

Abstract
Recent reports have linked neuronal cell death by necrosis to poly(ADP-ribose) polymerase-1 (PARP-1) hyperactivation. It is believed that under stress, the activity of this enzyme is up-regulated, resulting in extensive poly(ADP-ribosyl)ation of nuclear proteins, using NAD+ as its substrate, which, in turn, leads to the depletion of NAD+. In efforts to restore the level of NAD+, depletion of ATP occurs, resulting in the shutdown of ATP-dependent ionic pumps. This results in cell swelling and eventual loss of membrane selectivity, hallmarks of necrosis. Reports from in vitro and in vivo studies in the brain have shown that NMDA receptor activation stimulates PARP activity and that blockade of the enzyme provides substantial neuroprotection. The present study was undertaken to determine whether PARP activity is regulated by NMDA in the rat retina, and whether blockade of PARP activity provides protection against toxic effects of NMDA. Rat retinas exposed to intravitreal injections containing NMDA, with or without the PARP inhibitor N-(6-oxo-5, 6-dihydrophenanthridin-2-yl)-(N,-dimethylamino) acetamide hydrochloride (PJ-34), were assessed for changes in PARP-1 activity as evidenced by poly(ADP-ribosyl)ation (PAR), loss of membrane integrity, morphological indicators of apoptosis and necrosis, and ganglion cell loss. Results showed that: NMDA increased PAR formation in a concentration-dependent manner and caused a decline in retinal ATP levels; PJ-34 blockade attenuated the NMDA-induced formation of PAR and decline in ATP; NMDA induced the loss of membrane selectivity to ethidium bromide (EtBr) in inner retinal neurons, but loss of membrane selectivity was not prevented by blocking PARP activity; cells stained with EtBr, or reacted for TUNEL-labeling, displayed features characteristic of both apoptosis and necrosis. In the presence of PJ-34, greater numbers of cells exhibited apoptotic features; PJ-34 provided partial neuroprotection against NMDA-induced ganglion cell loss. These findings suggest that although blockade of PARP activity fully attenuates NMDA-induced PAR formation and loss of retinal ATP content, and improves the survival of select populations of ganglion cells, this approach does not provide full neuroprotection. In contrast, blockade of PARP activity promotes apoptotic-like cell death in the majority of cells undergoing cell death. Furthermore, these studies show that the loss of membrane selectivity is not dependent upon PAR formation or the resulting decline of ATP, and suggests that an alternative pathway, other than PARP activation, exists to mediate this event.
Keywords: ATP levels, ethidium bromide staining, fluorogold labeling, necrosis, poly(APD-ribose) polymerase, poly(ADP-ribosyl)ation, TUNEL labeling
Abbreviations used: BZ, benzamide; 3-BZ, 3-amino-benzamide; EtBr, ethidium bromide; FG, fluorogold; GCL, ganglion cell layer; INL, inner nuclear layer; PAR, poly(ADP-ribosyl)ation; PARP-1, poly(ADP-ribose) polymerase-1; PBS, phosphate-buffered saline; PJ-34, N-(6-oxo-5, 6-dihydrophenanthridin-2-yl)-(N, -dimethylamino) acetamide hydrochloride; PT, post-treatment; TUNEL, terminal dUTP nick end labelling; WGS, whole goat serum
Understanding the events that lead to the death of neuronal cells following an excitotoxic event has been the focus of extensive studies (Choi 1992; Lipton and Rosenberg 1994; Olney 1994). Historically, the characterization of cell death has been based upon distinct morphological features that assign cells to one of two pathways, either apoptosis or necrosis. In recent years, the determination of whether a neuron undergoes apoptosis or necrosis has proven to be more difficult (Pollard et al. 1994; Bonfoco et al. 1995; Ferrer et al. 1995; Gwag et al. 1995). Adding to the complexity is the additional view that considers apoptosis to be just one of several forms of programmed cell death (Sperandio et al. 2000, 2004). This has led to the notion that excitotoxic cell death is a continuum that ranges from apoptosis at one end to necrosis at the other (Portera-Cailliau et al. 1997; for review see Virág and Szabó 2002).
There is evidence that the level of cellular ATP dictates the mode of neuronal cell death, with apoptosis requiring high amounts of ATP and necrosis occurring when energy stores are low or depleted (Ha and Snyder 1999). Under certain stress-related conditions, depletion of ATP has been linked to hyperactivation of a nuclear protein, poly(ADP-ribose) polymerase-1 (PARP-1: EC 2.4.2.30). PARP-1 hydrolyzes nicotinamide adenine dinucleotide (NAD+) to nicotinamide to facilitate the transfer of ADP ribose units onto nuclear proteins (de Murcia et al. 1994), forming poly(ADP-ribose) polymers. It is hypothesized that following a strong insult, endogenous NAD+ pools are consumed by the increased activity of PARP-1, and that replenishment of NAD+, in turn, consumes endogenous ATP (Ha and Snyder 1999). The loss of ATP is thought to truncate the apoptotic pathway and to lead to the shutdown of ATP-dependent systems, such as transmembrane ion pumps. The loss of active ion transport results in ionic and osmotic imbalances within the stressed neuron, and promotes the characteristic cell swelling that leads to the loss of membrane selectivity, a hallmark of necrosis (Ha and Snyder 1999).
Activation of PARP-1 occurs in response to DNA strand breaks caused by oxidation or nitric oxide toxicity (Zhang et al. 1994; Szabó et al. 1996; Pieper et al. 2000). This activation promotes DNA repair, a beneficial and restorative effect. However, when activation of PARP-1 becomes too strong, resulting in depletion of NAD+ and ATP, then detrimental changes consistent with classical signs of necrosis are observed (Ha and Snyder 1999).
Evidence for PARP-1 involvement in mediating necrosis has been demonstrated in vitro where inhibitors of PARP provide partial or full neuroprotection against glutamate- and nitric oxide (NO)-induced toxicity (Sims et al. 1983; Takahashi and Greenburg 1999). In addition, PARP blockade attenuated the decline of endogenous NAD+ and ATP levels following excitotoxic challenge (Sims et al. 1983). Similarly, pre-administration of PARP antagonists has been shown to reduce significantly the volume of the infarct in rats following ischemia (Takahashi et al. 1997).
It has long been recognized that NMDA receptors mediate excitotoxic cell death via both apoptosis and necrosis, and that the degree of insult dictates the prevalence of one of the pathways (Bonfoco et al. 1995). However, little is known about the mechanism(s) involved in regulating these events in vivo. Interestingly, it has been shown that over-stimulation of NMDA receptors is linked to an increase in PARP-1 activity in the brain (Mandir et al. 2000; Pieper et al. 2000) and that PARP-1 activation is dependent upon glutamate or NMDA-receptor induction of neuronal nitric oxide synthase (nNos) (Dawson et al. 1991; Pieper et al. 2000). These studies also indicated that the NMDA receptor is unique with respect to the glutamate family of receptors, in that PARP-1 activation is directly linked to NMDA receptor activation, but not to AMPA receptor stimulation (Mandir et al. 2000).
Collectively, these findings imply that following an excitotoxic challenge, the regulation of PARP-1 plays a pivotal role in dictating whether a neuron will die by apoptosis or necrosis. The present study was undertaken to examine the role of PARP in NMDA-induced cell death in the rat retina, and to determine whether inhibition of PARP prevents cell death. Results indicate that the level of PARP activity is dependent upon the degree of NMDA insult, and that inhibition of PARP substantially attenuates the process of poly(ADP-ribosyl)ation. However, blocking ribosylation provides only partial protection of retinal ganglion cells.
Materials and methods
N-methyl-D-aspartate, anti-β-actin monoclonal clone AC-15, ethidium bromide (EtBr), N-(6-oxo-5, 6-dihydrophenanthridin-2-yl)-(N,-dimethylamino) acetamide hydrochloride (PJ-34), 3-amino-benzamide (3-BZ) and benzamide (BZ) were from Sigma (St Louis, MO, USA). The in situ cell death detection kit, rabbit anti-PARP-1 polyclonal antiserum, 4-nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl-phosphate (NBT/BCIP) and Lumilight western blot substrate were from Roche Molecular Biochemicals Inc. (Indianapolis, IN, USA). Rabbit anti-poly(ADP-ribosyl)ation (PAR) polyclonal antibody was from BD Biosciences Pharmingen (San Diego, CA, USA) and Slowfade mounting medium was from Molecular Probes (Eugene, OR, USA). Sheep anti-mouse-IgG-horseradish peroxidase (HRP) conjugate was from Amersham (Piscataway, NJ, USA), goat anti-rabbit-HRP conjugate from ICN (Costa Mesa, CA, USA) and bovine anti-rabbit-alkaline phosphate (AP) from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Fluorogold was from Fluorochrome (Denver, CO, USA).
Treatment conditions
To minimize animal-to-animal variations (Danias et al. 2002), comparisons were made between retinas in the same animal. The right eye of each animal was injected intravitreally with NMDA alone, or served as a control, and the left eye received the designated inhibitor in conjunction with NMDA treatment. The protocols described here were approved by the Wayne State University Animal Investigation Committee, and are in accordance with the recommendations of the Association for Research in Vision and Ophthalmology (ARVO) and NIH. Male Sprague–Dawley rats weighing 200–250 g were anesthetized with isoflurane, then given a 3 μL injection into the left eye containing the PARP inhibitor, PJ-34, using 0.24, 2.4 or 24 nmol (4, 40 and 400 μM, respectively, assuming a vitreal volume of 55 μL), 3-BZ (60, 120, 240 or 1200 nmol) or 120 nmol BZ in 0.01 M sterile phosphate-buffered saline (PBS) containing 2% dimethylsulfoxide (DMSO) (pH 7.2). All intravitreal injections were administered using a modified 32 gauge needle inserted in the superior-nasal quadrant at the ora serrata. Needle placement was monitored using a stereo dissecting microscope to assure that agents were injected into the center of the vitreal chamber. The contents were injected over 1 min, with the needle remaining in place for an additional minute before removal from the eye.
Two hours later, the animals were re-anesthetized and administered a second intravitreal injection; the left eyes received NMDA ranging from 2 to 200 nmol (40 μM to 4 m M, respectively) with the corresponding inhibitor and the right eyes receiving NMDA alone. To monitor the effect of exposure to inhibitor or vehicle, animals from the same group received an intravitreal injection of inhibitor alone in the left eye and were compared with vehicle only or untreated right eyes. Rats were killed by decapitation at designated times following the final injection, and the eyes were quickly removed for analysis, as described below.
Detection of poly(ADP-ribosyl)ation of proteins
Treated retinas were homogenized in 62.5 mM Tris-HCl, 6M urea, 10% glycerol, 2% sodium dodecyl sulfate (SDS), 5% β-mercaptoethanol and 200 μg/mL benzamide, and sonicated on ice. The samples were heated for 15 min at 65°C and centrifuged at 14 000 g, and the supernatant fluid was assessed for protein concentration according to the method of Henkel and Bieger (1994). Equal protein loads, ranging from 45 to 70 μg, were separated on 8% SDS–polyacrylamide gel electrophoresis (PAGE) gels, electrotransferred onto nitrocellulose membrane and processed for PAR immunoreactivity. Blots were blocked in 10 mM Tris-saline-0.5% Tween 20 (TST), containing 8% dried milk and 2% whole goat serum (WGS), for 1 h at room temperature (23°C), followed by an overnight incubation in a 1 : 2000 dilution of rabbit anti-PAR polyclonal antibody in TST containing 1% WGS. Following several TST washes, the blots were incubated in a 1 : 5000 dilution of a goat anti-rabbit-IgG AP conjugate. The immunoreactive proteins were visualized by incubating the blots in NBT/BCIP. They were then digitally scanned for densitometric analysis using SCION IMAGE/NIH IMAGE (Scion Corp., Frederick, MD, USA). To ensure equal loading of samples, the blots were re-probed for β-actin using a mouse anti-β-actin monoclonal antibody and a sheep anti-mouse-peroxidase-linked secondary antibody with visualization via chemiluminescence. Accounting for variability in PAR staining between individual western transfers, each gel contained an identical load of an internal PAR-immunoreactive sample that was used to normalize the resulting PAR densities.
Measuring PARP-1 levels in the retina
Eyes injected with 20 or 200 nmol NMDA were processed, in parallel with untreated eyes, to determine whether NMDA insult alters the level of PARP-1 protein expression in the retina, using western blot analysis. The freshly obtained retinas were homogenized as above in denaturing buffer. Total protein loads ranging from 35 to 70 μg were separated on an 8% SDS–polyacrylamide gel, transferred onto nitrocellulose membrane, blocked in 8% dried milk containing 2% WGS, then subjected to western blot analysis using a rabbit anti-PARP-1 IgG, followed by incubation in a 1 : 10 000 dilution of peroxidase-labeled goat anti-rabbit-IgG. Visualization of PARP-1 immunoreactivity was accomplished using Lumilight western blot enhanced chemiluminescence (ECL) substrate and captured on X-ray film.
Immunoprecipitation studies
Although it has been widely documented that PARP-1 is one of several large nuclear proteins that is poly(ADP-ribosyl)ated following an excitotoxic event (Ogata et al. 1981; de Murcia et al. 1994; Mandir et al. 2000), it has yet to be determined whether this occurs in the retina following NMDA insult. To assess this possibility, immunoprecipitation methodology was used to isolate PARP-1-immunoreactive protein from NMDA-treated retinas, and then determine its susceptibility to PAR. Briefly, retinas obtained 4 h after NMDA treatment were sonicated in cold lysis buffer [25 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1% Noniopt P740, 2.3 μg aprotonin, 1 μM leupeptin, 200 μM phenylmethylsulfonyl fluoride (PMSF) and 0.01% sodium azide]. Following centrifugation, the supernatant was immunoprecipitated with 0.5 μg of a rabbit anti-PARP-1 IgG for 18 h at 4°C. The antibody–protein complex was recovered with the addition of equilibrated Protein A Sepharose 4B gel (2 mg) and incubated overnight at 4°C. The recovered beads were rinsed through a series of low-salt and high-salt buffers, suspended in SDS loading dye and heat-denatured at 95°C for 10 min, as previously described (Goebel and Poosch 2001), prior to loading onto a 8% polyacrylamide–SDS gel. The separated proteins were electro-transferred and processed for PAR immunoreactivity (PAR-IR) as described above.
Ethidium bromide staining
Eyes injected as described above were dissected in cold PBS (0.01M sodium phosphate-0.85% NaCl, pH 7.2) to remove the cornea, lens, iris and ciliary body. The resulting eyecups were incubated in 100 μg/mL EtBr in PBS for 1 min, rinsed twice in PBS and fixed for 30 min in 4% paraformaldehyde. The stained retinas were oriented for proper mapping of the four retinal quadrants, removed from the eye cup and flat-mounted, ganglion cell side up, on glass slides. These retinas were coverslipped with Slowfade mounting medium, examined under epifluorescence using a rhodamine filter pack and digitally photographed using a Zeiss Apotome-equipped microscope (Jena, Germany).
Terminal dUTP nick end labelling (TUNEL)
Freshly-dissected eyecups were fixed overnight with 4% paraformaldehyde in 0.1M phosphate buffer, pH 7.2, rinsed in phosphate buffer and embedded in paraffin. Radial sections (10 μm) through the optic nerve were mounted on polylysine-coated slides, deparaffinized, subjected to proteinase K (PK) treatment (20 mg/mL PK in 10 mM Tris/HCl, pH 7.5) for 1 min at room temperature (23°C) and permeated with 0.1% Triton X 100 in PBS. The sections were then incubated for 1 h at 37°C in terminal transferase mixture using fluorescein-labeled dUTP (Roche in situ cell death labeling kit), rinsed, and coverslipped in Slowfade. The reacted retinas were digitally photographed using an FITC filter pack as above.
Measuring ATP levels
Treated and control eyes were rapidly removed and placed in ice-cold bicarbonate buffer. Within 15 s the retina was excised, cleaned of vitreal matter, homogenized in 0.4 mL 5% perchloric acid and centrifuged at 20 000 g for 10 min. ATP content was measured on diluted (200-fold with water) supernatant extracts using a Turner Systems luminometer (Sunnyvale, CA, USA), as described previously (Winkler et al. 2003). Sample values were compared against a standard curve for each experiment. Total protein in each retina was determined from the resulting pellet that was resuspended and neutralized in 0.5 mL 1 N NaOH (Henkel and Bieger 1994).
Measuring ganglion cell loss in the retina
Ganglion cell densities for untreated, NMDA-treated or PJ-34/NMDA-treated retinas were assessed using fluorogold retrograde labeling of the superior colliculus. Two sets of animals were used in this study, one group receiving 24 nmol PJ-34/20 nmol NMDA in the left eye and 20 nmol NMDA in the right, while a second set received 20 nmol NMDA in the left and no treatment in the right. One week later, the animals were anesthetized using ketamine/xylazine i.p. and then secured in a stereotaxic holder. Oval ports were drilled into the skull bilaterally at sites 1 mm lateral to the sagittal suture and spanning 0.5–2.5 mm rostrally from the lambda suture. A gelform slurry containing 2% fluorogold (FG) was loaded into an 18 gauge positive displacement needle and stereotaticaly positioned just above the superior colliculi. A 20 μL volume was injected over a 4 min period onto each colliculus, followed by a 4 min rest period prior to needle retraction. The scalp was sutured, and the animal was administered lactate Ringer’s (10–20 mL i.p.) twice daily to maintain adequate hydration. Four to seven days following this procedure the rats were killed and the eyes were removed and dissected, as above. Eyecups were fixed in 4% paraformaldehyde for 1 h, rinsed in PBS, then marked for orientation by a single radial incision from the margin to the optic nerve head to divide the superior-temporal and -nasal quadrants. The retinas were isolated, flat-mounted with the ganglion cell layer facing up, and coverslipped in Slowfade mounting medium. The entire retina was digitally photographed at 10×, in montage format, using an epifluorescence Zeiss Apotome system equipped with a 4′,6-diamidino-2-phenylindole (DAPI) filter pack. Cell counts were obtained using the PC version of NIH IMAGE (Scion Image, Scion Corp.), or by manual counting in regions where cell densities were too dense for computer counting. To ensure that computer-generated counts were accurate, random images were manually counted to verify these values. In all cases, we found good correspondence between the two counting methods. In agreement with Danias et al. (2002), we found no significant difference in the average cell densities, at a given distance from the optic nerve, among the four quadrants of the retina. As a result, cell densities were pooled from all four quadrants and assigned to bins of 1 mm increments, based on the distance from the optic nerve.
Results
NMDA induces PAR of proteins in a concentration-dependent manner
Retinas exposed to intravitreal injections of varying concentrations of NMDA were assessed at 4 h following treatment for PAR of proteins using western blot analysis (Figs 1a and b). The amount of PAR-IR increased in proportion to the level of NMDA (Figs 1a and b: data taken from 3–12 retinas per sample point). It was determined that 20 nmol NMDA provided a non-saturating PAR signal which optimized the monitoring of the pharmacological effects of the putative PARP inhibitors used in this study.
Fig. 1.
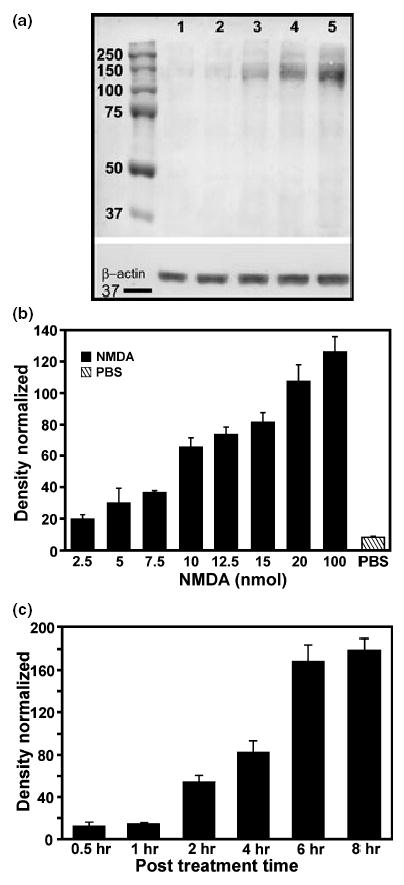
NMDA was injected into the vitreal chamber of rat eyes to determine whether NMDA receptor activation induces PARP activity and whether the visualization of the product, poly(ADP-ribose) (PAR), increases with the concentration of NMDA. Dose–response (a, b) and time course (c) studies show that NMDA induces PARP activity in the retina. (a) A representative western blot showing an increase in PAR-immunoreactivity (PAR-IR) as the concentration of NMDA increases. Lane 1: untreated; lane 2: PBS-treated; lane 3: 5 nmol NMDA; lane 4: 20 nmol NMDA; lane 5: 100 nmol NMDA. β-actin staining to confirm equal sample loading is also shown (Inset at bottom). (b) Dose–response histogram shows that an increase in NMDA, from 2.5 to 100 nmol, increases PAR-IR at 4 h following injections (n ≥3). Statistical analyses show significant differences in PAR-IR (p < 0.05) between 100 and 15 nmol, 20 and 12.5 nmol, 12.5 and 7.5 nmol, 10 and 5 nmol, 7.5 and 2 nmol, and 5 nmol NMDA and PBS as determined by one-way ANOVA. (c) Time course of the effects of injecting 20 nmol NMDA intravitreally on PAR-IR. Results show a sharp rise in PAR-IR between 1 and 2 h post-treatment (PT), with staining reaching a plateau by 8 h PT. Significant differences (p < 0.05) between 1 h and 2 h, 2 and 4 h, and 4 and 6 h were determined by one-way ANOVA
Following NMDA injection, the majority of PAR-IR was confined to proteins that were larger than 115 kDa (Fig. 1a). In most cases, PAR-IR was seen in a heavily stained band near 115 kDa and a broader, less intense series of bands ranging from 130 to 250 kDa. Although injection of PBS alone caused a slight elevation of PAR-IR in the retina, particularly in a band migrating at 115 kDa, a substantial increase in PAR was noted only in retinas exposed to NMDA at concentrations of 5 nmol or higher (Figs 1a and b).
In order to assess the effect of PARP inhibitors on the level of NMDA-induced PAR, it was first necessary to determine the time course of this event in the presence of 20 nmol of NMDA alone (Fig. 1c). Results show that PAR-IR was barely detectable and not significantly different from controls at 30 min and 1 h following treatment. However, by 2 h there was a significant elevation (p < 0.004, paired Student’s t-test, one-tail) in PAR-IR compared with controls, and these levels continued to rise, eventually reaching a plateau by 8 h (Fig. 1c). Four hours was chosen as the optimal time for the subsequent experiments whereby changes in PAR-IR could be easily monitored.
Numerous reports have shown that the enzyme PARP-1 is regulated by PAR (Ogata et al. 1981; de Murcia et al. 1994), and it is thought to be one of the first proteins to undergo this process following acute DNA damage (Ogata et al. 1981). Western blot comparisons of NMDA-treated retinas showed that the densely stained PAR-IR band that appeared at about 115 kDa (Fig. 2a: lanes 1 and 2) co-migrated with PARP-1-immunoreactive protein (Fig. 2a: lane 3). Immunoprecipitation studies confirmed this interpretation, as PARP-1-extracted proteins stained robustly for PAR-IR (Fig. 2b: lane 4), although the 2 day incubation process led to the hydrolysis of the 115 kDa molecule into its 89 kDa inactive form (Kaufmann et al. 1993). This was expected because inhibitors of caspase 3 and caspase 7, two of the major enzymes involved in the hydrolysis of PARP-1 (Germain et al. 1999), were not present during the initial 18 h immunoprecipitation of the whole retinal supernatant fraction. Despite the extensive cleavage of PARP-1, the PARP-1-immunoprecipitated protein also displays PAR-IR covering a broad size range (89–99 kDa), indicating that poly(ADP-ribosyl)ation of PARP-1 is extensive.
Fig. 2.

(a) Representative western blots of two retinas treated with 20 nmol NMDA (lanes 1 and 2) show similar staining intensities for PAR-IR. Note that the heavily stained PAR-IR band that appears at approximately 120 kDa (lanes 1 and 2, arrow heads) co-migrates with a PARP-1 immunoreactive band (lane 3, arrow) from the same retinal homogenate in lane 2. (b) To determine whether PARP-1 is a major recipient of poly(ADP-ribose) following NMDA treatment, individual retinal homogenates were immunoprecipitated using anti-PARP-1 antibody, separated on a polyacrylamide gel and reacted for PAR-immunoreactivity by western blot. Results in lane 4 show a broad band (arrow) between approximately 92–98 kDa that is PAR-IR. As inhibitors for caspase 3 and 7 (which are known to hydrolyze PARP; Germain et al. 1999) were not included in the immunoprecipitation mixture, we conclude that the PAR-IR band (arrow) is the 95 kDa hydrolyzed fragment of PARP-1 (lane 4). Denatured IgG of the PARP-1 (rabbit) antibody (indicated by * at approximately 50 kDa) reacts with the secondary antibody (peroxidase-labeled goat anti-rabbit IgG) used to visualize PAR-IR. Molecular weight standard (shown to the left of lane 4) is identical to that shown in (a). (c) To determine whether the increase in PAR-IR was due to a rise in PARP activity or an increase in PARP-1 protein levels, retinas treated with NMDA at either 20 nmol (lane 5), 100 nmol (lane 6) or 200 nmol NMDA (lane 7) were assessed for PARP-1 immunoreactivity. Results from three sets of retinas consistently show that although NMDA induces a rise in PAR-IR (see Fig. 1), it does not induce a change in PARP-1 protein levels. Samples were corrected against β-actin staining as shown in the lower bands (about 40 kDa) of lanes 5–7.
In a separate set of animals, PARP-1 levels were measured to determine whether NMDA altered the expression of this protein. Although there is a strong correlation between the amount of NMDA injected into the vitreous and the intensity of PAR-IR (Figs 1a and b), this was not the case with PARP-1 as there was essentially no difference in the level of PARP-1-IR following a 4 h exposure to 20 nmol (Fig. 2c: lane 5), 100 nmol (Fig. 2c: lane 6) or 200 nmol NMDA (Fig. 2c: lane 7).
Blockade of PARP down-regulates the induction of PAR by NMDA in a concentration-dependent manner
Selected inhibitors of PARP were assessed for their ability to block NMDA-induced PAR (Fig. 3a). Inhibition was expressed as the percentage reduction of PAR-IR compared with NMDA-treatment alone. 3-BZ did not have any significant effect on NMDA-induced PAR-IR at concentrations of 120 nmol and below. At 1200 nmol, 3-BZ caused a moderate reduction in PAR-IR and peaked at 2400 nmol, whereby a 52% reduction was obtained (shown in Fig. 3a). BZ was tested at doses up to 120 nmol (its solubility limit), but even at this dose, there was little inhibition of PAR-IR, in agreement with an earlier report (Eliasson et al. 1997). Results showed that PJ-34 was the most effective antagonist in blocking PAR. A 25% inhibition of PARP activity was observed with as little as 0.24 nmol PJ-34, and with 2.4 nmol, inhibition significantly increased to 84% (p < 0.0004, paired, one-tail Student’s t-test; n = 10) compared with NMDA treatment alone. Figure 3(b) shows that peak inhibition averaging 93% was achieved with 24 nmol PJ-34. Attempts to demonstrate complete blockade of PARP activity using higher concentrations of PJ-34 (72 nmol) were compromised by substantial swelling of the retina in the presence of inhibitor alone. Because of this complication, we focused on effects of 24 nmol PJ-34 in the subsequent experiments.
Fig. 3.
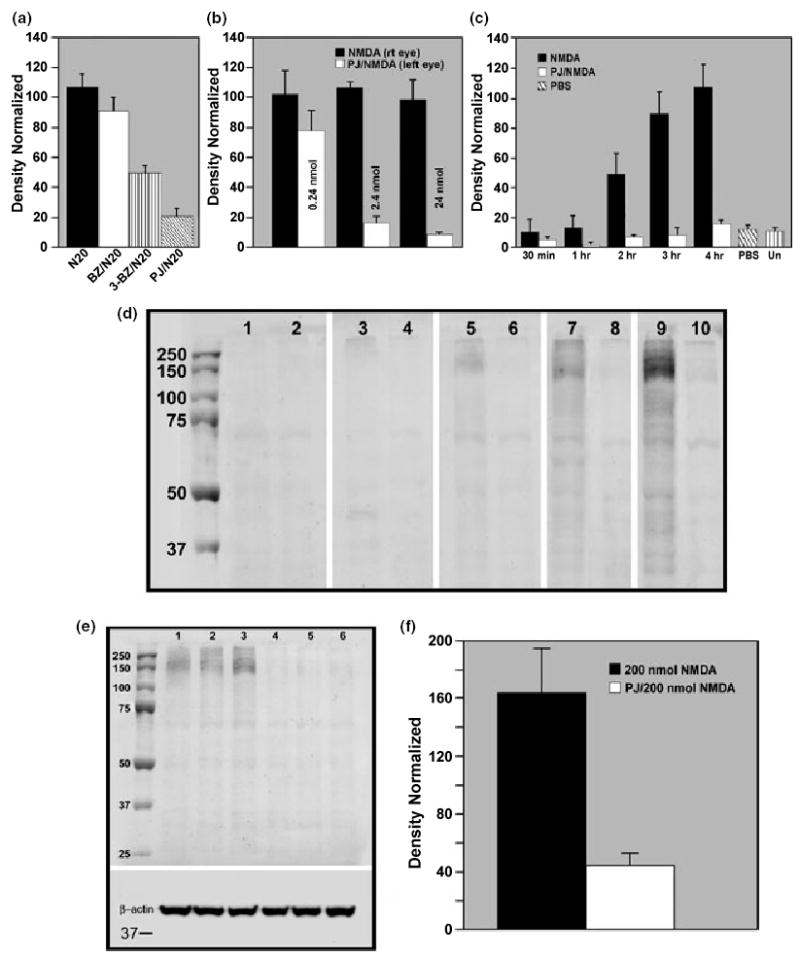
(a) The efficacy of selected PARP inhibitors in attenuating NMDA-induced PAR-IR were tested in vivo. The resulting histogram shows the peak level of suppression of PARP activity for each inhibitor tested. Benzamide (120 nmol BZ: white bar) was least effective in blocking PARP activity as PAR-IR levels were not statistically different from NMDA alone (N20: black bar). In contrast, 3-benzamide (3-BZ: 2400 nmol: vertical striped bar) and PJ-34 (PJ/N20; 2.4 nmol: diagonal striped bar) produced a significant reduction (52% and 84%, respectively) in PAR-IR (p < 0.001 for 3-BZ/N20 vs. N20; and p < 0.0005 for PJ/N20 vs. N20, as determined by one-way ANOVA). (b) Compiled data compare NMDA-induced PAR-IR in the presence or absence of increasing concentrations of PJ-34 and show that this inhibitor effectively blocks NMDA-induced PAR-IR in a concentration-dependent manner. The delivery of 24 nmol PJ-34 was selected for its ability to reduce NMDA-induced PAR-IR by an average of 90%. There was no significant difference (p > 0.05, one-way ANOVA) between NMDA and 0.24 nmol PJ-34/NMDA treatments. However, 2.4 and 24 nmol PJ-34 both resulted in a significant decrease (p < 0.001, n ≥ 6) in PAR-IR compared with NMDA treatment alone, and were also significantly different from each other (p < 0.04). (c) Comparison of retinas exposed to 20 nmol NMDA shows that PAR-IR is attenuated in the presence of PJ-34 up to 4 h PT, and that the levels of PAR-IR in these samples were not significantly different from untreated or vehicle-treated retinas (p > 0.05; one-way ANOVA; n ≥ 3). Further analysis determined that there was no significant differences between NMDA and PJ/NMDA retinas up to 1 h (p > 0.05). However, 2 h and beyond, PJ-34 significantly (p < 0.003) lowered PAR-IR compared with NMDA treatment alone. (d) Representative western blots showing PAR-IR in paired retinas, with the right eye receiving 20 nmol NMDA (odd numbered lanes) and the left eye given 24 nmol PJ-34 with 20 nmol NMDA (even numbered lanes), were examined at 30 min (lanes 1 and 2), 1 h (lanes 3 and 4), 2 h (lanes 5 and 6), 3 h (lanes 7 and 8) and 4 h (lanes 9 and 10) following NMDA. PAR-IR in the NMDA-treated retina is faintly detectable by 1 h (lane 3) and increases in intensity with time (lanes 5, 7 and 9), whereas PAR-IR in PJ-34/NMDA-treated retinas was attenuated at all time points measured (lanes 2, 4, 6, 8 and 10). (e) To determine the effectiveness of PJ-34 blockade, retinas were given a strong insult consisting of 200 nmol NMDA in the absence (lanes 1–3) or presence of 24 nmol PJ-34 (lanes 4–6). Results show that PJ-34 attenuates NMDA-induced PAR-IR following this high dose of NMDA. β-actin (insert) shows equivalent loading. (f) Data from five sets of retinas show that 24 nmol PJ-34 significantly reduces the level of PAR-IR by an average of 78% (p < 0.0006; by paired Student’s t-test) following a strong (200 nmol) NMDA insult.
Retinas treated with 20 nmol NMDA in the presence or absence of 24 nmol PJ-34 were assessed for PAR-IR at 0.5, 1, 2, 3 and 4 h following treatment (Figs 3c and d). Levels of PAR in NMDA-treated retinas sampled at 30 min and 1 h were not significantly different from untreated retinas or retinas exposed to vehicle (PBS with DMSO) alone. However, by 2 h, PAR-IR had increased nearly 4.5-fold and by 4 h, this increase amounted to more than 10-fold. In the presence of PJ-34, PAR-IR in NMDA-treated retinas was at or below control levels at all tested times, including 8 h (data not shown). The time course of the decrease in PAR-IR due to PJ-34 blockade of PARP activity is illustrated in the western blots shown in Fig. 3(d). Lanes 9 and 10, representing the 4 h post-NMDA time point, show the effect the best. Here, the intense band with NMDA (lane 9) is only faintly detectable in the presence of PJ-34 (lane 10). The effectiveness of PJ-34 in blocking PARP activity following a strong insult (200 nmol NMDA) is shown in Fig. 3(e and f). As can be seen, PJ-34 decreased NMDA-induced PAR-IR by 75%, further demonstrating the effectiveness of the inhibitor.
Measurement of ATP content
ATP levels were measured at selected time points in NMDA-treated, sham-treated and untreated retinas (Fig. 4). ATP content was similar between NMDA-treated and control retinas at 4 h. However, by 6 and 8 h there was a 20% decline in ATP content in NMDA- treated retinas. To determine whether blockade of PARP activity with PJ-34 prevented the decline in ATP at 8 h, animals were treated so that one eye of each animal received PJ-34/NMDA treatment while the companion eye received either NMDA or vehicle alone. Results from at least four matched pairs per condition (Fig. 4b) showed that ATP levels in the PJ-34/NMDA-treated retinas were significantly elevated (p < 0.04; paired, one-tail Student’s t-test) compared with retinas exposed to NMDA alone, and not different from the controls.
Fig. 4.
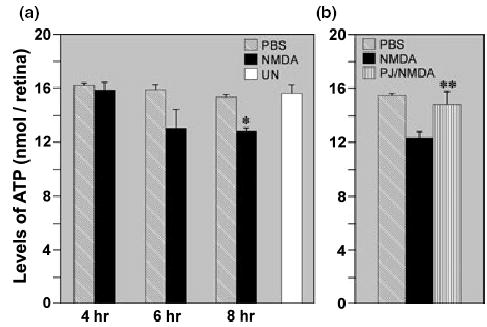
(a) Control (untreated/sham-treated) retinas and retinas treated with 20 nmol NMDA were assessed for ATP content at designated times. Results from at least five sets of retinas per time point show no significant differences in ATP content between NMDA- and sham-treated retinas at 4 and 6 h following treatment. However, by 8 h, the level of ATP in NMDA-treated retinas was significantly lower than in PBS-treated and untreated (UN) controls (*p < 0.005, paired Student’s t-test). Comparisons of ATP levels between sham-treated and untreated retinas showed that there were no significant differences at all three time points (p > 0.81, paired Student’s t-test). (b) Blockade of PARP activity with PJ-34 significantly attenuated the NMDA-induced loss of ATP content at 8 h (**p < 0.04, paired Student’s t-test). In addition, the levels of ATP in the PJ-34/NMDA-treated retinas were not significantly different from controls (p > 0.05, paired Student’s t-test). Data represent n = 4 for each of the following treatment conditions: NMDA vs. PJ34/NMDA; NMDA vs. PBS; PJ34/NMDA vs. PBS.
Effects of NMDA and PJ-34 on membrane selectivity in retinal cells
Retinas were exposed to increasing concentrations of NMDA (Figs 5a–c) and were assessed at 4 h, in comparison with controls (Figs 5d–f), for loss of plasma membrane selectivity to EtBr. After treatment with 5, 20 or 100 nmol NMDA, retinal whole mounts displayed a uniform distribution of EtBr-positive cells in the ganglion cell layer (GCL), in both central and peripheral areas. The high density of EtBr-labeled cells and broad range of cell sizes labeled with EtBr indicate that, even at low magnification, multiple cell types, i.e. ganglion cells and displaced amacrine cells, are susceptible to NMDA. In addition to the stained cells in the GCL, small diameter cells (5–8 μm) in the inner half of the inner nuclear layer (INL) (data not shown) were also stained with EtBr. Although identification of these cells was hindered by the intense fluorescence in the GCL, areas that were resolvable suggested that populations of amacrine cells and, possibly, bipolar cells were stained, and that their distribution was also uniform. Control retinas that were exposed to vehicle alone (Fig. 5d), treated with 2.4 nmol PJ-34 (Fig. 5e) or left untreated (Fig. 5f) showed only sparse EtBr labeling (arrows), indicating that, in the absence of NMDA, cell selectivity was intact.
Fig. 5.
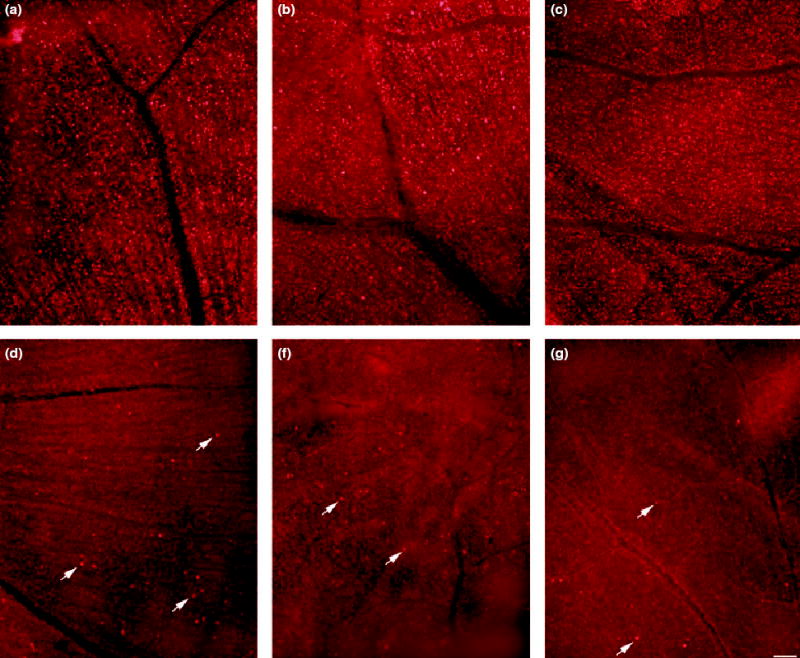
Retinas were assessed for the loss of membrane selectivity using ethidium bromide (EtBr) following treatment with (a) 5 nmol NMDA, (b) 20 nmol NMDA, (c) 100 nmol NMDA, (d) PBS-2% DMSO (vehicle), (e) 24 nmol PJ-34 (f) or left untreated. All retinas were processed at 4 h following injection. Flatmount views of the ganglion cell layer (GCL) show that NMDA causes the loss of membrane selectivity in numerous cells (a–c). Note that the staining in the NMDA-treated retinas (a–c) was uniformly distributed, whereas staining in the three control retinas (d–f) showed only random staining for EtBr (arrows). Horizontal bar shown in (f) = 100 μm applies to all panels.
To determine the time course of NMDA-induced loss of membrane selectivity to EtBr, cell densities were determined at 1–2 mm from the optic nerve head in retinas exposed to 20 nmol NMDA and processed at 30 min and 1, 2 and 3 h (Fig. 6a–d). Labeled and unlabeled cells in the GCL could easily be identified in retinas exposed to EtBr, allowing an accurate assessment of the percentage of stained cells. Loss of membrane selectivity was detectable as early as 30 min after NMDA treatment (Fig. 6a), accounting for 15.4 ± 0.6% of the cells. The percentage of EtBr-labeled cells increased to 23.00 ± 0.9% (n = 3) by 1 h (Fig. 6b), to 29.6 ± 1.3% (n = 3) at 2 h (Fig. 6c) and to 39.6 ± 1.3% (n = 6) at 3 h (Fig. 6d). By 4 h, the percentage of stained cells rose to 50.1 ± 0.6% (n = 10) (Fig. 7a) and remained unchanged at 6 h (data not shown). Note that rapid loss of membrane selectivity in NMDA-treated retinas (within 2 h of insult) precedes the appearance of PAR-IR (see Fig. 3c) and the decline in retinal ATP content (Fig. 4).
Fig. 6.

Retinas treated with 20 nmol NMDA were examined for EtBr staining at designated time points to map the progression of the loss of membrane selectivity in cells located in the GCL. (a) Flatmount views of the GCL show EtBr staining in medium- to large-sized cells 30 min following insult. A progressive rise in the density of stained cells is noted at 1 h (b), 2 h (c) and 3 h following NMDA (d), with the density reaching a plateau by 4 h (see Figs 7a and c). At the later time points (d) EtBr staining was seen in cells with diverse morphology, with some displaying apoptotic features, such as nuclear condensation and apoptotic bodies, and others showing homogeneous staining of the nucleus and cytoplasm, a feature that is consistent with necrosis. Horizontal bar in (d) = 10 μm and applies to all panels.
Fig. 7.
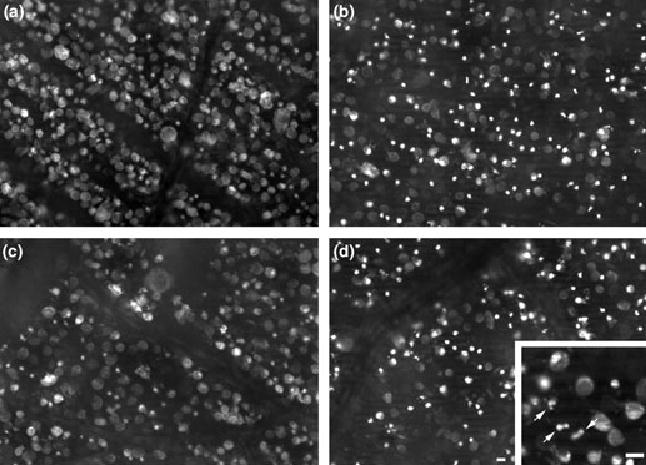
Comparison of paired retinas. One eye injected with 20 nmol NMDA alone (a and c) and the companion eye treated with 24 nmol PJ-34/20 nmol NMDA (b and d), were stained with EtBr and examined at 4 h. Views from the central (a and b) and peripheral regions (c and d) of both retinas are shown. There was no significant difference in the density of EtBr-labeled cells in the presence or absence of PJ-34 (p > 0.05 as determined by one-way ANOVA, n = 6). Staining in the NMDA-treated retina (a and c) reveals features that are characteristic of both apoptosis (cells with intensely stained nuclei) and necrosis (uniform staining of the cell and nucleus). Note that in retinas treated with PJ-34/NMDA (b and d), there are more EtBr-stained cells that display apoptotic features, including apoptotic bodies in both central (b) and peripheral (d) regions, than are found in NMDA-treated retinas (a, c). Inset of a higher magnification of the PJ-34/NMDA-treated retina shows apoptotic bodies (arrows). Horizontal bars = 10 μm. Bar in (d) applies to (a–c).
Although peak density of EtBr-labeling was achieved by 4 h, the morphological appearance of the stained cells changed progressively between 2 and 4 h (Figs 6c and d, 7a–c). Prior to 2 h, most of the EtBr-labeled cells were medium- to large-sized cells, measuring 15–40 μm in diameter. Labeling in these cells was mostly cytoplasmic, although a small number of cells were stained in the nucleus and cytoplasm (Figs 6a and b). However, by 2 h, smaller stained cells (less than 8 μm in diameter) were also evident (Fig. 6c) and their staining persisted at 3 h (Fig. 6d) and beyond (Figs 7a and c). At the later times, the number of cells displaying preferential staining of the nucleus increased (Figs 6c and d), although cells showing uniform staining in the nucleus and cytoplasm were still present (Fig. 6d, 7a–c).
Blockade of NMDA-induced PARP activity by PJ-34 failed to prevent loss of membrane selectivity. This effect was consistent among all six animals examined. Note that while there was no difference (p < 0.05; determined by one-way ANOVA) in the percentage of EtBr-stained cells between PJ-34/NMDA-treated and NMDA-treated retinas (see Fig. 7), there was a difference in the distinct pattern of staining within different cells. That is to say, cells treated with PJ-34 showed more intense staining of the nucleus and displayed more advanced apoptotic features, such as apoptotic bodies, in both central and peripheral regions compared with the pattern of staining seen with NMDA alone (Fig. 7d, inset).
TUNEL labeling
Retinas treated with 20 nmol NMDA or with 24 nmol PJ-34/20 nmol NMDA were processed for TUNEL labeling (Fig. 8). Control eyes treated with PBS-2% DMSO (Fig. 8f; n = 4), PBS alone or left untreated (data not shown) were TUNEL-negative. NMDA-treated retinas (Figs 8a and b) showed sparse TUNEL labeling in both the GCL and INL by 4 h (Fig. 8a) but by 6 h, an increase in the number of stained cells was seen in both layers (Fig. 8b) 0(n = 8 retinas sampled). Retinas treated with 24 nmol PJ-34/20 nmol NMDA (n = 8) showed stronger staining of cells in both layers at 6 h (Fig. 8c). These cells displayed apoptotic features that were more advanced in staging (Figs 8c and e) than those seen with NMDA alone (Figs 8b and d), and featured some cells with densely stained, irregularly-shaped nuclei and others displaying cell fragmentation and apoptotic bodies (Fig. 8e).
Fig. 8.

Retinas were assessed for TUNEL labeling following NMDA treatment in the presence or absence of PJ-34. (a) TUNEL labeling in the NMDA-treated retina shows a sparse distribution in both the GCL and INL at 4 h. Six hours following NMDA treatment (b), the frequency of TUNEL-positive cells was dramatically increased in both layers. Retinas exposed to 24 nmol PJ-34/20 nmol NMDA at 6 h (c) show a labeling pattern similar to that seen with NMDA treatment alone (b). However, there is a noticeable increase in the intensity of staining and in the number of cells that display advanced apoptotic features (arrowheads), as compared with NMDA treatment alone. NMDA treatment alone (d) and PJ-34/NMDA treatment (e) are shown at higher magnification to illustrate the differences in the morphological appearances of the TUNEL-positive cells. In the NMDA-treated retina (d), the TUNEL-positive cells show a diverse range of morphology, with some cells displaying densely stained nuclei while others show preferential staining of chromatin in the proximity of the nuclear envelope (a feature of early stage apoptosis). In contrast, most of the TUNEL-stained cells in the PJ-34/NMDA-treated retina show more advanced staging of apoptosis, as indicated by the presence of apoptotic bodies (arrowheads). Controls treated with PBS-2% DMSO (f) were TUNEL-negative at 6 h. Non-specific staining in the outer segments (OS) is noted in all retinas assayed. Abbreviations: outer nuclear layer (ONL), outer plexiform layer (OPL), inner plexiform layer (IPL). Horizontal bar in (c) = 10 μm and applies to (a) and (b); horizontal bar in (e) = 10 μm applies to (d).
Fluorogold retograde labeling of retinal ganglion cells
Glutamate- and NMDA-induced toxicity affects not only ganglion cells but also, cells in the INL (Sisk and Kuwabara 1985; Ullian et al. 2004). In the rat retina, the GCL consists of mixed populations of cells: ganglion, displaced amacrines, astrocytes and microglia. To assess the extent of loss of ganglion cells, and to document the extent of protection against NMDA by blockade of PARP, retrograde labeling of ganglion cells with FG was used. Results showed that NMDA, with or without PJ-34, caused a significant loss of ganglion cells 14 days post-treatment, amounting to 56% and 75%, respectively, in comparison with cell counts in control retinas (Fig. 9). Although there was some variation in ganglion cell densities among animals receiving the same treatment paradigm, blockade of PARP activity with PJ-34 significantly improved the retention of ganglion cells at all eccentricities, as measured in matched pairs from four different experiments (Fig. 10).
Fig. 9.

Retinas were assessed to determine whether blockade of PARP activity by PJ-34 provides neuroprotection against NMDA in retinal ganglion cells. Fluorogold labeling (FG) of ganglion cells obtained from identical regions of the retina (1.5 mm from the optic nerve head in the superior nasal quadrant; a–c) shows representative labeling patterns in the untreated retina (a), as compared with a matched paired set of retinas treated with NMDA alone (b) or PJ-34/NMDA (c) sampled at 14 days post-insult. In the untreated retina (a), robust labeling of ganglion cells is clearly noted. However, following NMDA treatment (b), the density of FG cells was reduced by an average of 75%. Comparison of the companion retina treated with PJ34/NMDA (c) shows an approximately 50% increase in ganglion cell density, as compared with NMDA treatment alone (b). However, blockade of PARP activity does not provide full neuroprotection, as the density of FG-positive cells in the PJ-34/NMDA-treated retina (b) is 50% less than observed in the untreated retina (a). Horizontal bar in (c) = 50 μm applies to all panels.
Fig. 10.
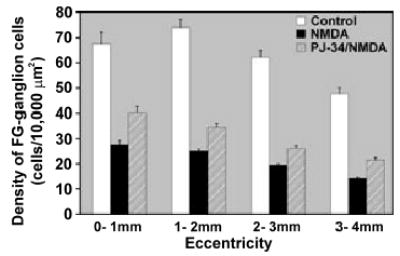
Blockade of PARP activity with PJ-34 provides partial neuroprotection in the retina. Data obtained from four retinal pairings with one eye treated with NMDA alone and the matching eye exposed to PJ-34/NMDA were assessed for ganglion cell survival using fluorogold (FG) retrograde labeling 14 days after NMDA insult. Values reported in this set of experiments were normalized to the average density value obtained from the NMDA-treated retinas (n = 6). Regional cell densities of FG-labeled ganglion cells were obtained from each retina at 1 mm increments from the optic nerve. Results indicate that retinas treated with NMDA, with or without inclusion of PJ-34, show a significant loss of ganglion cell densities in all four regions sampled (n = 4; p < 0.0001 Student’s t-test, unpaired). In comparing the FG-labeled ganglion cell densities between NMDA- and PJ-34/NMDA-treated retinas, blockade of PARP activity significantly improves survival of retinal ganglion cells by an average of 43.56% ± 7.03 over NMDA treatment alone (*p ≤0.0008, n = 4). Error bars reflect standard error of the mean.
Discussion
Neuronal cell death resulting from an excitotoxic challenge has been thought to occur through two distinct modes: apoptosis or necrosis (Leist and Nicotera 1997; Ientile et al. 2001). Newer studies have suggested that the pathway leading to cell death is dictated by the concentration of ATP, high levels supporting apoptosis and low or depleted levels resulting in necrosis (Portera-Cailliau et al. 1997; Koh et al. 2005). Numerous laboratories have explored these pathways using both in vivo and in vitro models. It is now hypothesized that hyperactivation of PARP, signaled through the NMDA receptor (Mandir et al. 2000) and/or by activation of nNos (Dawson et al. 1991; Pieper et al. 2000), plays an integral role in controlling the relative contributions of apoptotic and necrotic pathways (for review see Yu et al. 2003). Hyperactivation of PARP-1, the most major form of PARP (Pieper et al. 2000), is suggested to be the dominant effector of necrosis in the brain (Cole and Perez-Polo 2002) and, as shown in the present study, appears to have a similar role in the retina (Fig. 2).
Current understanding of how PARP-1 hyperactivation leads to necrosis involves the depletion of the PARP-1 substrate NAD+ from the cytoplasmic pool. In vitro studies have shown that when NAD+ levels fall, due to poly(ADP-ribosyl)ation of nuclear proteins by PARP-1, so do the endogenous stores of ATP (Ha and Snyder 1999). The drop in ATP has been shown to be the result of cells attempting to replenish NAD+ (Ha and Snyder 1999). Cells thus become energy starved, which leads to a failure in ATP-dependent active ion transport, cell swelling and loss of membrane permeability properties, i.e. necrosis (Schwartz et al. 1993).
In the retina, NMDA causes a concentration-dependent increase in the level of PAR-IR of large molecular weight proteins (Fig. 1a). This process is the direct result of PARP activity, as evidenced by the attenuation of NMDA-induced PAR-IR in retinas treated with the PARP-antagonist, PJ-34 (Fig. 3). These findings are consistent with those reported by Mandir et al. (2000) in the brain. Although PJ-34 reduces PAR-IR by approximately 90% following NMDA treatment, it is not completely abolished as a faint band, corresponding in size to PARP-1, remains (Fig. 3d: lane 10). Residual PAR-IR following PJ-34 treatment in the brain has also been reported (Abdelkarim et al. 2001; Besson et al. 2005), and in the present study was faintly detectable in PBS-treated and untreated retinas (Figs 1a and b, 3c), as well as in retinas exposed to PJ-34 alone (data not shown). The latter finding suggests that an isoform of PARP or an alternative pathway for PAR of proteins exists that is insensitive to PJ-34 blockade.
With the level of PAR-IR correlating to the degree of NMDA insult (Figs 1a and b), we addressed the question as to whether this effect corresponds to an increased level of PARP-1 protein expression. Results consistently showed that the NMDA-induced increase in PAR is not due to a rise in the level of the PARP-1 protein (Fig. 2b), as levels of PARP-1-IR in retinas treated with increasing concentrations of NMDA were indistinguishable. Immunoprecipitation studies showed that PARP-1 is highly poly(ADP-ribosyl)ated (Fig. 2a) in the retina following NMDA, as has been shown in the brain (Virág and Szabó 2002). At this time, it is not known if the elevation of PAR-IR is the direct result of an increase in PARP activity (Skidmore et al. 1979; Wielckens et al. 1982), an alteration in the hydrolysis of PAR by poly(ADP-ribose) glycohydrolase (PARG; Davidovic et al. 2001), or a combination of both processes.
A striking result obtained in the present study is that blockade of PARP activity enhances apoptotic features in PJ-34/NMDA-treated retinas. This was shown in a series of matched-pair retinas in which EtBr- (Fig. 7) and TUNEL-stained (Fig. 8) PJ-34/NMDA-treated retinas revealed more advanced apoptotic features (Figs 7b and d, 8c and e) than companion retinas treated with NMDA alone (Figs 7a and c, 8b and d). In the PJ-34/NMDA-treated retinas, many of the labeled cells exhibited nuclei that were fragmented into apoptotic bodies (see Fig. 7d, inset, and Fig. 8e) or displayed nuclei that were highly condensed and intensely stained. These findings are consistent with studies showing that when PARP activity is inhibited, fewer cells display necrotic features while greater numbers of dying cells express apoptotic features (Ha and Snyder 1999; Moroni et al. 2001). This suggests that as long as ATP is available in the dying cells, the apoptotic pathway can progress (Ünal-Çevik et al. 2004).
The above observations are also consistent with the reports of Bonfoco et al. (1995) and, more recently, of Meli et al. (2004), which show that the degree of NMDA insult dictates whether neurons display apoptotic or necrotic features. This is clearly demonstrated in Fig. 1(a), where retinas treated with 2 nmol NMDA show very low levels of PAR-IR and those exposed to a strong insult (100 nmol NMDA) show intense PAR-IR at 4 h (Fig. 1a). Again, this would predict that ATP reserves are retained after a low-level insult, favoring apoptosis, whereas under a strong insult, endogenous ATP stores are exhausted, resulting in a reduction or truncation of the apoptotic process and leaving the cell vulnerable to necrosis. On this basis, it is expected that ATP levels would decline to low levels in the affected cells and that this decline would be detected within 4 h following NMDA, because at this time, the density of cells that have lost their membrane selectivity to EtBr has reached its peak (Figs 6 and 7). However, ATP levels in retinas sampled 4 and 6 h after 20 nmol NMDA treatment were not significantly different from untreated or PBS controls (p > 0.05; n ≥5 per sample point) (Fig. 4a). In contrast, a measurable decline (20%) in ATP was found in retinas sampled 8 h following NMDA (Fig. 4a). Thus, the loss of membrane selectivity precedes any major decline in ATP content, as measured in whole retinas.
The timing of the biochemical events linked to necrosis appears to lag behind the timing of the loss of membrane selectivity to EtBr, suggesting that this event may not be the result of PAR or ATP loss. Additional support for this argument is demonstrated by PARP blockade with PJ-34, whereby the induction of PAR-IR and the decline in ATP by NMDA was prevented (Figs 3 and 4b, respectively). However, loss of membrane selectivity to EtBr was not attenuated (Figs 7b and d). Similar observations have been reported following NMDA treatment in the hippocampus and cerebral cortex (Gilliams-Francis et al. 2003). Thus, our data do not support the view that PARP-dependent depletion of ATP or the formation of PAR is the primary cause of the NMDA-induced loss of membrane selectivity.
Although the present study shows that PJ-34 effectively inhibits PAR-formation in the retina by PARP, this treatment provides only partial neuroprotection against ganglion cell loss. In the four matched pairs of retinas that were retrogradely labeled with FG, PJ-34 showed a 56% loss in ganglion cells compared with 71% in retinas treated with NMDA alone (Figs 9 and 10). Mandir et al. (2000) reported that PARP−/− mice were completely resistant to NMDA-induced toxicity in the striatum; this result differs from the present work where we achieved only partial protection, despite inhibiting fully PARP-dependent PAR activity. The possibility that PJ-34 did not adequately diffuse into the retina is unlikely because PJ-34 consistently depressed PAR formation to levels that were indistinguishable from control values following exposure of retinas to a low and high dose of NMDA (Figs 3b, d–f). Furthermore, ATP is maintained at a normal level in PJ-34/NMDA-treated retinas (Fig. 4). This suggests that if PARP plays a role in the loss of membrane integrity in NMDA-treated retinas, it is not the result of PAR. What this process may involve is presently unknown.
In summary, NMDA-induced toxicity in the rat retina causes an increase in PAR formation in a dose-dependent manner that is effectively blocked by the PARP inhibitor, PJ-34. Blockade of PARP activity attenuates the decline in endogenous ATP stores and appears to accelerate the process of apoptosis in cells in the INL and GCL. However, it does not block the loss of membrane selectivity, a process that is linked to necrosis. Although pharmacological blockade of PARP activity does not fully prevent ganglion cell loss following NMDA-induced neurotoxicity, the fact that partial neuroprotection is observed offers some promise that this approach can be used in conjunction with other putative neuroprotective agents to prevent propagation of the cell death signal in neurons.
Acknowledgments
We thank Ms Eren D. Berberoglu and Ms Catherine A. Starnes for their expert technical assistance on this project. This work was supported by Grants from the National Eye Institute, including EY014430 (DJG), EY10015 (BSW) and Vision Core Grant EY-04068 (DJG), and by Vision Research Infrastructure Development Grant EY R24-014803 (BSW).
References
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, Endres M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–260. [PubMed] [Google Scholar]
- Besson VC, Zsengeller Z, Plotkine M, Szabó C, Marchand-Verrecchia C. Beneficial effects of PJ34 and INO-1001, two novel water-soluble poly(ADP-ribose) polymerase inhibitors, on the consequences of traumatic brain injury in rat. Brain Res. 2005;1041:149–156. doi: 10.1016/j.brainres.2005.01.096. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: Two distinct events induced, respectively by mild and intense insults with N-methyl-D aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Cole KK, Perez-Polo JR. Poly(ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H2O2 injury. J Neurochem. 2002;82:19–29. doi: 10.1046/j.1471-4159.2002.00935.x. [DOI] [PubMed] [Google Scholar]
- Danias J, Shen F, Goldblum D, Chen B, Ramos-Esteban J, Podos SM, Mittag T. Cytoarchitecture of the retinal ganglion cells in the rat. Invest Ophthal Vis Sci. 2002;43:587–594. [PubMed] [Google Scholar]
- Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in controlling poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJL, Sampei K, Mandir AS, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Martin F, Serrano T, Reiriz J, Pérez-Navarro E, Alberch J, Macaya A, Planas AM. Both apoptosis and necrosis occur following intrastriatal administration of excitotoxins. Acta Neuropathol. 1995;90:504–510. doi: 10.1007/BF00294812. [DOI] [PubMed] [Google Scholar]
- Germain M, Affar E-B, D’Amours D, Dixit VM, Salvesen GS, Poirier GG. Cleavage of automodified Poly(ADP-ribose) polymerase during apoptosis. J Biol Chem. 1999;274:28 379–28 384. doi: 10.1074/jbc.274.40.28379. [DOI] [PubMed] [Google Scholar]
- Gilliams-Francis KL, Quaye AA, Naegele JR. PARP cleavage, DNA fragmentation and pyknosis during excitotoxin-induced neuronal death. Exp Neurol. 2003;184:359–372. doi: 10.1016/j.expneurol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Goebel DJ, Poosch MS. Transient down-regulation of NMDA receptor subunit gene expression in the rat retina following NMDA-induced neurotoxicity is attenuated in the presence of the non-competitive NMDA receptor antagonist MK-801. Exp Eye Res. 2001;72:547–558. doi: 10.1006/exer.2001.0981. [DOI] [PubMed] [Google Scholar]
- Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96:13 978–13 982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel AW, Bieger SC. Quantification of proteins dissolved in an electrophoresis sample buffer. Anal Biochem. 1994;223:329–331. doi: 10.1006/abio.1994.1595. [DOI] [PubMed] [Google Scholar]
- Ientile R, Macaione V, Teletta M, Pendale S, Torre V, Macaione S. Apoptosis and necrosis occurring in excitotoxic cell death in isolated chick embryo retina. J Neurochem. 2001;79:71–78. doi: 10.1046/j.1471-4159.2001.00532.x. [DOI] [PubMed] [Google Scholar]
- Kaufmann S, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL. Of cell death by poly(ADP-ribose) polymerase-1. Pharm Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Leist M, Nicotera P. The shape of cell death. Biochem Biophys Res Commun. 1997;236:1–9. doi: 10.1006/bbrc.1997.6890. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, Dawson VL. NMDA but not non-NMDA excitotoxicity is mediated by poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. doi: 10.1523/JNEUROSCI.20-21-08005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meli E, Pangallo M, Picca R, Baronti R, Moroni F, Pellegrini-Giampietro E. Differential role of poly(ADP-ribose) polymerase-1 in apoptotic and nercrotic neuronal death induced by mild or intense NMDA exposure in vitro. Mol Cell Neurosci. 2004;25:172–180. doi: 10.1016/j.mcn.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, Romagnoli P, Pellicciari R, Pellegrini-Giampietro DE. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ. 2001;8:921–932. doi: 10.1038/sj.cdd.4400884. [DOI] [PubMed] [Google Scholar]
- de Murcia G, Schreiber V, Molinete M, Saulier B, Poch O, Masson M, Niedergang C, Menissier de Murcia J. Structure and function of poly(ADP-ribose) polymerase. Mol Cell Biochem. 1994;138:15–24. doi: 10.1007/BF00928438. [DOI] [PubMed] [Google Scholar]
- Ogata N, Ueda K, Kawaichi M, Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J Biol Chem. 1981;256:4135–4137. [PubMed] [Google Scholar]
- Olney JW. Excitatory transmitter neurotoxicity. Neurobiol Aging. 1994;15:259–260. doi: 10.1016/0197-4580(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Pieper AW, Blackshaw S, Clements EE, et al. Poly(ADP-ribosyl)ation basally activated by DNA strand breaks reflects glutamate-nitric oxide neurotransmission. Proc Natl Acad Sci USA. 2000;97:1845–1850. doi: 10.1073/pnas.97.4.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;6:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for apoptosis-necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- Schwartz LM, Smith SW, Jones MEE, Osborne BA. Do all programed cell deaths occur via apoptosis? Proc Natl Acad Sci USA. 1993;90:980–984. doi: 10.1073/pnas.90.3.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JL, Berger SJ, Berger NA. Poly(ADP-ribose) polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5′-triphosphate pools in DNA-damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochemistry. 1983;22:5188–5194. doi: 10.1021/bi00291a019. [DOI] [PubMed] [Google Scholar]
- Sisk DR, Kuwabara T. Histological changes in the inner retina of albino rats following intravitreal injection of monosodium glutamate. Graefes Arch Clin Exp Ophthalmol. 1985;223:250–258. doi: 10.1007/BF02153655. [DOI] [PubMed] [Google Scholar]
- Skidmore CJ, Davies MI, Goodwin PM, Halldorsson H, Lewis PJ, Shall S, Siaee AA. The involvement of poly(ADP-ribose) polymerase in the degradation of NAD+ caused by gamma-radiation and N-methyl-N-nitrosourea. Eur J Biochem. 1979;101:135–142. doi: 10.1111/j.1432-1033.1979.tb04225.x. [DOI] [PubMed] [Google Scholar]
- Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14 376–14 381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, Bredesen DE. Paraptosis: mediation by MAP kinase and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11:1066–1075. doi: 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- Szabó C, Zingarelli B, O’Connor M, Salzman AL. DNA strand breakage, activation of poly(ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity in macrophage and smooth muscle cells exposed to peroxynitrate. Proc Natl Acad Sci USA. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Greenburg JH. The effect of reperfusion on neuroprotection using an inhibitor of poly(ADP-ribose) polymerase. Neuropharmacology. 1999;10:2017–2022. doi: 10.1097/00001756-199907130-00005. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1137–1142. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Barkis WB, Diamond JS, Barres BA. Invulnerability of retinal ganglion cells to NMDA excitotoxicity. Mol Cell Neurosci. 2004;26:544–557. doi: 10.1016/j.mcn.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Ünal-Çevik I, Kilinç M, Can A, Gürsoy-Özdemir Y, Dalkara T. Apoptotic and necrotic mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:2189–2194. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Wielckens K, Schmidt A, George B, Bredehorst R, Hilz H. DNA fragmentation and NAD+ depletion. J Biol Chem. 1982;257:12 872–12 877. [PubMed] [Google Scholar]
- Winkler BS, Pourcho RG, Starnes C, Slocum J, Slocum N. Metabolic mapping in mammalian retina: a biochemical and 3H-2-deoxyglucose autoradiographic study. Exp Eye Res. 2003;77:327–337. doi: 10.1016/s0014-4835(03)00147-7. [DOI] [PubMed] [Google Scholar]
- Yu SW, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]