

Abstract
Constitutive overexpression and activation of NPM-ALK fusion protein [t(2:5)(p23;q35)] is a key oncogenic event that drives the survival and proliferation of anaplastic large-cell lymphomas (ALCLs). We have identified a highly potent and selective small-molecule ALK inhibitor, NVP-TAE684, which blocked the growth of ALCL-derived and ALK-dependent cell lines with IC50 values between 2 and 10 nM. NVP-TAE684 treatment resulted in a rapid and sustained inhibition of phosphorylation of NPM-ALK and its downstream effectors and subsequent induction of apoptosis and cell cycle arrest. In vivo, NVP-TAE684 suppressed lymphomagenesis in two independent models of ALK-positive ALCL and induced regression of established Karpas-299 lymphomas. NVP-TAE684 also induced down-regulation of CD30 expression, suggesting that CD30 may be used as a biomarker of therapeutic NPM-ALK kinase activity inhibition.
Keywords: anaplastic large-cell lymphoma, CD30, kinase inhibitor
Anaplastic large-cell lymphomas (ALCLs) are a subtype of the high-grade non-Hodgkin's family of lymphomas with distinct morphology, immunophenotype, and prognosis (1, 2). ALCLs are postulated to arise from T cells and, in rare cases, can also exhibit a B cell phenotype. In addition, there are ≈40% of cases for which the cell of origin remains unknown and that are classified as “null.” First described as a histological entity by Stein et al. (3, 4) based on the expression of CD30 (Ki-1), ALCL presents as a systemic disease afflicting skin, bone, soft tissues, and other organs, with or without the involvement of lymph nodes. ALCL can be subdivided into at least two subtypes, characterized by the presence or absence of chromosomal rearrangements between the anaplastic lymphoma kinase (ALK) gene locus and various fusion partners such as nucleophosmin (NPM) (5). Approximately 50–60% of cases of ALCL are associated with the t(2;5;)(p23;q35) chromosomal translocation, which generates a hybrid gene consisting of the intracellular domain of the ALK tyrosine kinase receptor juxtaposed with NPM. The resulting fusion protein, NPM-ALK has constitutive tyrosine kinase activity and has been shown to transform various hematopoietic cell types in vitro and support tumor formation in vivo (1, 6). Other less frequent ALK fusion partners, e.g., tropomyosin-3 and clathrin heavy chain, have also been identified in ALCL as well as in CD30-negative diffuse large-cell lymphoma (1, 2, 7, 8). Despite subtle differences in signaling and some biological functions, all fusions appear to be transforming to fibroblasts and hematopoietic cells (9). ALK fusion proteins have also been detected in a rare form of malignancy called inflammatory myofibroblastic tumor (10). Extensive analysis of the leukemogenic potential of NPM-ALK in animal models has further corroborated the importance of NPM-ALK and other ALK rearrangements in the development of ALK-positive ALCL and other diseases (11).
In this work, we developed a cell proliferation assay using NPM-ALK-transformed murine pre-B cell line (Ba/F3) to identify a highly potent and selective inhibitor of ALK kinase activity, NVP-TAE684. TAE684 blocked proliferation and survival of Ba/F3 NPM-ALK, SU-DHL-1 and Karpas-299 cells with 50% inhibitory concentrations (IC50) between 2 and 5 nM. This inhibition was accompanied by a rapid and sustained reduction in ALK autophosphorylation, inactivation of NPM-ALK downstream signaling proteins, and the down-regulation of CD30 expression, a hallmark of ALCL. Finally, TAE684 inhibited lymphomagenesis in vivo in two independent models of ALK-positive ALCL.
Results
TAE684 Selectively Inhibits Proliferation of NPM-ALK Fusion Kinase-Dependent Cell Lines in Vitro.
To identify a selective small-molecule kinase inhibitor of ALK, a cellular screen was used to search for compounds that were selectively cytotoxic to Ba/F3 NPM-ALK, but not to nontransformed parental Ba/F3 cells. This effort led to the identification of TAE684, a 5-chloro-2,4-diaminophenylpyrimidine (Fig. 1A) from a kinase-directed small-molecule library assembled from several different medicinal chemistry programs. TAE684 inhibited the proliferation of Ba/F3 NPM-ALK cells with an IC50 of 3 nM, without affecting the survival of parental Ba/F3 cells at concentrations up to 1 μM [see supporting information (SI) Fig. 7].
Fig. 1.

Effects of TAE684 on NPM-ALK-dependent cell proliferation in vitro. (A) Structure of TAE684. (B) TAE684 inhibits cell proliferation of Karpas-299 and SU-DHL-1 cell lines. Cell proliferation was assayed by using the Bright-Glo Luciferase Assay System after 72 h of treatment with serial dilutions of TAE684. Obtained relative luminescence values were normalized to values from corresponding DMSO-treated wells and displayed as percent survival ± SE. (C) TAE684 blocks NPM-ALK autophosphorylation in Karpas-299 and Ba/F3 NPM-ALK cells after 4 h of treatment. (D) Effect of TAE684 on InsR signaling in H-4-II-E rat hepatoma cells. H-4-II-E cells were preincubated for 30 min with TAE684 at concentrations indicated before stimulation with recombinant insulin. Treatment of Ba/F3 NPM-ALK cells with TAE684 was performed in parallel. IC50 concentrations for InsR and ALK signaling inhibition were determined through the quantification of bands by using Bio-Rad's QuantityOne software package.
Next, we assessed the potency of TAE684 against established human ALCL cell lines expressing NPM-ALK. TAE684 inhibited proliferation of Karpas-299 and SU-DHL-1 cell lines with an IC50 range of 2–5 nM (Fig. 1B). Growth inhibition of NPM-ALK-dependent cell lines correlated with a dose-dependent reduction of NPM-ALK (Y664; equivalent to Y1604 of full-length ALK) autophosphorylation in both Karpas-299 and SU-DHL-1 cells (Fig. 1C and data not shown) as well as Ba/F3 NPM-ALK cells (Fig. 1 C and D). A significant reduction of ALK phosphorylation was observed with an IC50 lower than 10 nM after treatment of cells with the inhibitor for 4 h.
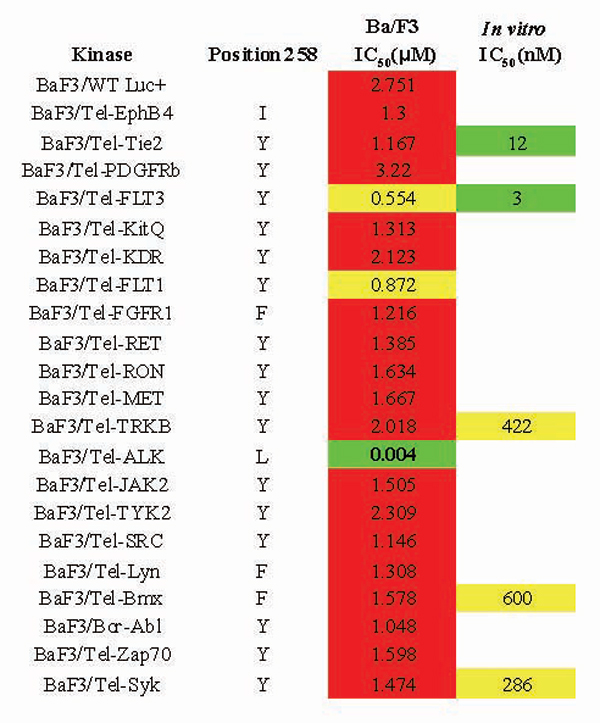
To further evaluate the selectivity of TAE684, we tested the compound against a panel of 35 Ba/F3 cells transformed by various tyrosine kinases constitutively activated by fusion to TEL (12). As shown in SI Fig. 7, the inhibitory activity of TAE684 is highly selective for ALK-driven cell proliferation, requiring a 100- to 1,000-fold higher concentration to inhibit other tyrosine kinases included in the panel. IC50 values between 0.5 and 3 μM were observed for the various cell lines tested.
TAE684 Is Selective for ALK over InsR.
ALK shares high sequence homology with the insulin receptor kinase (InsR) and the insulin-like growth factor receptor (IGF1R). To evaluate the potential of TAE684 to inhibit InsR kinase activity and signaling, the activity of TAE684 was assessed against both recombinant InsR enzyme and full-length InsR in a cellular assay. Indeed, when TAE684 was tested against recombinant InsR in an in vitro kinase assay an IC50 of ≈10–20 nM was obtained in various independent experiments. Similar results where obtained for IGF1R. To assess the potency of TAE684 against InsR in a cellular assay, H-4-II-E rat hepatoma cells were stimulated with purified bovine insulin after preincubation of cells with either DMSO or increasing concentrations of TAE684. As shown in Fig. 1D, stimulation of H-4-II-E cells with insulin led to a several-fold increase in phosphorylation of InsR (Y1146) as well as of both Akt (S473) and FKHR (T24), two key downstream molecules of InsR signal transduction. In marked contrast to the enzymatic data, a concentration of >1 μM TAE684 was required to block insulin-induced phosphorylation of InsR, Akt, and FKHR, which is ≈100-fold higher than the concentration required to inhibit cellular NPM-ALK activity (Fig. 1D). The IC50 for blocking InsR phosphorylation was determined to be 1.2 μM, based on protein band intensity. IC50 data for reduction of Akt and FKHR phosphorylation could not be determined because of insufficient curve fitting but were between 1.1 and 3.3 μM. This discrepancy between the cellular and in vitro biochemical assay is reminiscent of data recently published by Garcia-Echeverria et al. (13), demonstrating selectivity of a small-molecule inhibitor of IGF1R, NVP-AEW564, over InsR in cellular assays, but not in biochemical assays. To examine whether this phenomenon was observed for more recombinant kinases in addition to InsR, we determined the IC50 of TAE684 against a variety of other kinases in biochemical assays. As shown in SI Fig. 7, IC50 values as low as 3 and 12 nM were found for Flt3 and Tie2, respectively, in biochemical assays. As was observed for InsR, the cellular potency of TAE684 against Ba/F3 Tel-Flt3 (IC50 = 554 nM) and Ba/F3 Tel-Tie2 (IC50 >1 μM) were much higher than those observed in biochemical assays.
These results indicate that, at least in cellular systems at its therapeutic IC50, TAE684 is a potent and selective NPM-ALK kinase inhibitor, without exhibiting significant cross-reactivity against other kinases tested in this study, including the highly homologous InsR.
Structural Basis for the Selectivity of TAE684.
Inhibitors that bind to the “DFG-out” conformation of kinases, by filling a hydrophobic cavity adjacent to the ATP-binding site, may more readily achieve higher kinase selectivity than compounds that simply bind to the ATP pocket (14, 15). Access to this hydrophobic pocket seems to be regulated by multiple factors including the identity of the “gatekeeper” amino acid, amino acid sequence upstream of the activation loop preceding the highly conserved DFG motive, and the phosphorylation state of the kinase. For example, imatinib (STI571), a specific inhibitor of Abl, c-kit, and PDGFR binds to the inactive conformation of Abl by using the “DFG-out” conformation, thereby giving the piperazinylbenzamide functionality access to the allosteric pocket (14, 15). To investigate the structural basis for the high selectivity of TAE684 in cellular assays, a model of ALK in complex with TAE684 was built based on the published crystal structure of InsR in an “active” or “DFG-in” conformation (16, 17). As shown in Fig. 2, TAE684 is expected to bind to the ATP-binding site by using the ubiquitously observed bidentate hydrogen-bonding pair to the kinase “hinge” region of ALK but should not extend into the hydrophobic binding pockets. This result is consistent with the fact that TAE684 does not possess any of the pharmacophoric features characteristic of compounds that bind to the DFG-out kinase conformation. Interestingly, the orthomethoxy group attached to the 2-aniline substitutent projects into a small groove located between the side chains of residues L258 and M259. Sequence alignments of kinases available in the Ba/F3 panel revealed that most kinases have bulkier residues at this position (SI Fig. 7). Molecular modeling revealed that bulkier amino acids at this position would lead to a steric clash with TAE684, suggesting that L258 may be one of the major kinase-selectivity determinants for TAE684. InsR, like ALK, also possesses a leucine at position 258; however, a 100-fold difference in the IC50 between ALK and InsR has been observed in cellular assays, suggesting that additional unknown structural features, above all differences in the three-dimensional structure, rather than the amino acid sequence may contribute to the selectivity of TAE684. Analysis of cocrystal structures of ALK and InsR with TAE684 could resolve this question.
Fig. 2.

Structural basis for ALK kinase inhibition selectivity by TAE684. (A) Model of ALK in complex with TAE684, developed based on the published crystal structure of InsR in an “active” conformation by using homology modeling (MOE). TAE684 is expected to bind to the ATP-binding site by using a bidentate hydrogen bonding pair to the kinase “hinge” region of ALK. The orthomethoxy group attached to the 2-aniline substitutent is anticipated to project into a small groove located between the side chains of residues L258 and M259. (B) Position L258 is one of the major determinants for ALK selectivity of TAE684. Substitution of leucine with a bulkier amino acid such as phenylalanine (F258) induces a conformational change that leads to a steric clash of the hinge region with TAE684.
TAE684 Inhibits Signaling Downstream of NPM-ALK.
STAT transcription factor signaling has been shown to play an essential role in transformation and lymphomagenesis mediated by the NPM-ALK fusion. Several investigators have independently shown that STAT3 and/or STAT5 are activated by NPM-ALK (18–20). Using either a Cre/Lox system or antisense knockdown, Chiarle et al. (20) could show that loss of STAT3 in NPM-ALK transformed T cells isolated from transgenic mice induces apoptosis and blocks growth in s.c. tumor models. To further corroborate the involvement of STAT3 and/or STAT5 in signaling downstream of NPM-ALK, we performed Western blot analysis on lysates of NPM-ALK-positive cells treated with either DMSO or increasing concentrations of TAE684. As demonstrated in Fig. 3A, TAE684 inhibited STAT3 and STAT5 phosphorylation in a dose-dependent manner in both Ba/F3 NPM-ALK and Karpas-299 cells. Similar results were obtained by using SU-DHL-1 cells (data not shown). After 4 h of treatment with TAE684, STAT3 and STAT5 phosphorylation levels decreased significantly at concentrations as low as 10 nM and were completely inhibited at concentrations >50 nM. We also performed kinetic experiments with TAE684 at a concentration of 50 nM to determine the time required to achieve full inhibition of NPM-ALK and STAT3. A significant reduction in the phosphorylation of NPM-ALK and STAT3 (Fig. 3B) was seen as early as 15 min after incubation and was sustained up to 48 h (data not shown). A direct correlation between time and concentration was seen for inhibition of both NPM-ALK and STAT3. The impact of NPM-ALK inhibition on both RAS/RAF/MAPK and PI3K/Akt signaling was investigated by using p-ERK and p-Akt as surrogate markers for these pathways. As shown in Fig. 3C, inhibition of NPM-ALK by TAE684 led to a dose-dependent reduction in phosphorylation of both ERK and Akt in Karpas-299 cells. These results reconfirm that NPM-ALK is an activator of STAT, RAS/RAF/MAPK, and PI3K/Akt in both transformed Ba/F3 NPM-ALK cells and NPM-ALK-positive ALCL cell lines. Although the analysis of the signaling pathways downstream of NPM-ALK is by far not exhaustive, these data demonstrate that TAE684 is not only a potent inhibitor of NPM-ALK, but also a physiological modulator of its crucial downstream signaling intermediates.
Fig. 3.

Effects of TAE684 on NPM-ALK downstream signaling events. (A and B) TAE684 inhibits NPM-ALK and STAT3 activation in Ba/F3 NPM-ALK and Karpas-299 cells in a concentration- and time-dependent manner (A and B, respectively). (C) Effects of TAE684 on ERK and Akt phosphorylation in Karpas-299 cells after 4 h of treatment.
TAE684 Induces Cell Cycle Arrest and Apoptosis in ALCL and Ba/F3 NPM-ALK Cell Lines.
To further study the biological effects of inhibition of NPM-ALK on the growth and survival of ALCL cell lines, we performed cell cycle and apoptosis analyses on cells treated with either TAE684 or DMSO. Ba/F3, Ba/F3 NPM-ALK, SU-DHL-1, and Karpas-299 cells were treated with various concentrations of TAE684 for 72 h and were assessed for induction of apoptosis and growth arrest by flow cytometry every 24 h. Treatment with TAE684 increased the number of Annexin V-positive Ba/F3 NPM-ALK cells in a dose- and time-dependent manner, without affecting the survival of the parental Ba/F3 cell line. At 48 h after incubation with TAE684, 85–95% of cells stained Annexin V-positive in several independent experiments. In contrast, no increase in the number of Annexin V-positive cells was seen for parental Ba/F3 cells grown in the presence of IL-3 (Fig. 4A and B). Similar to our results obtained by using Ba/F3 NPM-ALK cells, SU-DHL-1 cells appeared to be sensitive to TAE684-mediated apoptosis induction, with 70–80% of cells staining positive for Annexin V after 48 h of treatment (Fig. 4 A and B). Intriguingly, Karpas-299 did not undergo apoptosis to a similar degree as did SU-DHL-1 and Ba/F3 NPM-ALK cells despite Karpas-299 cell growth being inhibited by TAE684 with an IC50 of 3 nM (Fig. 1B). After 72 h of treatment with a 50 nM concentration of TAE684, only 20–30% of Karpas-299 cells stained positive for Annexin V (data not shown).
Fig. 4.

TAE684 induces apoptosis and G1 phase arrest in NPM-ALK-expressing Ba/F3 cells and ALCL patient cell lines. (A) Ba/F3, Ba/F3 NPM-ALK, and SU-DHL-1 cells were treated with either DMSO or TAE684 50 nM for 48 h. Induction of apoptosis was assayed with Annexin V and 7-AAD staining. (B) Total percent of Annexin V-positive cells was determined after 48-h treatment with DMSO or increasing concentrations of TAE684. (C) Karpas-299 cells were pretreated with DMSO or 50 nM TAE684 for 48 h, fixed, and stained with PI for cell cycle analysis. (D) Cell cycle distribution of Karpas-299 cells after 48-h treatment with TAE684. Representative graph from one of three separate experiments is shown.
The lack of apoptosis in ≈70% of cells suggested a profound effect of TAE684 on cell cycle progression in Karpas-299 cells. To investigate the impact of TAE684 on cell cycle in more detail, TAE684-treated Karpas-299 cells were stained with propidium iodide (PI) and analyzed for cell cycle distribution. As shown in Fig. 4 C and D, TAE684 induced G1 phase arrest in a time-dependent manner. After 72 h of treatment with TAE684 (25 nM), 72% of Karpas-299 cells were arrested in G1 phase compared with 26% of cells in G1 phase in DMSO-treated controls. The number of cells in S phase was reduced from 60% to 14% (Fig. 4 C and D). Collectively, these data suggest that TAE684 inhibits the growth of ALCL cells by both inhibiting the progression of cell cycle and induction of apoptosis. These data also suggest that NPM-ALK-positive cell lines respond differently to NPM-ALK inhibition. Differences in the behavior of SU-DHL-1 and Karpas-299 cells had been described previously and have been suggested to correlate with acquired secondary mutations. These differences are also apparent in the different potential of these cell lines to induce lymphoma in mice. Although Karpas-299 cells readily give rise to a lymphoma-like disease in immunocompromised mice, no engraftment was seen with SU-DHL-1 cells after both s.c. and i.v. implantation of up to five million cells (data not shown). A more detailed and global analysis of signaling downstream of NPM-ALK as well as investigation of additional cell lines is warranted and may be useful in predicting clinical outcomes to ALK inhibition.
TAE684 Inhibits the Development of NPM-ALK-Driven Lymphomas in Vivo.
We validated the potential of TAE684 to inhibit the growth of ALCL in a newly established, clinically relevant lymphoma model. To develop a model that would allow us to follow systemic ALCL development and would resemble clinical disease progression as closely as possible, we engineered a luciferized Karpas-299 cell line, which could be monitored in vivo with the highly sensitive Xenogen bioluminescence imaging system. Six- to 8-week-old SCIDbeige mice were injected i.v. with one million Karpas-299-luc cells and were monitored for disease progression by measuring bioluminescence and palpable lymphoma development. Seven days after inoculation, a strong bioluminescence signal was detected in the nasal-associated lymphoid tissue, which then spread to the lymph nodes after 2 weeks. Lymph node infiltration was most prominent but not limited to nuchal and peritoneal lymph nodes. Histological analysis of the enlarged excised lymph nodes revealed strong infiltration of CD246- (ALK) and CD30-positive Karpas-299 cells (Fig. 5A). TAE684 displayed appreciable bioavailability and half-life in vivo (data not shown). Seven hours after an oral dose of 20 mg/kg of TAE684 (formulated as a solution in 10% 1-methyl-2-pyrrolidinone/90% PEG 300) a maximum plasma level (Cmax) of 800–1,000 nM was measured, with a bioavailability (BAV) ranging between 60% and 70% and an elimination half-life (T1/2) of ≈12 h. To demonstrate the feasibility of targeting NPM-ALK in vivo without causing toxicity, TAE684 was administered at 1, 3, and 10 mg/kg once daily by oral gavage to mice starting 72 h after Karpas-299 i.v. injection. After 2 weeks of treatment, we observed a 100-fold reduction in bioluminescence signal in the 3- and 10-mg/kg treatment groups. Although the compound was not efficacious at 1 mg/kg, after 4 weeks of treatment with TAE684 at 3 and 10 mg/kg, there was a significant (P = 0.005) delay in lymphoma development and 100- to 1,000-fold reduction in luminescence signal (Fig. 5 B–D). The TAE684- (10 mg/kg) treated group appeared healthy and did not display any signs of compound- or disease-related toxicity.
Fig. 5.

Effects of orally administered TAE684 on disease progression in an in vivo Karpas-299 lymphoma model. (A) Histopathology of an excised enlarged lymph node from a Fox Chase SCIDBeige mouse 4 weeks after an i.v. injection of one million Karpas-299 cells. Representative sections stained with H&E and against CD246 (ALK) or CD30 antigens are shown (magnification ×60). Images demonstrate strong infiltration of anaplastic, CD30- and ALK-positive Karpas-299 into the lymph node architecture. (B) Dose–response of the Karpas-299 lymphomas to 1, 3, and 10 mg/kg TAE684 or vehicle solution administered once daily. Dosing was initiated 3 days after mice received an i.v. injection of luciferase-expressing Karpas-299 cells. Representative low- and high-sensitivity bioluminescence images after 4 weeks of dosing are shown (n = 8 mice per group). (C) Effects of TAE684 or vehicle treatment on disease progression, estimated by weekly increases in bioluminescence signal with the Xenogen Imaging System (±SD). (D) Bioluminescence signal readout ± SD for mice shown in B after 4 weeks of treatment with either TAE684 or vehicle solution.
To further validate that the observed in vivo effects of ALCL inhibition were not the consequence of unanticipated off-target effects, we examined the response of Ba/F3 NPM-ALK- and Ba/F3 BCR-ABL-induced lymphoid disease to TAE684 (10 mg/kg) treatment. Although no difference in light emission was observed in mice transplanted with Ba/F3 BCR-ABL cells after 2 weeks of treatment, we found a >99% difference between vehicle- and TAE684-treated mice allografted with Ba/F3 NPM-ALK cells (SI Fig. 8). Ba/F3 NPM-ALK-induced disease did not affect spleen weights to the same extent as Ba/F3 BCR-ABL disease burden; nevertheless, we observed a significant 80% reduction of spleen weight with TAE684 treatment (P < 0.005) in Ba/F3 NPM-ALK-injected mice. These data demonstrate the specificity of TAE684 therapeutic effects, further corroborating the selectivity of this compound at the therapeutic doses chosen.
TAE684 Inhibits NPM-ALK Fusion Kinase-Mediated Signaling and Induces Regression of Established Lymphomas in Vivo.
To determine whether TAE684 treatment would induce regression of established lymphomas, in a separate experiment dosing was initiated 12 days after injection of Karpas-299 cells. Before the start of treatment, disease progression was confirmed by bioluminescence imaging, as evidenced by strong signal in the nasal-associated lymphoid tissue as well as nuchal, inguinal, and peritoneal lymph nodes (Fig. 6A). Mice with validated early stages of lymphoma were assigned to three treatment groups and one control group. The control group continued to develop signs of disease progression and had to be killed on day 19 because of disease burden and signs of premorbidity. In contrast, TAE684- (3, 5, and 10 mg/kg p.o. daily) treated mice responded to treatment in a dose-dependent manner, displayed significant signs of improvement, and had a 1,000-fold reduction in bioluminescence signal after 2 weeks of dosing (Fig. 6A).
Fig. 6.

TAE684 treatment induced disease regression in established Karpas-299 lymphomas. (A) Treatment with 3, 5, and 10 mg/kg (mpk) TAE684 was initiated 12 days after Karpas-299 inoculation and disease establishment as evidenced by bioluminescent imaging, obtained before (day 12) and after 2 weeks (day 26) of dosing. (B and C) Mice (m1–m6) with established palpable Karpas-299 lymphomas were treated for 3 days with either 10 mg/kg TAE684 (m4–m6) or vehicle solution (m1–m3). Four hours after the third dose, mice were killed, and excised lymph nodes were analyzed for in vivo effects of TAE684 treatment on NPM-ALK and Stat3 phosphorylation by immunoblotting (B) or for CD30 expression by immunohistochemistry (C).
As a follow-up study, we examined the immediate molecular effects of short-term TAE684 treatment on established lymphomas. Treatment was delayed until 3.5 weeks after Karpas-299 cell injection, at which point mice had displayed signs of established disease and had developed palpable lymphomas. The mice were then treated with either TAE684 (10 mg/kg) or vehicle solution for 3 days. Immunoblotting analysis of protein from extracted inguinal lymph nodes revealed a reduction in the phosphorylation levels of NPM-ALK and its downstream target, STAT3 (Fig. 6B). Histological examination confirmed high infiltration of the lymph node tissue by the anaplastic, CD246-positive Karpas-299 cells (data not shown). CD30 receptor expression appeared to vary between lymph node sections from vehicle- and TAE684-treated groups. Vehicle-treated groups displayed high levels of CD30, as previously observed during model development; however, CD30 expression was significantly reduced in lymph nodes from TAE684-treated mice (Fig. 6C). We were able to replicate these results in vitro, where an ≈80% reduction in the expression of CD30 receptor was observed on the cell surface of Karpas-299 24 h after the addition of TAE684 to the culture media (SI Fig. 9). It is currently unknown whether high CD30 expression on ALCL cells reflects the phenotype of the cell of origin transformed by NPM-ALK or whether it is directly induced as a consequence of NPM-ALK's kinase activity. Watanabe et al. (21) have recently demonstrated that CD30 promoter activity is controlled by JunB, expression of which is regulated by the CD30-ERK1/2 MAPK signaling axis. NPM-ALK expression by itself can also induce strong activation of the MEK/ERK signaling pathway independently of c-RAF in NPM-ALK-transformed Ba/F3 cells. TAE684-mediated NPM-ALK inhibition leads to a significant reduction of ERK phosphorylation in Karpas-299 cells (Fig. 3C), which may, in turn, affect CD30 promoter activation. These data indicate that the down-regulation of CD30 expression through the inhibition of NPM-ALK kinase activity is a clinically relevant event and correlates with disease regression. CD30 receptor expression can be easily assayed for in the clinic and could be used as a pharmacodynamic marker of therapeutic NPM-ALK inhibition.
Conclusions
NPM-ALK and related ALK fusion proteins possess transforming and lymphomagenic potential, likely to be mediated by constitutive kinase activity. Although NPM-ALK-positive lymphomas have a rather benign prognosis, ≈40–45% of patients do not respond or relapse after standard therapy. In addition, standard therapy is associated with considerable toxicity, a problem specifically bothersome in pediatric patients. Therefore, a highly effective and targeted therapy would be beneficial and highly warranted not only for relapsed patients but also as first-line therapy if well tolerated and efficacious.
NPM-ALK-positive cells show activation of signaling pathways, such as PI3K/Akt (22, 23), JAK/STAT (18–20), and Src kinases (24), which are reminiscent of, but not completely overlapping with, those activated in BCR-ABL-transformed cells (25). Several studies have suggested that signaling molecules within these pathways could serve as therapeutic targets in the absence of a specific small-molecule inhibitor targeting NPM-ALK (20, 24, 26–30). However, given the enormous redundancy in signal transduction, it has become clear that no single pathway downstream of an activated kinase is as suitable a target as the activated oncogene itself. Given the homology between the oncogenic transformation induced by BCR-ABL and NPM-ALK and the success of ABL targeting small-molecule inhibitors such as imatinib in the clinic, we endeavored to develop a selective small-molecule inhibitor of ALK kinase activity, which would inhibit the proliferation and survival of NPM-ALK-positive cells both in vitro and in vivo. Two recent studies have described small-molecule inhibitors of NPM-ALK that are capable of blocking both ALK kinase activity and signal transduction, demonstrating the feasibility of this approach (31, 32). It was shown that these inhibitors blocked the proliferation of NPM-ALK-transformed cells in a concentration-dependent manner and that an ALK-specific inhibitor would have the potential to become a therapeutic agent for the treatment of ALK-positive ALCL and other conditions associated with the expression of activating ALK gene rearrangements. However, neither kinase selectivity nor in vivo data have been published for these compounds, suggesting that further optimization may be necessary before these compounds can be used to specifically target ALK in vivo.
In this study, we have identified and characterized TAE684, a highly potent and specific inhibitor of NPM-ALK. TAE684 inhibited the growth of NPM-ALK-transformed cells with an IC50 of ≈3 nM and was highly selective against several other tyrosine kinases tested. Although potent on the highly homologous InsR kinase in vitro, we demonstrated that in cellular assays TAE684 is ≈100-fold more potent against ALK when compared with InsR. This finding might be explained by differences in the three-dimensional structure of the truncated enzyme compared with the full-length receptor in a cellular system or by differences in the activity of the compound at the ATP concentration used in the enzyme assays versus the physiological cellular ATP concentration. It will be interesting to see whether a similar difference can be found for IGF1R, a potential target for anticancer therapeutics, and future effort will need to be focused on elucidating the potency of TAE684 against IGF1R-dependent cell lines and tumor models. It will also be important to study in more detail the activity of TAE684 against the native full-length ALK receptor. Although ALK knockout mice have been reported to show no significant phenotype (1), several lines of evidence have suggested a role of full-length ALK in different tumor types including glioblastoma (2). TAE684 could be a valuable tool to study the role of ALK in various tumors in vivo, if the activity observed for NPM-ALK could be confirmed against the full-length receptor.
In summary, TAE684 displayed favorable pharmacokinetic properties in mice, including high bioavailability, decent half-life and sufficient distribution into tissues. Using a murine model of ALCL, we could demonstrate the feasibility of therapeutically targeting NPM-ALK in vivo. TAE684 prevented the development of Karpas-299-driven lymphoma if dosed early after injection of cells and led to the regression of established lymphoma, which was associated with inhibition of phosphorylation of NPM-ALK and STAT3 in infiltrated lymph nodes. Collectively, these data greatly support efforts to pursue the clinical development of small-molecule NPM-ALK inhibitors as a treatment strategy for therapy of refractory and relapsed ALK-positive lymphomas.
Materials and Methods
See SI Materials and Methods for a more detailed description of assay methods.
Cell Lines.
The murine pro-B cell line Ba/F3 and the human t(2, 5)-positive Karpas-299 and SU-DHL-1 ALCL cell lines (DSMZ, Berlin, Germany) were maintained in RPMI medium 1640 supplemented with 10% FBS (Sigma–Aldrich, St. Louis, MO). Ba/F3 cells were grown in the presence of IL-3 (10 ng/ml) (R & D Systems, Minneapolis, MN). Cell lines expressing luciferase alone or in combination with NPM-ALK, BCR-ABL, and TEL-kinase fusion constructs were generated by retroviral transduction of cells with pMSCV IRES puro/Luc vector.
Cell Proliferation Assays.
Luciferase-expressing Karpas-299, SU-DHL-1, and Ba/F3 cells and transformed Ba/F3 stably expressing NPM-ALK, BCR-ABL, or TEL-kinase fusion constructs were plated in 384-well plates (25,000 cells per well) and incubated with serial dilutions of TAE684 or DMSO for 2–3 days. Luciferase expression was used as a measure of cell proliferation/survival and was evaluated with the Bright-Glo Luciferase Assay System (Promega, Madison, WI). IC50 values were generated by using XLFit software.
Flow Cytometry.
Ba/F3 NPM-ALK, Karpas-299, and SU-DHL-1 cells were treated with DMSO or various concentrations of TAE684 for 24, 48, and 72 h before analysis of cell cycle distribution and apoptosis by flow cytometry. Samples were analyzed on a Becton-Dickinson LSRII Flow Cytometer (BD Biosciences, San Jose, CA).
In Vivo Experiments.
For in vivo compound efficacy studies, treatment was initiated 72 h after tail vein injection of 1 × 106 Karpas-299-, Ba/F3 NPM-ALK- or BCR-ABL-expressing cells into female Fox Chase SCIDBeige mice. Mice (n = 10 per group) were administered either TAE684 resuspended in 10% 1-methyl-2-pyrrolidinone/90% PEG 300 (Sigma) solution at 1, 3, and 10 mg/kg once daily for 3 weeks or the vehicle solution at the same dosing schedule. Disease progression and compound efficacy was monitored weekly with bioluminescence imaging. To determine the efficacy of TAE684 on established disease, dosing was initiated on day 12, at which time the disease confirmed to be widespread by bioluminescence imaging. For analysis of downstream molecular effects in vivo, mice with established lymphomas were administered vehicle solution or TAE684 (10 mg/kg) for 3 days. At the end of treatment, mice were killed, and lymph nodes were extracted for immunoblotting and histological analysis.
Homology Modeling and Docking.
The ALK model was developed by using the homology modeling function in the Molecular Operating Environment (MOE 2005.06) program (Chemical Computing Group, Montreal, QC, Canada). The InsR kinase [Protein Database (PDB) ID code: 1IR3] was used as the template for ALK homology modeling, because of the 45% sequence identity between the InsR and ALK kinase domains. TAE684 was docked into the ALK model by using GOLD [version 1.3 (Cambridge Crystallographic Data Center, Cambridge, U.K.)] with the standard default settings. All atom types and charges were assigned in GOLD. One hundred thousand independent genetic algorithm (GA) runs were performed for TAE684 flexible ligand docking. The radius of the search in the docking was set to 10 Å.
Supplementary Material
Abbreviations
- ALCL
anaplastic large-cell lymphoma
- ALK
anaplastic lymphoma kinase
- InsR
insulin receptor kinase
- NPM
nucleophosmin.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0609412103/DC1.
References
- 1.Duyster J, Bai RY, Morris SW. Oncogene. 2001;20:5623–5637. doi: 10.1038/sj.onc.1204594. [DOI] [PubMed] [Google Scholar]
- 2.Pulford K, Morris SW, Turturro F. J Cell Physiol. 2004;199:330–358. doi: 10.1002/jcp.10472. [DOI] [PubMed] [Google Scholar]
- 3.Kadin ME, Carpenter C. Semin Hematol. 2003;40:244–256. doi: 10.1016/s0037-1963(03)00138-0. [DOI] [PubMed] [Google Scholar]
- 4.Coluccia AM, Gunby RH, Tartari CJ, Scapozza L, Gambacorti-Passerini C, Passoni L. Expert Opin Ther Targets. 2005;9:515–532. doi: 10.1517/14728222.9.3.515. [DOI] [PubMed] [Google Scholar]
- 5.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 6.Turner SD, Alexander DR. Leukemia. 2005;19:1128–1134. doi: 10.1038/sj.leu.2403797. [DOI] [PubMed] [Google Scholar]
- 7.Gascoyne RD, Lamant L, Martin-Subero JI, Lestou VS, Harris NL, Muller-Hermelink HK, Seymour JF, Campbell LJ, Horsman DE, Auvigne I, et al. Blood. 2003;102:2568–2573. doi: 10.1182/blood-2003-03-0786. [DOI] [PubMed] [Google Scholar]
- 8.Chikatsu N, Kojima H, Suzukawa K, Shinagawa A, Nagasawa T, Ozawa H, Yamashita Y, Mori N. Mod Pathol. 2003;16:828–832. doi: 10.1097/01.MP.0000081729.40230.1F. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong F, Duplantier MM, Trempat P, Hieblot C, Lamant L, Espinos E, Racaud-Sultan C, Allouche M, Campo E, Delsol G, Touriol C. Oncogene. 2004;23:6071–6082. doi: 10.1038/sj.onc.1207813. [DOI] [PubMed] [Google Scholar]
- 10.Pulford K, Morris SW, Mason DY. Curr Opin Hematol. 2001;8:231–236. doi: 10.1097/00062752-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Chiarle R, Gong JZ, Guasparri I, Pesci A, Cai J, Liu J, Simmons WJ, Dhall G, Howes J, Piva R, Inghirami G. Blood. 2003;101:1919–1927. doi: 10.1182/blood-2002-05-1343. [DOI] [PubMed] [Google Scholar]
- 12.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, et al. Proc Natl Acad Sci USA. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, et al. Cancer Cell. 2004;5:231–239. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 14.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 15.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard SR, Wei L, Ellis L, Hendrickson WA. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 17.Hubbard SR. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, Levy DE, Inghirami G. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 19.Nieborowska-Skorska M, Slupianek A, Xue L, Zhang Q, Raghunath PN, Hoser G, Wasik MA, Morris SW, Skorski T. Cancer Res. 2001;61:6517–6523. [PubMed] [Google Scholar]
- 20.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G. Nat Med. 2005;11:623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 21.Marzec M, Kasprzycka M, Liu X, Raghunath PN, Wlodarski P, Wasik MA. Oncogene. 2006 doi: 10.1038/sj.onc.1209843. [DOI] [PubMed] [Google Scholar]
- 22.Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, Morris SW, Wasik MA, Skorski T. Cancer Res. 2001;61:2194–2199. [PubMed] [Google Scholar]
- 23.Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Blood. 2000;96:4319–4327. [PubMed] [Google Scholar]
- 24.Cussac D, Greenland C, Roche S, Bai RY, Duyster J, Morris SW, Delsol G, Allouche M, Payrastre B. Blood. 2004;103:1464–1471. doi: 10.1182/blood-2003-04-1038. [DOI] [PubMed] [Google Scholar]
- 25.Warmuth M, Danhauser-Riedl S, Hallek M. Ann Hematol. 1999;78:49–64. doi: 10.1007/s002770050473. [DOI] [PubMed] [Google Scholar]
- 26.Bonvini P, Dalla Rosa H, Vignes N, Rosolen A. Cancer Res. 2004;64:3256–3264. doi: 10.1158/0008-5472.can-03-3531. [DOI] [PubMed] [Google Scholar]
- 27.Bonvini P, Gastaldi T, Falini B, Rosolen A. Cancer Res. 2002;62:1559–1566. [PubMed] [Google Scholar]
- 28.Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, Rassidakis GZ, O'Connor SL, McDonnell TJ, Lai R. Oncogene. 2003;22:5399–5407. doi: 10.1038/sj.onc.1206849. [DOI] [PubMed] [Google Scholar]
- 29.Amin HM, McDonnell TJ, Ma Y, Lin Q, Fujio Y, Kunisada K, Leventaki V, Das P, Rassidakis GZ, Cutler C, et al. Oncogene. 2004;23:5426–5434. doi: 10.1038/sj.onc.1207703. [DOI] [PubMed] [Google Scholar]
- 30.Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, Claret FX, Medeiros LJ, Amin HM. Blood. 2005;105:827–829. doi: 10.1182/blood-2004-06-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marzec M, Kasprzycka M, Ptasznik A, Wlodarski P, Zhang Q, Odum N, Wasik MA. Lab Invest. 2005;85:1544–1554. doi: 10.1038/labinvest.3700348. [DOI] [PubMed] [Google Scholar]
- 32.Wan W, Albom MS, Lu L, Quail MR, Becknell NC, Weinberg LR, Reddy DR, Holskin BP, Angeles TS, Underiner TL, et al. Blood. 2005;107:1617–1623. doi: 10.1182/blood-2005-08-3254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}