

Abstract
The nitric oxide (NO) receptor, soluble guanylyl cyclase (sGC), is commonly manipulated pharmacologically in two ways. Inhibition of activity is achieved using 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-l-one (ODQ) which oxidizes the haem prosthetic group to which NO binds, while the compound 3-(5-hydroxymethyl-2-furyl)-1-benzylindazole (YC-1) is considered an ‘allosteric' activator. Knowledge of how these agents function and interact in a normal cellular environment is limited. These issues were addressed using rat cerebellar cells.
Inhibition by ODQ was not simply competitive with NO. The rate of onset was ODQ concentration-dependent and developed in two kinetic phases. Recovery from inhibition occurred with a half-time of ∼5 min.
YC-1 slowed the rate at which sGC deactivated on removal of NO by 45 fold, consistent with YC-1 increasing the potency of NO for sGC. YC-1 also enhanced the maximal response to NO by 2 fold. Furthermore, when added to cells in which sGC was 90% desensitized, YC-1 abruptly enhanced sGC activity to a degree that indicated partial reversal of desensitization.
After pre-exposure to YC-1, sGC became resistant to inhibition by ODQ. In addition, YC-1 rapidly reversed inhibition by ODQ in cells and for purified sGC, suggesting that YC-1 either increases the NO affinity of the oxidized sGC haem or reverses haem oxidation.
It is concluded that the actions of ODQ and YC-1 on sGC are broadly similar in cells and purified preparations. Additionally, YC-1 transiently reverses sGC desensitization in cells. It is hypothesized that YC-1 has multiple actions on sGC, and thereby both modifies the NO binding site and enhances agonist efficacy.
Keywords: Nitric oxide, soluble guanylyl cyclase, YC-1, ODQ, kinetics, cerebellum
Introduction
Soluble guanylyl cyclase (sGC) is the major physiological receptor for NO. When activated, sGC catalyses the synthesis of cyclic GMP, which modulates the activity of several intracellular targets including cyclic GMP-dependent kinases (Francis & Corbin, 1994), cyclic nucleotide gated ion channels (Zagotta & Siegelbaum, 1996), and phosphodiesterases (Juilfs et al., 1999). Through this transduction pathway, NO engages in numerous physiological processes, including vasodilatation, neural signalling and platelet disaggregation.
The mechanistic steps underlying activation of sGC by NO are uncertain, but initially NO binds to the haem group of the protein, which is linked to a histidine residue on the β subunit, forming a six-coordinate complex with the haem iron. Destabilization of the iron-histidine bond follows and subsequent bond scission (forming a five-coordinate nitrosyl complex) is associated with an increase in catalytic activity, presumably because of a conformational change that propagates to the active site.
Two pharmacological tools are frequently used to manipulate the NO-cyclic GMP signalling pathway at the level of sGC: an inhibitor, 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-l-one (ODQ; Garthwaite et al., 1995) and an ‘allosteric' activator, 3-(5-hydroxymethyl-2-furyl)-1-benzylindazole (YC-1; Ko et al., 1994). The inhibitory mechanism of ODQ has been characterized for the purified enzyme as a slow, irreversible oxidation of the sGC haem iron (Schrammel et al., 1996). YC-1 was originally proposed to be an NO-independent activator of sGC, although it cannot directly activate sGC (as NO does), inasmuch as it does not appear, by spectroscopic methods, to interact with sGC-haem (Friebe & Koesling, 1998). YC-1 increases both the potency of NO for activation of purified sGC and the catalytic efficiency of the enzyme in the presence of maximal NO levels, increasing Vmax by ∼40% (Friebe et al., 1996; Denninger et al., 2000) and decreasing the Km for GTP by 3 fold (Denninger et al., 2000). The increase in the potency of NO elicited by YC-1 reflects a decrease in the rate of dissociation of NO from the haem group of sGC (Friebe & Koesling, 1998). It is generally assumed, consequently, that YC-1 binds at an allosteric site on sGC and propagates a conformational change which alters the haem environment such that the binding of NO is of higher affinity (Friebe et al., 1996; 1999; Denninger et al., 2000).
The actions of YC-1 extend beyond sGC activation, however, as the compound also inhibits phosphodiesterases (PDE), the enzymes that hydrolyse cyclic GMP. This action of YC-1 has been demonstrated on a broad spectrum of PDEs (PDE 1, 2, 3, 4 and 5) in extracts of aorta (Galle et al., 1999) and in platelets (Friebe et al., 1998).
Although widely applied to tissue preparations, the mechanisms and kinetics of the actions of ODQ and YC-1 on sGC have not been studied in detail within intact cells, where their pharmacological effects will perturb an intact signal transduction pathway. Understanding how they influence the decoding of NO signals by cells is necessary in order to evaluate the benefits and limitations of applying drugs of this type therapeutically (Stasch et al., 2001). The relevance of information gathered on purified sGC to the way the enzyme operates within cells also needs to be tested, particularly as the kinetic profile of sGC activity in intact cells varies substantially from that of the purified cyclase. In particular sGC within cells desensitizes rapidly (Bellamy et al., 2000), a phenomenon that, by analogy with numerous other receptors (Jones & Westbrook, 1996), may be attributable to the existence of an additional desensitized state of the NO receptor.
In this study we characterize the potency and kinetics of ODQ and YC-1 in cells isolated from the cerebellum, the functional interactions between these two agents, and the influence of YC-1 on sGC desensitization.
Methods
Cerebellar cell suspensions were prepared from 8 – 9-day old rats as previously described (Bellamy & Garthwaite, 2001a), in line with British Home Office and local ethics committee regulations. The suspension (20×106 cells ml−1) was incubated at 37°C in buffer (pH 7.4) containing (mM): NaC1 130; KC1 3; MgSO4 1.2; Na2HPO4 1.2; Tris/HC1 15; CaC12 1.5 and glucose 11. Except where stated, L-nitroarginine (L-NA, 100 μM) was also included to inhibit NO synthase. To examine reversal of inhibition by ODQ, aliquots of cells were pre-incubated (10 min) with 0.3 μM ODQ, then diluted 1 : 10 in incubation buffer supplemented by 1% BSA, and then exposed to DEA/NO after various times. Control cells were pre-incubated (10 min) with 0.3 μM or 0.03 μM ODQ, diluted 1 : 10 into BSA-supplemented buffer containing the same concentrations of ODQ, and left for 10 min before applying 1 μM DEA/NO.
Purified sGC (Alexis Biochemicals, Nottingham, U.K.) was diluted 1 : 50 with ice-cold buffer at pH 7.4 containing 10 mM Tris/HC1, 1 mM dithiothreitol and 0.5% BSA, and stored on ice. For experiments, 2 μl of this stock solution was added to 200 μl of reaction buffer containing: Tris/HC1 50 mM, MgC12 3 mM, GTP 1 mM and 0.5% BSA, at pH 7.4 and 37°C. The final concentration of sGC in the reaction mixture was 50 ng ml−1 (∼0.33 nM).
The NO donor diethylamine-NONOate (DEA/NO; Alexis Biochemicals, Nottingham, U.K.) was prepared in 10 mM NaOH, stored on ice, and added at a dilution of 1 : 100 to give final concentrations of 1 μM for cell experiments, and 100 μM for purified sGC. ODQ and YC-1 (both from Tocris Cookson, Bristol, U.K.) were dissolved in DMSO, and added at a 1 : 100 dilution. Solvent alone had no effect on cellular or purified responses to DEA/NO (not shown). L-NA (Tocris Cookson, Bristol, U.K.) was dissolved in equimolar NaOH and added at 1 : 100 dilution. Reduced (ferrous) Hb was prepared according to the method of Martin et al. (1985) and stored at −80°C until use.
NO concentrations were measured using an electrochemical probe (ISO-NO electrode, World Precision Instruments, Herts, U.K.).
Results
Characterization of sGC inhibition by ODQ in cerebellar cells
Following a 10 min pre-incubation with ODQ the cyclic GMP accumulation evoked by 1 μM DEA/NO in cerebellar cells was inhibited in a concentration-dependent way (Figure 1a). The half-maximal inhibitory concentration (IC50) was ∼100 nM, a value comparable with other estimates in cerebellar slices and endothelial cells (Garthwaite et al., 1995; Schrammel et al., 1996). When cells were pre-incubated with 0.3 μM ODQ for 20 min, the maximum cyclic GMP response was reduced by about 70% (Figure 1b), confirming that ODQ was not acting as a simple competitive inhibitor. Nevertheless, the concentration of DEA/NO required to elicit half-maximal activation of sGC (EC50) was increased by ODQ (from ∼0.08 – 0.2 μM), indicating a mixed (partially competitive, partially not) type of inhibition. These results are compatible with the effects of ODQ on purified sGC (Schrammel et al., 1996).
Figure 1.
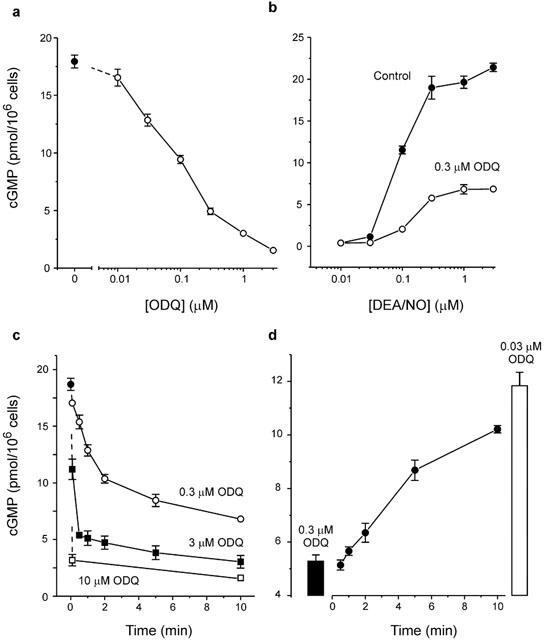
Inhibition of sGC by ODQ in cerebellar cells. (a) Concentration-response curve for ODQ in cells exposed to 1 μM DEA/NO for 2 min. Cells were preincubated with ODQ for 10 min. (b) Concentration-response curve for DEA/NO following 20 min preincubation in the absence (control) and presence of 0.3 μM ODQ. (c) Time courses for onset of inhibition. Cells were preincubated with ODQ at the concentrations and for the times illustrated, before exposure to 1 μM DEA/NO for 2 min. (d) Recovery of cells from ODQ inhibition. Cells were preincubated for 10 min with 0.3 μM ODQ, then diluted 1 in 10 into buffer lacking ODQ. After various times the cells were exposed to 1 μM DEA/NO for 2 min. Recovery was assessed by comparison with the cyclic GMP response in cells treated with 0.3 μM and 0.03 μM ODQ when diluted into buffer containing an equal concentration of ODQ (columns).
The time course for onset of ODQ inhibition was determined by pre-incubating cells with the inhibitor for different periods before applying 1 μM DEA/NO for 2 min and measuring the cellular cyclic GMP response. With 0.3 μM ODQ, the degree of inhibition progressively increased over 10 min but at a 10 fold higher concentration the onset of inhibition was more rapid (Figure 1c). At 10 μM ODQ, ∼90% inhibition was achieved with a pre-incubation of 5 s. The onset of inhibition appeared complex, as two distinct kinetic phases were observed: an initial rapid phase, followed by a more gradual decline. This was most apparent at the higher ODQ concentrations where the rapid initial phase comprised the majority of total inhibition (Figure 1c).
In cerebellar slices and endothelial cultures, inhibition by ODQ is reversible (Garthwaite et al., 1995; Schrammel et al., 1996) whereas, for purified sGC, it is irreversible (Schrammel et al., 1996). To assess the kinetics of recovery from inhibition, cells were pre-incubated (10 min) with 0.3 μM ODQ and then diluted 10 fold. After various periods, 1 μM DEA/NO was applied for 2 min and the time for recovery from inhibition was determined (Figure 1d). Cellular sGC progressively recovered from inhibition over 10 min, with 50% recovery taking 5 min.
Effect of YC-1 on cellular sGC activity
YC-1 was originally described as a NO-independent activator of sGC (Ko et al., 1994). In cerebellar cells incubated without a NO synthase inhibitor, YC-1 (10 min exposure) increased cyclic GMP in a concentration-dependent manner (Figure 2) to reach a level (at 100 μM YC-1) equivalent to the maximum achieved with 1 μM DEA/NO after 2 min. The EC50 value for YC-1 was ∼20 μM.
Figure 2.
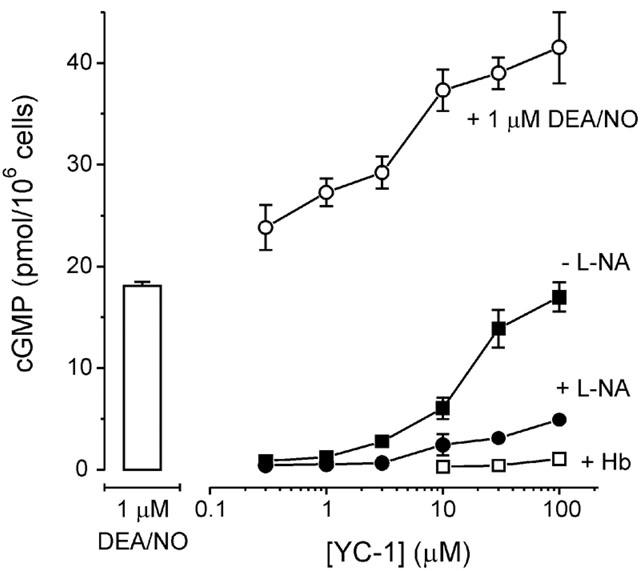
Effects of YC-1 on sGC. YC-1 was applied at various concentrations for 10 min to control (untreated) cells, cells preincubated (10 min) with 100 μM L-NA, and cells preincubated (10 min) with 10 μM Hb, as indicated. Also shown are the cyclic GMP responses of cells that were preincubated with 100 μM L-NA (10 min) and YC-1 (5 s) and then exposed to 1 μM DEA/NO for 2 min. Column indicates response to 1 μM DEA/NO alone.
YC-1 increases the potency of NO for activating sGC (Friebe et al., 1996) and so to examine the contribution of basal NO synthase activity to the cyclic GMP elevation evoked by YC-1, cells were incubated with the inhibitor L-nitroarginine (L-NA; note that L-NA at 100 μM is present in cell incubation buffer for all other experiments). In the presence of 100 μM L-NA, the cyclic GMP response to YC-1 (10 min exposure) was substantially diminished, but a modest increase in cyclic GMP was still observed. Pre-incubation of the cells with the NO scavenger haemoglobin (Hb, 10 μM) decreased the response further, although a statistically significant increase in cellular cyclic GMP levels remained (0.79±0.09 pmol/106 cells at 100 μM concentration; n=3; P<0.001, Student's t-test), this being ∼0.5% of the response evoked by YC-1 in the absence of NO synthase inhibitor and Hb. The cyclic GMP levels of cells that were not exposed to DEA/NO were decreased by 10 min incubation with either 100 μM L-NA (0.28±0.04 pmol/106 cells) or 10 μM Hb (0.27±0.06 pmol/106 cells), relative to control cells (0.47±0.05 pmol/106 cells), indicating tonic NO formation and sGC activation within the suspension.
The ability of YC-1 to enhance cellular sGC activity beyond maximal NO stimulation was tested by adding 1 μM DEA/NO for 2 min after the 10 min pre-incubation with YC-1 (in the presence of L-NA). YC-1 enhanced the DEA/NO-evoked cyclic GMP accumulation concentration-dependently (Figure 2), an effect that levelled off by ∼30 μM when cyclic GMP levels were about 2 fold greater than with DEA/NO alone. The EC50 for this effect was about 3 μM, suggesting that YC-1 was more potent in the presence of a maximally active NO concentration than with the low concentrations produced by the combination of cellular NO synthase and environmental NO (EC50 ∼20 μM; Figure 2).
Effect of YC-1 on sGC deactivation, PDE activity, and inhibition by ODQ
The effect of YC-1 on the rate of NO dissociation from sGC in cells was explored using Hb to deactivate sGC (Bellamy & Garthwaite, 2001a), 5 s after stimulation with DEA/NO (Figure 3a). Under control conditions, the cyclic GMP accumulation effectively ceased immediately and thereafter the levels declined to basal values. In contrast, when cells were pre-incubated with YC-1, Hb no longer caused a sudden cessation of cyclic GMP synthesis. Instead, cyclic GMP levels continued to rise, diverging from the accumulation in the absence of Hb over a period of 20 to 30 s, before levelling off. The rate of sGC deactivation was therefore substantially decreased by YC-1 treatment. To assess this decrease quantitatively, the data following Hb addition were fitted with an exponential function. This assumes that the influence of PDE activity on cyclic GMP levels is negligible over the relevant time period and so the time taken for sGC deactivation corresponds to the time taken for cyclic GMP accumulation to level off (the validity of this assumption is explored below). Deactivation of sGC occurred with a time constant (τ) of 11.7 s, 45 fold slower than deactivation of sGC in the absence of YC-1 (τ=0.27 s; Bellamy & Garthwaite, 2001b).
Figure 3.
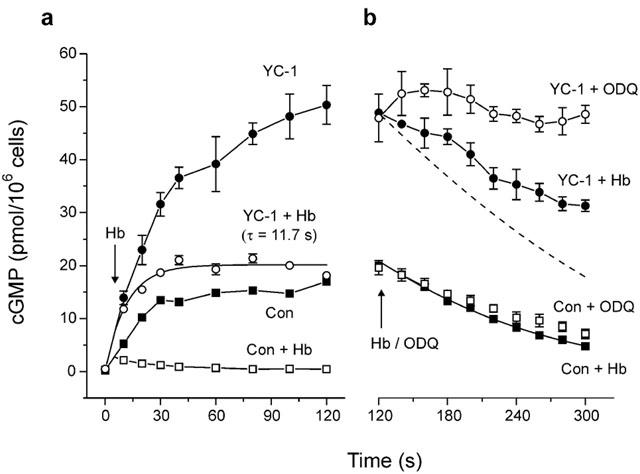
Effect of YC-1 on the rate of deactivation of sGC. (a) Cells were exposed to 1 μM DEA/NO at t=0, in the absence (control) or presence of 100 μM YC-1 (5 s preincubation) as indicated. After 5 s (arrow) 10 μM Hb was added to effect deactivation of sGC. The decline in cyclic GMP accumulation in YC-1-treated cells is fitted with an exponential. (b) Addition of Hb or ODQ (arrow) to control and YC-1-treated cells after 2 min DEA/NO exposure. The continuous line is a fit to control data with the integrated Michaelis-Menten equation. Broken line is the predicted decline in cyclic GMP in YC-1 treated cells given the Michaelis-Menten parameters determined for control cells.
YC-1 has been shown to inhibit PDEs in both platelets and aorta (Galle et al., 1999). The strategy previously adopted for monitoring PDE activity in cerebellar cells quantitatively was to follow the decline in cyclic GMP levels after sudden deactivation of sGC and fit the curve with the integrated form of the Michaelis-Menten equation (Bellamy & Garthwaite, 2001a). In the presence of YC-1, however, the decreased rate of deactivation would mean that, following addition of Hb, a lag would precede the onset of the decline in cyclic GMP. Accordingly, addition of Hb to cells treated with YC-1 and exposed for 2 min to DEA/NO showed a disruption in the profile of cyclic GMP decline (Figure 3b). Nevertheless, after sufficient time for sGC to become completely deactivated had elapsed (1 min), the fall of cyclic GMP was less rapid than predicted from the behaviour of control cells (Figure 3b, dashed line), indicating that PDE was partially inhibited by YC-1. Moreover, this experiment showed that the PDE activity in the presence of YC-1 was sufficiently slow not to have interfered significantly with the measurement of sGC deactivation (see above).
An alternative strategy for monitoring PDE activity is to inhibit sGC directly with ODQ and then follow the decline in cyclic GMP levels. In normal cells, this approach gives essentially the same result as applying Hb (Bellamy et al., 2000; Figure 3b). When ODQ was applied to YC-1 treated cells, however, cyclic GMP levels did not decline but remained stably elevated during the following 3 min (Figure 3b). As ODQ did not significantly inhibit PDE activity, this result indicates that YC-1 prevented ODQ from inhibiting sGC over the period examined.
Effects of YC-1 at steady-state
When exposed to a sustained supramaximal concentration of NO, cerebellar cells respond with a high-amplitude cyclic GMP accumulation that reaches a steady-state plateau (Garthwaite & Garthwaite, 1987). It was concluded from earlier studies (Bellamy et al., 2000; Bellamy & Garthwaite, 2001b) that this profile is determined largely by the desensitizing profile of sGC such that, at steady-state, the rate of cyclic GMP synthesis by sGC has fallen to about 10% of the peak and is matched by the rate of cyclic GMP hydrolysis by PDEs.
The aim of the next experiment was to examine the influence of YC-1 on sGC after steady-state had been established, allowing examination of its effect once sGC had desensitized. In light of the ability of pre-exposure to YC-1 to influence the inhibition by ODQ (see above), the converse effect of YC-1 on cells pre-exposed to ODQ was also examined.
Three populations of cells were treated to attain steady-state cyclic GMP concentrations: control cells, cells pre-treated (∼5 s) with 10 μM ODQ, and cells pre-treated (∼5 s) with 100 μM YC-1. All were stimulated with 1 μM DEA/NO for 2 min and then another 1 μM DEA/NO was added to maintain NO at supramaximal levels for a further 3 min (Figure 4, solid symbols). The peak NO concentration resulting from this second addition was about 2 fold higher than from the first (Figure 4 inset), reflecting the fact that the cells' capacity to consume NO can be overwhelmed (Griffiths & Garthwaite, 2001). Steady-state cyclic GMP levels were maintained in all cases (i.e. control, ODQ and YC-1 pre-treated cells), as expected from desensitization of sGC (Bellamy et al., 2000).
Figure 4.
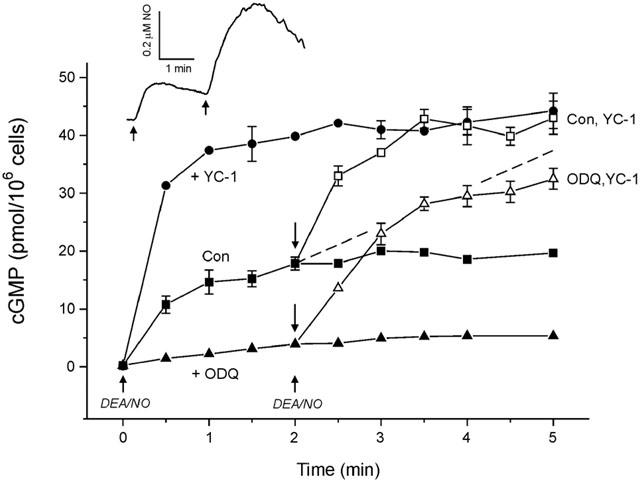
Effect of YC-1 at steady-state cyclic GMP levels. Steady-state cyclic GMP levels were achieved in untreated (control), YC-1 pre-treated (100 μM, 5 s), and ODQ pre-treated (10 μM, 5 s) cells by addition of DEA/NO (ascending arrows) at 0 and 2 min. Inset shows the corresponding NO concentration profile. At steady-state, 100 μM YC-1 was added (at descending arrows) to control and ODQ-treated cells. Dashed line indicates a 2 fold enhancement of the 10% of sGC that is non-desensitized at steady-state, assuming zero PDE activity and no further desensitization (i.e. the maximum possible rate of cyclic GMP synthesis predicted for a simple mechanism of YC-1 action; see Discussion).
Once these steady-state conditions had been established, 100 μM YC-1 was added to control and ODQ-treated cells simultaneously with the second application of DEA/NO at 2 min and cyclic GMP levels monitored for a further 3 min (Figure 4). In the control cells, YC-1 caused a rapid increase in cyclic GMP accumulation. The initial rate was much greater than predicted by just a 2 fold enhancement of active sGC when 90% is desensitized (Figure 4, dashed line). After ∼1.5 min the cyclic GMP level settled at a level equivalent to the plateau reached when YC-1 was added before DEA/NO, meaning that potentiation of sGC by YC-1 after a steady-state had been established resulted in a final equilibrium position equivalent to that obtained when YC-1 was added before DEA/NO.
Addition of YC-1 at steady-state to cells preincubated with ODQ resulted in sGC inhibition being rapidly counteracted and, over the next 3 min, cyclic GMP levels rose beyond the steady-state level evoked by DEA/NO alone, but levelled off below the plateau reached with YC-1 in the absence of ODQ (Figure 4).
Interplay between YC-1 and ODQ on purified sGC
To test if YC-1 both blocked (Figure 3b) and reversed (Figure 4) sGC inhibition by ODQ in cells by acting at the level of sGC itself, experiments were carried out using the purified enzyme. Pre-incubation of purified sGC with 100 μM YC-1 caused a 31% increase in the mean rate of cyclic GMP synthesis (Figure 5a), consistent with other reports (Friebe et al., 1996; Denninger et al., 2000). Addition of 1 μM ODQ to the YC-1-treated enzyme 2 min after 100 μM DEA/NO did not influence the rate of cyclic GMP accumulation over the subsequent 4 – 5 min but, thereafter, sGC activity decreased (Figure 5a). Pre-incubation (30 s) of sGC with 1 μM ODQ caused a 35% decrease in sGC activity (Figure 5b). Addition of YC-1 after 2 min exposure to DEA/NO caused a sudden increase in the rate of cyclic GMP synthesis. The increase was greater than that predicted by a simple 31% enhancement of uninhibited sGC activity by YC-1 (Figure 5b dashed line) and approached that of maximal (YC-1 enhanced) sGC activity (Figure 5a), indicating that the inhibition due to ODQ had been counteracted.
Figure 5.
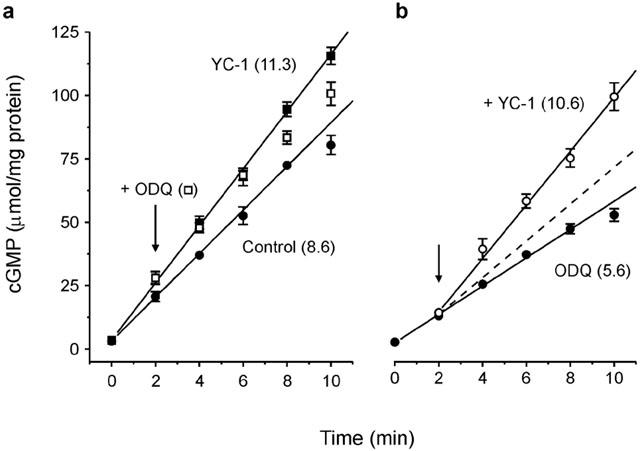
Interaction between YC-1 and ODQ on purified sGC. (a) Time course of the activity of purified sGC (50 ng ml−1) exposed to 100 μM DEA/NO in the absence (control) and presence of 100 μM YC-1, and following addition (arrow) of ODQ (1 μM) in the presence of YC-1. (b) Time course of sGC activity in the presence of 1 μM ODQ with or without addition of 100 μM YC-1 (arrow). The broken line represents the slope predicted if the degree of potentiation was the same as that found for uninhibited sGC. Numbers in brackets are rates in μmol cyclic GMP.mg sGC−1.min−1.
Discussion
Mechanism and kinetics of ODQ inhibition in cells
The mechanism by which ODQ inhibits sGC in its purified form has been examined in some detail (Garthwaite et al., 1995; Schrammel et al., 1996). ODQ binds to the sGC haem group, presumably competitively (explaining the lowering of the EC50 for DEA/NO), and thereafter causes oxidation of the haem iron to the ferric (Fe3+) form. Ferric sGC has low affinity for NO (Stone et al., 1996) and so NO activation is prevented (not competitive and irreversible in the absence of reductants). These findings fit the experimental observations in cells. Inhibition by ODQ caused a modest increase in the EC50 of DEA/NO and, more obviously, reduced the maximal response (Figure 1b). At 0.3 μM ODQ, the kinetics of onset of inhibition was comparable with that found with the same ODQ concentration on the purified enzyme (Schrammel et al., 1996) but it appeared biphasic at higher ODQ concentrations (Figure 1c). This may reflect a more complex interaction of ODQ with sGC in cells. Inhibition by ODQ in the cells reversed with a half time of around 5 min (Figure 1d), so the complexity may arise from the action of cellular reductants. If the cellular capacity to re-reduce sGC did not follow simple first order kinetics (e.g. if the mechanism was inducible), complex kinetics of inhibition would be observed. Whether recovery from inhibition is due to a low molecular weight reductant (e.g. NAD(P)H) or an enzyme analogous to methaemoglobin reductase, remains to be determined.
Effect of YC-1 on sGC in cerebellar cells
YC-1 caused a 45 fold decrease in the rate of sGC deactivation following removal of free NO with Hb (Figure 2), consistent with qualitative observations made using the purified enzyme (Friebe & Koesling, 1998). This finding implies an increase in the potency of NO as an activator of sGC, which explains the origin of the cyclic GMP response evoked by YC-1 in the absence of added NO, as inhibition of basal NO synthase and NO scavenging with Hb together eliminated >99% of the YC-1 evoked cyclic GMP accumulation. An effect of Hb beyond that produced by NO synthase inhibition implies that part of the cyclic GMP response to YC-1 was brought about by a potentiation of sGC activation by environmental NO. The cyclic GMP increase remaining in the presence of Hb may reflect NO-independent activation of sGC by YC-1, but could also be explained by inhibition of PDE activity. On the basis of these data, therefore, there is no reason to suspect that YC-1 is able to activate sGC appreciably in the absence of NO. These conclusions are at odds with the description of YC-1 as a NO-independent activator of sGC (Ko et al., 1994) but are in close agreement with those of a recent study in endothelial cells (Schmidt et al., 2001).
YC-1 increased the maximum NO-evoked cyclic GMP response by about 2 fold, a degree greater than its potentiation of purified sGC activity (1.3 fold). A possible mechanism for the greater potentiation of cyclic GMP responses in cells is inhibition of PDEs. YC-1 did cause a decrease in PDE activity in cerebellar cells, although the extent of inhibition could not be evaluated quantitatively (Figure 3b). The PDE activity is low in cerebellar cells in comparison to many other tissues, however, and so contributes little to the profile of cyclic GMP accumulation, which is instead largely determined by sGC activity (Bellamy et al., 2000). The enhancement is thus mainly attributable to increased cellular sGC activity. That this is greater than for purified sGC has been noted in other cell types (Schmidt et al., 2001; Friebe et al., 1998). A possible explanation has emerged from the finding that cell extracts can enhance the potentiation of purified sGC by YC-1 (Schmidt et al., 2001). The evidence from cerebellar cells supports the notion of a cellular modulatory factor that enhances the effect of YC-1.
Effect of YC-1 on desensitized sGC
We have hypothesized that the fall in sGC activity that occurs with time of exposure to NO in cells is due to the receptor progressively adopting a distinct desensitized form such that, at steady-state, there is a distribution of the total receptor population into (at least) four distinct states: unbound, bound-but-inactive, active, and desensitized (Figure 6; Bellamy & Garthwaite, 2001b). This is analogous to models for classical receptors (Colquhoun & Hawkes, 1995). The relative distribution of sGC between these states would thus determine the rate of cyclic GMP synthesis and, at steady-state, about 90% is desensitized. The large and rapid rise in cyclic GMP that occurred when YC-1 was added to the cells in which a steady-state had been pre-established, however, was disproportionately high for simple enhancement of 10% active sGC by the 2 fold maximally achievable (Figure 4), indicating additional actions, possibly on the activation step (discussed below).
Figure 6.
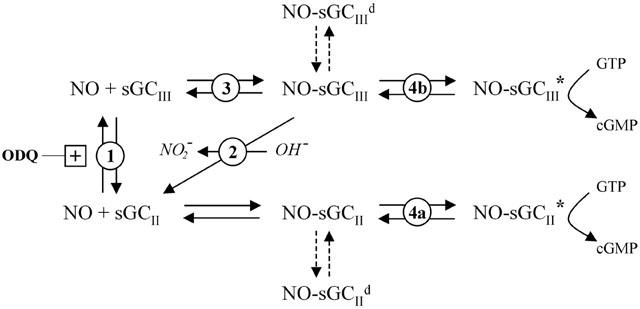
Model for the interaction of the NO receptor, sGC, with agonist (NO) and pharmacological modulators (ODQ and YC-1). The NO receptor exists in equilibrium between free (NO+sGC), bound (NO-sGC), active (NO-sGC*), and desensitized states (NO-sGCd), and also between ferrous (sGCII) and ferric (sGCIII) states. The inhibitor ODQ oxidizes ferrous sGC to the ferric form. YC-1 has multiple effects on sGC function, illustrated for simplicity as modulation of the transitional steps between states (as numbered; see Discussion for details). Dashed lines illustrate hypothesized routes to the desensitized form.
From studies on human platelets treated with PDE inhibitors, Mullershausen et al. (2001) have speculated that the time-dependent decline in sGC activity results, not from receptor desensitization itself, but from depletion of substrate, GTP. This is unlikely to contribute significantly to the desensitization of sGC in the cerebellar cells. As we have shown (Bellamy et al., 2000), the desensitization profile is unchanged despite 10 fold variations in cyclic GMP accumulation, corresponding to a similar range of rates of GTP utilization by sGC. Moreover, to cause a 90% decrease in the rate of cyclic GMP synthesis (see Bellamy et al., 2000), cellular GTP would have to fall to around 3 μM (given a Km of 30 μM; Lewicki et al., 1982; Denninger et al., 2000). If this were to occur, there would not be sufficient GTP available for YC-1 to subsequently evoke a doubling of the steady-state cyclic GMP level (which is ∼800 μM under control conditions). For GTP limitation to account for desensitization, the Km of cellular sGC would have to be substantially higher than that of purified sGC. While this is possible, it means that the factor that results in a shift of the Km in cells is, in effect, the proposed ‘desensitization factor' (Bellamy et al., 2000).
YC-1 counteracts ODQ inhibition
That YC-1 can both reverse and prevent (or retard) inhibition of sGC by ODQ in the cerebellar cells was unexpected and indicates an interaction in the mechanism of action of the two compounds. Importantly, similar findings were made using purified sGC (Figure 5), indicating that the effect is exerted on the enzyme itself. ODQ acts at the sGC haem site and so the implication is that YC-1 interacts with sGC haem at some level. If YC-1 promoted a reduction of the sGC haem oxidized by ODQ back to the ferrous (Fe2+) form (Figure 6, site 1), or was able to accelerate reduction of the NO-bound ferric (Fe3+) form (Zhao et al., 2000; Figure 6, site 2), then a reduced effectiveness of ODQ would be the outcome. Alternatively, oxidized sGC could, in principle, form an active enzyme (Makino et al., 1999) and so it is possible that YC-1 increases the affinity of oxidized sGC for NO (Figure 6; site 3) and/or increases the efficacy of this pathway (Figure 6; site 4b). Should a significant amount of oxidized sGC normally exist in cells, any of these three mechanisms could also explain the increased maximal activation by NO in the presence of YC-1 (i.e. a normally inert pool of enzyme would possess activity). These alternatives could be tested directly by monitoring sGC haem absorbance to distinguish ferric and ferrous iron. Unfortunately, with the limited amounts of purified enzyme available, we could not do this experiment, but YC-1 (100 μM) was unable to directly reduce another ferric haemoprotein, methaemoglobin (authors' unpublished observation), suggesting that haem reduction is not a general feature of YC-1 action.
The ability of YC-1 to counteract ODQ inhibition was not complete at the concentrations tested. In cells, the steady-state cyclic GMP response to NO that followed addition of YC-1 to ODQ-inhibited cells was less than the response of cells untreated with ODQ (Figure 4). Similarly, inhibition appeared to set in 4 – 6 min after YC-1-treated purified sGC was exposed to ODQ (Figure 5). These findings suggest that YC-1 and ODQ have opposing actions that depend on their relative concentrations and exposure times. This would be consistent with YC-1 facilitating conversion of ferric sGC to the ferrous form (Figure 6; site 1 or 2), rather than sensitizing ferric sGC to NO (Figure 6; site 3), as the latter mechanism should be independent of ODQ concentration or time of exposure. This notion is in keeping with several published studies where ODQ has been used to inhibit sGC ‘activation' by YC-1. In these cases, the potency of ODQ appears compromised relative to its effect on purified sGC lacking YC-1 (Seitz et al., 1999; Feelisch et al., 1999) and complete inhibition is apparently not possible (Martin et al., 2001).
Additional sites of action of YC-1
From the possible ways discussed above by which YC-1 might affect inhibition by ODQ it would not be predicted that YC-1 would reverse sGC desensitization or inhibit sGC deactivation. It would also not be predicted that YC-1 would become more potent with increasing NO concentration (Figure 3), an observation that agrees with data for purified sGC (Hoenicka et al., 1999). Hence, additional mechanisms need to be invoked. An explanation for the effect of YC-1 on sGC desensitization would be that the compound disrupts the equilibrium between sGC in the active and desensitized states at steady-state (Figure 6, site 4a,b), such that more active sGC is formed on addition of YC-1. The formation of a new cyclic GMP steady-state at a higher concentration would then imply that cells can compensate for the effect of YC-1 by increasing the degree of sGC desensitization. We have recently found a correlation between the degree of sGC desensitization and the cellular cyclic GMP level (V. Wykes et al., manuscript submitted). That the disruption of desensitization by YC-1 could be compensated for at a higher cyclic GMP level is consistent with the idea that cyclic GMP influences the degree of desensitization. An action of YC-1 at this step would also predict a decreased rate of deactivation on removal of NO and, if YC-1 bound preferentially to the liganded protein, a potentiation of YC-1 activity with increasing NO concentration. Moreover, this hypothesis could be consistent with a potentiation of maximal sGC activity by YC-1, but only if NO is an agonist of low efficacy. These possibilities could be addressed by determining the kinetics and equilibrium constants for sGC activation by NO, although preliminary attempts to achieve this aim have proven equivocal (Bellamy et al., 2002).
Conclusions
Despite the fact that sGC has long been recognized as a major receptor for NO, a pharmacology of the protein has begun to develop only recently. The inhibitor ODQ appears to act similarly in cells and on the purified enzyme except that, in cells, the inhibition is reversible, a property that may contribute to the complex kinetics of the onset of inhibition. The actions of YC-1 on cellular sGC are also broadly consistent with those on the purified enzyme, including its ability to influence the inhibitory action of ODQ, but YC-1 also transiently reverses sGC desensitization, a phenomenon so far only observed in cells. The properties of YC-1 cannot be explained on the basis of any simple interaction with the receptor-enzyme and so a combination of effects is implied. This may either reflect a YC-1 binding site that is able to influence both the haem region and regions involved in protein isomerization (i.e. activation) or, more likely, from multiple binding sites. The further analysis of the mechanism of action of these tools will be valuable for understanding the molecular events governing the interactions between NO and sGC.
Acknowledgments
This work was supported by a Wellcome Trust Programme Grant and a Medical Research Council Studentship.
Abbreviations
- DEA/NO
diethylamine-NONOate
- Hb
haemoglobin
- L-NA
NG-nitro-L-arginine
- ODQ
1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-l-one
- PDE
phosphodiesterase
- sGC
soluble guanylyl cyclase
- YC-1
3-(5-hydroxymethyl-2-furyl)-1-benzylindazole
References
- BELLAMY T.C., GARTHWAITE J. ‘cAMP-specific' phosphodiesterase contributes to cGMP degradation in cerebellar cells exposed to nitric oxide. Mol. Pharmacol. 2001a;59:54–61. doi: 10.1124/mol.59.1.54. [DOI] [PubMed] [Google Scholar]
- BELLAMY T.C., GARTHWAITE J. Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells. J. Biol. Chem. 2001b;276:4287–4292. doi: 10.1074/jbc.M006677200. [DOI] [PubMed] [Google Scholar]
- BELLAMY T.C., WOOD J., GARTHWAITE J. On the activation of soluble guanylyl cyclase by nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 2002;99:507–510. doi: 10.1073/pnas.012368499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELLAMY T.C., WOOD J., GOODWIN D.A., GARTHWAITE J. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2928–2933. doi: 10.1073/pnas.97.6.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLQUHOUN D., HAWKES A.G.The principles of the stochastic interpretation of ion-channel mechanisms Single-Channel Recording 1995New York: Plenum Press; 397–482.ed. B. Sakmann & E. Neher. pp [Google Scholar]
- DENNINGER J.W., SCHELVIS J.P., BRANDISH P.E., ZHAO Y., BABCOCK G.T., MARLETTA M.A. Interaction of soluble guanylate cyclase with YC-1: kinetic and resonance raman studies. Biochemistry. 2000;39:4191–4198. doi: 10.1021/bi992332q. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., KOTSONIS P., SIEBE J., CLEMENT B., SCHMIDT H.H. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol. Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- FRANCIS S.H., CORBIN J.D. Progress in understanding the mechanism and function of cyclic GMP-dependent protein kinase. Adv. Pharmacol. 1994;26:115–170. doi: 10.1016/s1054-3589(08)60053-8. [DOI] [PubMed] [Google Scholar]
- FRIEBE A., KOESLING D. Mechanism of YC-1-induced activation of soluble guanylyl cyclase. Mol. Pharmacol. 1998;53:123–127. doi: 10.1124/mol.53.1.123. [DOI] [PubMed] [Google Scholar]
- FRIEBE A., MULLERSHAUSEN F., SMOLENSKI A., WALTER U., SCHULTZ G., KOESLING D. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol. Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- FRIEBE A., RUSSWURM M., MERGIA E., KOESLING D. A point-mutated guanylyl cyclase with features of the YC-1-stimulated enzyme: implications for the YC-1 binding site. Biochemistry. 1999;38:15253–15257. doi: 10.1021/bi9908944. [DOI] [PubMed] [Google Scholar]
- FRIEBE A., SCHULTZ G., KOESLING D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- GALLE J., ZABEL U., HUBNER U., HATZELMANN A., WAGNER B., WANNER C., SCHMIDT H.H. Effects of the soluble guanylyl cyclase activator, YC-1, on vascular tone, cyclic GMP levels and phosphodiesterase activity. Br. J. Pharmacol. 1999;127:195–203. doi: 10.1038/sj.bjp.0702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARTHWAITE J., GARTHWAITE G. Cellular origins of cyclic GMP responses to excitatory amino acid receptor agonists in rat cerebellum in vitro. J. Neurochem. 1987;48:29–39. doi: 10.1111/j.1471-4159.1987.tb13123.x. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- GRIFFITHS C., GARTHWAITE J. The shaping of nitric oxide signals by a cellular sink. J. Physiol. 2001;536:855–862. doi: 10.1111/j.1469-7793.2001.00855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOENICKA M., BECKER E.M., APELER H., SIRICHOKE T., SCHRODER H., GERZER R., STASCH J.P. Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: stimulation by YC-1, nitric oxide, and carbon monoxide. J. Mol. Med. 1999;77:14–23. doi: 10.1007/s001090050292. [DOI] [PubMed] [Google Scholar]
- JONES M.V., WESTBROOK G.L. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- JUILFS D.M., SODERLING S., BURNS F., BEAVO J.A. Cyclic GMP as substrate and regulator of cyclic nucleotide phosphodiesterases (PDEs) Rev. Physiol. Biochem. Pharmacol. 1999;135:67–104. doi: 10.1007/BFb0033670. [DOI] [PubMed] [Google Scholar]
- KO F.N., WU C.C., KUO S.C., LEE F.Y., TENG C.M. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- LEWICKI J.A., BRANDWEIN H.J., MITTAL C.K., ARNOLD W.P., MURAD F. Properties of purified soluble guanylate cyclase activated by nitric oxide and sodium nitroprusside. J. Cyclic Nucleotide Res. 1982;8:17–25. [PubMed] [Google Scholar]
- MAKINO R., MATSUDA H., OBAYASHI E., SHIRO Y., IIZUKA T., HORI H. EPR characterization of axial bond in metal center of native and cobalt-substituted guanylate cyclase. J. Biol. Chem. 1999;274:7714–7723. doi: 10.1074/jbc.274.12.7714. [DOI] [PubMed] [Google Scholar]
- MARTIN E., LEE Y.C., MURAD F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12938–12942. doi: 10.1073/pnas.231486198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN W., VILLANI G.M., JOTHIANANDAN D., FURCHGOTT R.F. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J. Pharmacol. Exp. Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- MULLERSHAUSEN F., RUSSWURM M., THOMPSON W.J., LIU L., KOESLING D., FRIEBE A. Rapid nitric oxide-induced desensitization of the cGMP response is caused by increased activity of phosphodiesterase type 5 paralleled by phosphorylation of the enzyme. J. Cell Biol. 2001;155:271–278. doi: 10.1083/jcb.200107001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMIDT K., SCHRAMMEL A., KOESLING D., MAYER B. Molecular mechanisms involved in the synergistic activation of soluble guanylyl cyclase by YC-1 and nitric oxide in endothelial cells. Mol. Pharmacol. 2001;59:220–224. doi: 10.1124/mol.59.2.220. [DOI] [PubMed] [Google Scholar]
- SCHRAMMEL A., BEHRENDS S., SCHMIDT K., KOESLING D., MAYER B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol. Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- SEITZ S., WEGENER J.W., RUPP J., WATANABE M., JOST A., GERHARD R., SHAINBERG A., OCHI R., NAWRATH H. Involvement of K+ channels in the relaxant effects of YC-1 in vascular smooth muscle. Eur. J. Pharmacol. 1999;382:11–18. doi: 10.1016/s0014-2999(99)00574-9. [DOI] [PubMed] [Google Scholar]
- STASCH J.P., BECKER E.M., ALONSO-ALIJA C., APELER H., DEMBOWSKY K., FEURER A., GERZER R., MINUTH T., PERZBORN E., PLEISS U., SCHRODER H., SCHROEDER W., STAHL E., STEINKE W., STRAUB A., SCHRAMM M. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- STONE J.R., SANDS R.H., DUNHAM W.R., MARLETTA M.A. Spectral and ligand-binding properties of an unusual hemoprotein, the ferric form of soluble guanylate cyclase. Biochemistry. 1996;35:3258–3262. doi: 10.1021/bi952386+. [DOI] [PubMed] [Google Scholar]
- ZAGOTTA W.N., SIEGELBAUM S.A. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- ZHAO Y., BRANDISH P.E., DIVALENTIN M., SCHELVIS J.P., BABCOCK G.T., MARLETTA M.A. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]