyoIIA are related to a variety of functional activities of SP-A on cell motility and migration (25, 26), lipid secretion, lymphocyte proliferation (46), and phagocytosis (13, 17, 21, 22, 82).
The mediation of SP-A function by MyoXVIIIA/SP-R210 could occur directly by transversing the cell membrane or indirectly by binding to additional surface molecules. The topology of long and short MyoXVIIIA/SP-R210 isoforms on the cell membrane requires additional study. In recent studies a theme has emerged regarding the ability of SP-A to stimulate a variety of surface molecules indirectly. For example, it has been demonstrated that SP-A augments the activity of the phagocytic scavenger and mannose receptors indirectly (13, 16, 19), although the acting SP-A-binding site stimulating these receptors is not known. Two more examples linking SP-A function to more than one surface molecule indirectly are CD14 (42) and calreticulin (23). Thus, binding of SP-A to CD14 could be responsible for the indirect activation or inhibition of TLR4 and the binding of SP-A to calreticulin bridges SP-A-mediated apoptotic cell clearance to CD91. It becomes compelling to determine the role of MyoXVIIIA/SP-R210 as a primary SP-A-binding site on linking the internalization of SP-A-opsonized pathogens to macrophage phagocytic and signaling receptors.
It was postulated above that extra cellular MyoXVIIIA/SP-R210 links to MyoIIA inside the cell. However, Wright and co-workers (87) also demonstrated that SP-A interacts with MyoIIA, and their results indicated a role for SP-A in the clearance of cellular myosin released from dead cells. The identification of MyoIIA in the present report is consistent with this finding, but our immunoprecipitation results also indicate that MyoXVIIIA and MyoIIA are intimately linked at the plasma membrane, having exofacial and intracellular locations, respectively. However, there is evidence that MyoIIA can be accessible to cell-surface iodination of intact cells (88). Conditions that may expose MyoIIA to the cell surface may occur during membrane repair as a consequence of injury to the cell membrane. On this note, it was recently demonstrated that MyoIIA facilitates exocytosis-dependent cell membrane sealing (68), but this process in live cells is too rapid to allow detection of intracellular MyoIIA by bulky proteins such as antibodies and SP-A. The cells would have to sustain significant damage before SP-A could gain access to intracellular MyoIIA. On the other hand, MyoIIA is subject to specific degradation by caspases in apoptotic cells (89) and consequently may have a role in SP-A-mediated clearance of apoptotic cells as well (22). Thus, it is conceivable that externalization of MyoIIA peptides on the cell surface marks apoptotic cells for recognition by SP-A and clearance by bridging MyoIIA-tagged apoptotic cells to MyoXVIIIA/SP-R210-expressing macrophages.
In summary, we have identified SP-R210 as cell-surface MyoXVIIIA. Additional studies are required to dissect the functional consequences of SP-A binding to MyoXVIIIA expressed on macrophages and other immune cells. The distinct expression of MyoXVIIIA/SP-R210 suggests that this receptor has more functions in hematopoietic and immune cells. Understanding the expression patterns of MyoXVIIIA isoforms is critical for the generation of appropriate animal models. The mechanism of interaction between MyoXVIIIA/SP-R210, SP-A, and other collectins requires additional investigation.
Footnotes
This work was supported by NHLBI Grants HL068127 (to Z. C. C.) and GM37537 (to D. F. H.) and NHLBI SCOR Grant HL56387 (to J. A. W.) from the National Institutes of Health and a Parker Francis Fellowship grant (to Z. C. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- SP-A
- surfactant protein A
- SP-R210
- surfactant protein A receptor 210
- MyoXVIIIA
- myosin 18A
- MyoIIA
- nonmuscle myosin 2A
- SP-R210S and SP-R210L
- short and long SP-R210 isoforms of MyoXVIIIA, respectively
- MyoXVIIIAn
- MyoXVIIIA neck domain
- MyoXVIIIAct
- MyoXVIIIA C-terminal domain
- LRP
- lipoprotein receptor-related protein
- BSA
- bovine serum albumin
- UTR
- untranslated region
- mAM
- murine alveolar monocytes
- IP
- immunoprecipitation
- PBS
- phosphate-buffered saline
- MALDI
- matrix-assisted laser desorption ionization
- TOF
- time of flight
- HPLC
- high performance liquid chromatography
- MS
- mass spectrometry
- CHAPS
- 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonic acid
- MES
- 4-morpho-lineethanesulfonic acid
- DMEM
- Dulbecco’s modified Eagle’s medium
- NHS
- N-hydroxysuccinimide
- PE
- phycoerythrin
- ER
- endoplasmic reticulum.
C-H. Yang and Z. C. Chroneos, unpublished data.
REFERENCES
- 1.McCormack FX, Whitsett JA. J. Clin. Investig. 2002;109:707–712. doi: 10.1172/JCI15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Wetering JK, van Golde LM, Batenburg JJ. Eur. J. Biochem. 2004;271:1229–1249. doi: 10.1111/j.1432-1033.2004.04040.x. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y. J. Immunol. 2003;171:417–425. doi: 10.4049/jimmunol.171.1.417. [DOI] [PubMed] [Google Scholar]
- 4.Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, SiTahar M. J. Immunol. 2002;168:5989–5992. doi: 10.4049/jimmunol.168.12.5989. [DOI] [PubMed] [Google Scholar]
- 5.Hohlfeld JM, Erpenbeck VJ, Krug N. Pathobiology. 2002;70:287–292. doi: 10.1159/000070744. [DOI] [PubMed] [Google Scholar]
- 6.Murakami S, Iwaki D, Mitsuzawa H, Sano H, Takahashi H, Voelker DR, Akino T, Kuroki Y. J. Biol. Chem. 2002;277:6830–6837. doi: 10.1074/jbc.M106671200. [DOI] [PubMed] [Google Scholar]
- 7.Hickman-Davis JM, Fang FC, Nathan C, Shepherd VL, Voelker DR, Wright JR. Am. J. Physiol. 2001;281:L517–L523. doi: 10.1152/ajplung.2001.281.3.L517. [DOI] [PubMed] [Google Scholar]
- 8.Borron P, McIntosh JC, Korfhagen TR, Whitsett JA, Taylor J, Wright JR. Am. J. Physiol. 2000;278:L840–847. doi: 10.1152/ajplung.2000.278.4.L840. [DOI] [PubMed] [Google Scholar]
- 9.Stamme C, Walsh E, Wright JR. Am. J. Respir. Cell Mol. Biol. 2000;23:772–779. doi: 10.1165/ajrcmb.23.6.4083. [DOI] [PubMed] [Google Scholar]
- 10.Song M, Phelps DS. Infect. Immun. 2000;68:6611–6617. doi: 10.1128/iai.68.12.6611-6617.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasula R, Wright JR, Kachel DL, Martin WJ., II J. Clin. Investig. 1999;103:483–490. doi: 10.1172/JCI2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcorn JF, Wright JR. Am. J. Physiol. 2004;286:L129–L136. doi: 10.1152/ajplung.00427.2002. [DOI] [PubMed] [Google Scholar]
- 13.Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota SI, Takahashi H, Fujii N, Suzuki H, Kodama T, Abe S, Kuroki Y. J. Biol. Chem. 2004;279:21421–21430. doi: 10.1074/jbc.M312490200. [DOI] [PubMed] [Google Scholar]
- 14.Oberley RE, Ault KA, Neff TL, Khubchandani KR, Crouch EC, Snyder JM. Am. J. Physiol. 2004;287:L296–L306. doi: 10.1152/ajplung.00440.2003. [DOI] [PubMed] [Google Scholar]
- 15.van Iwaarden F, Welmers B, Verhoef J, Haagsman HP, van Golde LM. Am. J. Respir. Cell Mol. Biol. 1990;2:91–98. doi: 10.1165/ajrcmb/2.1.91. [DOI] [PubMed] [Google Scholar]
- 16.Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. J. Immunol. 1995;155:5343–5351. [PubMed] [Google Scholar]
- 17.Weikert LF, Edwards K, Chroneos ZC, Hager C, Hoffman L, Shepherd VL. Am. J. Physiol. 1997;272:L989–L995. doi: 10.1152/ajplung.1997.272.5.L989. [DOI] [PubMed] [Google Scholar]
- 18.Madan T, Eggleton P, Kishore U, Strong P, Aggrawal SS, Sarma PU, Reid KB. Infect. Immun. 1997;65:3171–3179. doi: 10.1128/iai.65.8.3171-3179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beharka AA, Gaynor CD, Kang BK, Voelker DR, McCormack FX, Schlesinger LS. J. Immunol. 2002;169:3565–3573. doi: 10.4049/jimmunol.169.7.3565. [DOI] [PubMed] [Google Scholar]
- 20.Lopez JP, Clark E, Shepherd VL. J. Leukocyte Biol. 2003;74:523–530. doi: 10.1189/jlb.0103027. [DOI] [PubMed] [Google Scholar]
- 21.Tino MJ, Wright JR. Am. J. Physiol. 1996;270:L677–L688. doi: 10.1152/ajplung.1996.270.4.L677. [DOI] [PubMed] [Google Scholar]
- 22.Schagat TL, Wofford JA, Wright JR. J. Immunol. 2001;166:2727–2733. doi: 10.4049/jimmunol.166.4.2727. [DOI] [PubMed] [Google Scholar]
- 23.Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, Greene KE. J. Immunol. 2002;169:3978–3986. doi: 10.4049/jimmunol.169.7.3978. [DOI] [PubMed] [Google Scholar]
- 24.Kramer BW, Jobe AH, Bachurski CJ, Ikegami M. Am. J. Respir. Crit. Care Med. 2001;163:158–165. doi: 10.1164/ajrccm.163.1.2005084. [DOI] [PubMed] [Google Scholar]
- 25.Schagat TL, Wofford JA, Greene KE, Wright JR. Am. J. Physiol. 2003;284:L140–L147. doi: 10.1152/ajplung.00125.2002. [DOI] [PubMed] [Google Scholar]
- 26.Wright JR, Youmans DC. Am. J. Physiol. 1993;264:L338–L344. doi: 10.1152/ajplung.1993.264.4.L338. [DOI] [PubMed] [Google Scholar]
- 27.Borron P, Veldhuizen RA, Lewis JF, Possmayer F, Caveney A, Inchley K, McFadden RG, Fraher LJ. Am. J. Respir. Cell Mol. Biol. 1996;15:115–121. doi: 10.1165/ajrcmb.15.1.8679215. [DOI] [PubMed] [Google Scholar]
- 28.Wang JY, Shieh CC, You PF, Lei HY, Reid KB. Am. J. Respir. Crit. Care Med. 1998;158:510–518. doi: 10.1164/ajrccm.158.2.9709111. [DOI] [PubMed] [Google Scholar]
- 29.Borron PJ, Mostaghel EA, Doyle C, Walsh ES, McHeyzer-Williams MG, Wright JR. J. Immunol. 2002;169:5844–5850. doi: 10.4049/jimmunol.169.10.5844. [DOI] [PubMed] [Google Scholar]
- 30.Brinker KG, Garner H, Wright JR. Am. J. Physiol. 2003;284:L232–L241. doi: 10.1152/ajplung.00187.2002. [DOI] [PubMed] [Google Scholar]
- 31.Linke MJ, Harris CE, Korfhagen TR, McCormack FX, Ashbaugh AD, Steele P, Whitsett JA, Walzer PD. J. Infect. Dis. 2001;183:943–952. doi: 10.1086/319252. [DOI] [PubMed] [Google Scholar]
- 32.LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, Korfhagen TR. J. Immunol. 2000;165:3934–3940. doi: 10.4049/jimmunol.165.7.3934. [DOI] [PubMed] [Google Scholar]
- 33.LeVine AM, Kurak KE, Bruno MD, Stark JM, Whitsett JA, Korfhagen TR. Am. J. Respir. Cell Mol. Biol. 1998;19:700–708. doi: 10.1165/ajrcmb.19.4.3254. [DOI] [PubMed] [Google Scholar]
- 34.LeVine AM, Kurak KE, Wright JR, Watford WT, Bruno MD, Ross GF, Whitsett JA, Korfhagen TR. Am. J. Respir. Cell Mol. Biol. 1999;20:279–286. doi: 10.1165/ajrcmb.20.2.3303. [DOI] [PubMed] [Google Scholar]
- 35.Hickman-Davis JM, Gibbs-Erwin J, Lindsey JR, Matalon S. Am. J. Respir. Cell Mol. Biol. 2004;30:319–325. doi: 10.1165/rcmb.2003-0246OC. [DOI] [PubMed] [Google Scholar]
- 36.Haddad IY, Milla C, Yang S, Panoskaltsis-Mortari A, Hawgood S, Lacey DL, Blazar BR. Am. J. Physiol. 2003;285:L602–L610. doi: 10.1152/ajplung.00088.2003. [DOI] [PubMed] [Google Scholar]
- 37.Hawgood S, Ochs M, Jung A, Akiyama J, Allen L, Brown C, Edmondson J, Levitt S, Carlson E, Gillespie AM, Villar A, Epstein CJ, Poulain FR. Am. J. Physiol. 2002;283:L1002–L1010. doi: 10.1152/ajplung.00118.2002. [DOI] [PubMed] [Google Scholar]
- 38.Yang S, Milla C, Panoskaltsis-Mortari A, Hawgood S, Blazar BR, Haddad IY. Am. J. Respir. Cell Mol. Biol. 2002;27:297–305. doi: 10.1165/rcmb.2002-0035OC. [DOI] [PubMed] [Google Scholar]
- 39.Li G, Siddiqui J, Hendry M, Akiyama J, Edmondson J, Brown C, Allen L, evitt S, Poulain F, Hawgood S. Am. J. Respir. Cell Mol. Biol. 2002;26:277–282. doi: 10.1165/ajrcmb.26.3.4584. [DOI] [PubMed] [Google Scholar]
- 40.Chabot S, Koumanov K, Lambeau G, Gelb MH, Balloy V, Chignard M, Whitsett JA, Touqui L. J. Immunol. 2003;171:995–1000. doi: 10.4049/jimmunol.171.2.995. [DOI] [PubMed] [Google Scholar]
- 41.LeVine AM, Hartshorn K, Elliott J, Whitsett J, Korfhagen T. Am. J. Physiol. 2002;282:L563–L572. doi: 10.1152/ajplung.00280.2001. [DOI] [PubMed] [Google Scholar]
- 42.Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. J. Biol. Chem. 2000;275:22442–22451. doi: 10.1074/jbc.M001107200. [DOI] [PubMed] [Google Scholar]
- 43.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. Cell. 2003;115:13–23. doi: 10.1016/s0092-8674(03)00758-x. [DOI] [PubMed] [Google Scholar]
- 44.Chroneos ZC, Abdolrasulnia R, Whitsett JA, Rice WR, Shepherd VL. J. Biol. Chem. 1996;271:16375–16383. doi: 10.1074/jbc.271.27.16375. [DOI] [PubMed] [Google Scholar]
- 45.Weikert LF, Lopez JP, Abdolrasulnia R, Chroneos ZC, Shepherd VL. Am. J. Physiol. 2000;279:L216–L223. doi: 10.1152/ajplung.2000.279.2.L216. [DOI] [PubMed] [Google Scholar]
- 46.Borron P, McCormack FX, Elhalwagi BM, Chroneos ZC, Lewis JF, Zhu S, Wright JR, Shepherd VL, Possmayer F, Inchley K, Fraher LJ. Am. J. Physiol. 1998;275:L679–L686. doi: 10.1152/ajplung.1998.275.4.L679. [DOI] [PubMed] [Google Scholar]
- 47.Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman AR, Metzker ML, Caskey CT, Todd JA, Hess JF. Biochem. Biophys. Res. Commun. 1998;248:879–888. doi: 10.1006/bbrc.1998.9061. [DOI] [PubMed] [Google Scholar]
- 48.Hey PJ, Twells RC, Phillips MS, Yusuke N, Brown SD, Kawaguchi Y, Cox R, Guochun X, Dugan V, Hammond H, Metzker ML, Todd JA, Hess JF. Gene (Amst.) 1998;216:103–111. doi: 10.1016/s0378-1119(98)00311-4. [DOI] [PubMed] [Google Scholar]
- 49.Haagsman HP, Sargeant T, Hauschka PV, Benson BJ, Hawgood S. Biochemistry. 1990;29:8894–8900. doi: 10.1021/bi00490a003. [DOI] [PubMed] [Google Scholar]
- 50.Zhu Q, Zelinka P, White T, Tanzer ML. Biochem. Biophys. Res. Commun. 1997;232:354–358. doi: 10.1006/bbrc.1997.6195. [DOI] [PubMed] [Google Scholar]
- 51.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 52.Jensen ON, Wilm M, Shevchenko A, Mann M. In: 2-D Proteome Analysis Protocols. Andrew JL, editor. Vol. 112. Humana Press Inc.; Totowa, NJ: 1999. pp. 487–512. [Google Scholar]
- 53.Wilkins MR, Williams KL. J. Theor. Biol. 1997;186:7–15. doi: 10.1006/jtbi.1996.0346. [DOI] [PubMed] [Google Scholar]
- 54.Sanchez JC, Hochstrasser D, Rabilloud T. Methods Mol. Biol. 1999;112:221–225. doi: 10.1385/1-59259-584-7:221. [DOI] [PubMed] [Google Scholar]
- 55.Shabanowitz J, Settlage RE, Marto JA, Christian RE, White FM, Russo PS, Martin SE, Hunt DF. In: Mass Spectrometry in Biology and Medicine. Burlingame AL, Carr SA, Baldwin MA, editors. Humana Press; Totowa, NJ: 2000. pp. 163–177. [Google Scholar]
- 56.Yates JR, III, Eng JK, McCormack AL. Anal. Chem. 1995;67:3202–3210. doi: 10.1021/ac00114a016. [DOI] [PubMed] [Google Scholar]
- 57.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofmann K, Stoffel W. Biol. Chem. Hoppe-Seyler. 1993;374:166. doi: 10.1515/bchm3.1993.374.7-12.507. [DOI] [PubMed] [Google Scholar]
- 59.Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DM, Ausiello G, Brannetti B, Costantini A, Ferre F, Maselli V, Via A, Cesareni G, Diella F, Superti-Furga G, Wyrwicz L, Ramu C, McGuigan C, Gudavalli R, Letunic I, Bork P, Rychlewski L, Kuster B, HelmerCitterich M, Hunter WN, Aasland R, Gibson TJ. Nucleic Acids Res. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szeliga J, Jordan J, Yang C-H, Sever-Chroneos Z, Chroneos ZC. Anal. Biochem., 2005 doi: 10.1016/j.ab.2005.07.021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furusawa T, Ikawa S, Yanai N, Obinata M. Biochem. Biophys. Res. Commun. 2000;270:67–75. doi: 10.1006/bbrc.2000.2377. [DOI] [PubMed] [Google Scholar]
- 62.Mori K, Furusawa T, Okubo T, Inoue T, Ikawa S, Yanai N, Mori KJ, Obinata M. J. Biochem. (Tokyo) 2003;133:405–413. doi: 10.1093/jb/mvg053. [DOI] [PubMed] [Google Scholar]
- 63.Berg JS, Powell BC, Cheney RE. Mol. Biol. Cell. 2001;12:780–794. doi: 10.1091/mbc.12.4.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Cunto F, Calautti E, Hsiao J, Ong L, Topley G, Turco E, Dotto GP. J. Biol. Chem. 1998;273:29706–29711. doi: 10.1074/jbc.273.45.29706. [DOI] [PubMed] [Google Scholar]
- 65.Ghosh S, Hevi S, Chuck SL. Blood. 2004;103:2369–2376. doi: 10.1182/blood-2003-09-3050. [DOI] [PubMed] [Google Scholar]
- 66.Cross M, Csar XF, Wilson NJ, Manes G, Addona TA, Marks DC, Whitty GA, Ashman K, Hamilton JA. Biochem. J. 2004;380:243–253. doi: 10.1042/BJ20031978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagase T, Seki N, Ishikawa K, Ohira M, Kawarabayasi Y, Ohara O, Tanaka A, Kotani H, Miyajima N, Nomura N. DNA Res. 1996;3:321–329. doi: 10.1093/dnares/3.5.321. [DOI] [PubMed] [Google Scholar]
- 68.Togo T, Steinhardt RA. Mol. Biol. Cell. 2004;15:688–695. doi: 10.1091/mbc.E03-06-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mori K, Matsuda K, Furusawa T, Kawata M, Inoue T, Obinata M. Biochem. Biophys. Res. Commun. 2005;326:491–498. doi: 10.1016/j.bbrc.2004.11.058. [DOI] [PubMed] [Google Scholar]
- 70.Isogawa Y, Kon T, Inoue T, Ohkura R, Yamakawa H, Ohara O, Sutoh K. Biochemistry. 2005;44:6190–6196. doi: 10.1021/bi0475931. [DOI] [PubMed] [Google Scholar]
- 71.Rezniczek GA, Abrahamsberg C, Fuchs P, Spazierer D, Wiche G. Hum. Mol. Genet. 2003;12:3181–3194. doi: 10.1093/hmg/ddg345. [DOI] [PubMed] [Google Scholar]
- 72.Kim KY, Kovacs M, Kawamoto S, Sellers JR, Adelstein RS. J. Biol. Chem. 2005;280:22769–22775. doi: 10.1074/jbc.M503488200. [DOI] [PubMed] [Google Scholar]
- 73.Xu B, deWaal RM, Mor-Vaknin N, Hibbard C, Markovitz DM, Kahn ML. Mol. Cell. Biol. 2004;24:9198–9206. doi: 10.1128/MCB.24.20.9198-9206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Nat. Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 75.Furmanek A, Hofsteenge J. Acta Biochim. Pol. 2000;47:781–789. [PubMed] [Google Scholar]
- 76.Nakayama K. Biochem. J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, Takio K, Adachi H, Tsujimoto M, Inoue K. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6538–6543. doi: 10.1073/pnas.100114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tino MJ, Wright JR. Am. J. Physiol. 1999;276:L164–L174. doi: 10.1152/ajplung.1999.276.1.L164. [DOI] [PubMed] [Google Scholar]
- 79.Pasternak C, Spudich JA, Elson EL. Nature. 1989;341:549–551. doi: 10.1038/341549a0. [DOI] [PubMed] [Google Scholar]
- 80.Xu XS, Lee E, Chen T, Kuczmarski E, Chisholm RL, Knecht DA. Dev. Biol. 2001;232:255–264. doi: 10.1006/dbio.2000.0132. [DOI] [PubMed] [Google Scholar]
- 81.Knecht DA, Loomis WF. Science. 1987;236:1081–1086. doi: 10.1126/science.3576221. [DOI] [PubMed] [Google Scholar]
- 82.Swanson JA, Johnson MT, Beningo K, Post P, Mooseker M, Araki N. J. Cell Sci. 1999;112:307–316. doi: 10.1242/jcs.112.3.307. [DOI] [PubMed] [Google Scholar]
- 83.Cox D, Berg JS, Cammer M, Chinegwundoh JO, Dale BM, Cheney RE, Greenberg S. Nat. Cell Biol. 2002;4:469–477. doi: 10.1038/ncb805. [DOI] [PubMed] [Google Scholar]
- 84.Wulfing C, Davis MM. Science. 1998;282:2266–2269. doi: 10.1126/science.282.5397.2266. [DOI] [PubMed] [Google Scholar]
- 85.Rey M, Vicente-Manzanares M, Viedma F, Yanez-Mo M, Urzainqui A, Barreiro O, Vazquez J, Sanchez-Madrid F. J. Immunol. 2002;169:5410–5414. doi: 10.4049/jimmunol.169.10.5410. [DOI] [PubMed] [Google Scholar]
- 86.Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. J. Biol. Chem. 2004;279:2800–2808. doi: 10.1074/jbc.M309981200. [DOI] [PubMed] [Google Scholar]
- 87.Michelis D, Kounnas MZ, Argraves WS, Sanford ED, Borchelt JD, Wright JR. Am. J. Respir. Cell Mol. Biol. 1994;11:692–700. doi: 10.1165/ajrcmb.11.6.7946398. [DOI] [PubMed] [Google Scholar]
- 88.Olden K, Willingham M, Pastan I. Cell. 1976;8:383–390. doi: 10.1016/0092-8674(76)90150-1. [DOI] [PubMed] [Google Scholar]
- 89.Kato M, Fukuda H, Nonaka T, Imajoh-Ohmi S. J. Biochem. (Tokyo) 2005;137:157–166. doi: 10.1093/jb/mvi015. [DOI] [PubMed] [Google Scholar]