

Abstract
Herpes simplex virus type 1 elicits a strong host inflammatory response following corneal infection. The purpose of the current study was to compare the production of chemokines induced by virus infection at sites known to harbor virus following ocular inoculation in order to determine the relationship between virus load and chemokine expression. Using highly resistant IFN-α1 transgenic mice whose transgene is under the control of the glial fibrillary acidic protein promoter in comparison to the more sensitive wild type counterparts, we compared the expression of chemokines versus the amount of infectious virus recovered from the anterior segment of the eye and nervous system. Consistent with our predicted outcome, the level of infectious virus recovered in the iris, trigeminal ganglia, and brain stem of resistant versus sensitive mice correlated with chemokine production; that is, the less virus recovered the less chemokine (CCL2, CCL3, CCL5, CXCL9, and CXCL10) produced. In contrast to the nervous system and iris, there was no correlation between chemokine expression and level of infectious virus recovered in the cornea. We interpret these results to suggest chemokine expression within the cornea in response to herpes simplex virus type 1 infection is driven by factors other than antigenic stimulation.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) is a neurotropic virus that upon infection of the eye replicates locally and traffics to the sensory ganglion (trigeminal ganglion, TG) by retrograde transport ultimately establishing a latent infection (6). From an immunologic perspective, the infection is not truly latent since a persistent, localized immune response is evident during latency (8, 22) and such a response can be significantly reduced upon treating latent mice with acyclovir (9). It is the immune response that results in tissue pathology ultimately leading to herpetic keratitis characterized by infiltration of the cornea by leukocytes and angiogenesis of the normally avascular cornea (3, 27). It is thought chemokines generated locally within the cornea in response to the pathogen are the most likely mediators of leukocyte recruitment as evidence suggest the absence or neutralization of specific chemokines reduces the incidence of infiltrating cells or the development of herpetic keratitis (4, 24). Although chemokines are only one family of proteins involved in orchestrating the host response following ocular HSV-1 infection, understanding the induction of these soluble mediators may provide a better understanding for the development of strategies to reduce collateral damage of the visual axis as a result of the inflammatory process associated with the infection.
In the current study, the relationship between virus titer and chemokine production in infected tissue was assessed using a highly resistant transgenic mouse that expresses the murine IFN-α1 transgene under the control of the glial fibrillary acidic protein (GFAP) promoter (referred to as GIFN mice). GIFN mice are highly resistant to virus infection (1) including HSV-1 (5) which allows for the direct comparison between these mice and the more sensitive B6/129 wild type (WT) mouse strain. The results from the present study show a tissue-specific response with virus levels correlated with chemokine production in the iris and nervous system but not in the cornea following HSV-1 infection.
MATERIALS AND METHODS
Virus and cell line
Vero cells originally obtained from the American Type Tissue Culture Collection (ATCC, Manassas, VA) were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and antibiotic/antimycotic solution (Invitrogen) at 37° C, 5% CO2, and 95% humidity. HSV-1 stock (McKrae strain) was prepared as previously described (8) and maintained at a concentration of 1 × 108 plaque forming unit (pfu)/ml at −80° C until use.
Mice
The construction of the GFAP IFN-α1 fusion gene and generation and screening of transgenic mice has previously been described (1). Offspring from the heterozygous male GIFN mice crossed with female B6/129 were genotyped by PCR as previously described (5). Non-transgenic offspring were used as WT controls for the GIFN mice.
Infection of mice
The corneas of male and female GIFN and WT anesthetized mice (6-10 weeks of age) were scarified using a 25-gauge needle, and HSV-1 (1,000 pfu) was applied in a volume of 3 μl in RPMI-1640. At the indicated time post infection (pi), the mice were anesthetized (6.6 mg/kg xylazine and 100 mg/kg ketamine, intraperitoneal administration) and perfused with 20 ml of PBS (pH 7.4). The corneas, irises, TG, and brain stems were removed and placed in RPMI-1640 medium for determination of virus quantity by plaque assay or placed into PBS containing a protease inhibitor cocktail (Set I, Calbiochem, San Diego, CA) for subsequent analysis of chemokine/IFN-γ content by ELISA. In a survival study, GIFN and WT male and female mice (8-12 weeks of age) were infected with HSV-1 (1,500 – 6,000 pfu/eye) as described above and monitored for survival over 30 days. All procedures were approved by The University of Oklahoma Health Sciences Center and the Dean A. McGee Eye Institute animal care and use committees.
Virus plaque assay
Tissues (corneas, irises, TG, and brain stems) from HSV-1 infected mice were placed into 0.5 ml of RPMI-1640 and homogenized using a tissue homogenizer at a setting of 4 (Fisher Scientific, Pittsburgh, PA). Supernatants were clarified (12,000 × g, 1 min) and subsequently assessed for virus content using Vero cell monolayers.
ELISA
Detection of CCL2, CCL3, CCL5, CXCL9, CXCL10, and IFN-γ were measured in duplicate using commercially available kits (Quantikine kits, R&D Systems, Minneapolis, MN) with a sensitivity of 20-25 pg per tissue. The ELISA was carried out according to the manufacturer's instructions and analyzed at an absorbance of 450 nm using a FL600 microplate fluorescence reader (Bio-Tek Instruments, Inc., Winooski, VT).
Statistics
Analysis of data was carried out by one-way analysis of variance and Scheffe multiple-comparison test to determine statistically significant differences (p<.05) between the WT and GIFN groups.
RESULTS
GIFN mice are highly resistant to ocular HSV-1 infection
To evaluate the sensitivity of GIFN mice to ocular HSV-1 infection, WT and GIFN mice were infected with increasing amounts of HSV-1. The results show GIFN mice are much less susceptible to HSV-1-mediated mortality compared to WT controls based on cumulative survival (Fig. 1). Only the highest dose of virus (6,000 pfu/eye) resulted in mortality of GIFN mice. Consistent with these results, virus recovered in the cornea, iris, TG, and brain stem was significantly higher in the WT compared to the GIFN mice during acute infection (Fig. 2). In fact, no infectious virus was detectable in the brain stem of GIFN mice throughout the time points chosen during acute infection (Fig. 2). Taken together, these results reinforce the observation that GIFN mice are highly resistant to virus infection including HSV-1.
FIGURE 1.
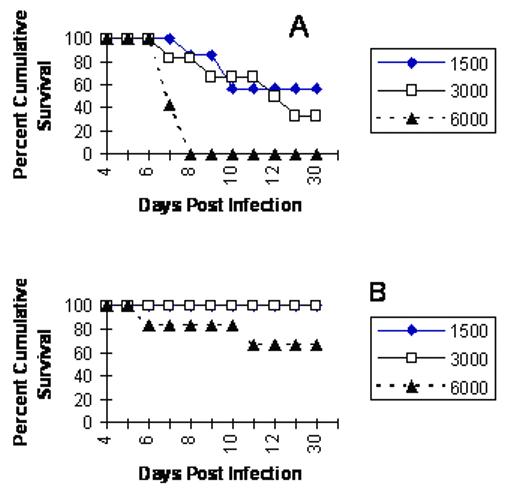
GIFN transgenic mice are less sensitive to acute HSV-1 infection compared to wild type mice. Wild type (Panel A) and GIFN transgenic (Panel B) mice (n=6-7 mice/group) were infected with increasing doses of HSV-1 (1500-6000 pfu/eye) and assessed for cumulative survival out to 30 days.
FIGURE 2.
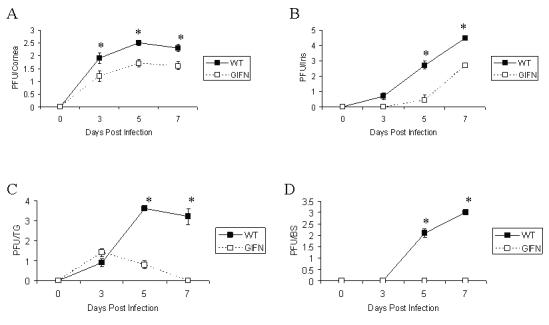
Virus titer in infected tissue following ocular infection of GIFN transgenic and wild type mice. Wild type (WT) and GIFN transgenic mice (n=9-13 mice) were infected with HSV-1 (1,500 pfu/eye) and subsequently anesthetized and perfused at the indicated time post infection. The corneas (panel A), irises (panel B), trigeminal ganglia (panel C), and brain stems (panel D) were removed, homogenized, and quantified for virus by plaque assay. The results expressed as the mean ± SEM are the summary of three separate experiments with 3-5 mice/group/experiment. *p<.05 comparing the WT to GIFN mice for each tissue within each time point.
Chemokine levels are similar in the cornea but not nervous system of WT and GIFN mice
One of the hallmarks following ocular HSV-1 infection includes a frank inflammatory response (13, 23). Since the amount of infectious virus is elevated in all tissues sampled comparing WT to GIFN mice, we assessed the inflammatory response as measured by expression of soluble mediators focusing on chemokines and IFN-γ as a means to determine the relationship between antigenic stimulus and cytokine/chemokine production. We found the increased levels of HSV-1 in the iris, TG, and brain stem of WT mice correlated with an increase in CCL2, CCL3, CCL5, CXCL9, CXCL10, and IFN-γ (Fig. 3). Likewise, the reduced infectious virus recovered in the iris, TG, and brain stem of GIFN mice was reflected by a drop in cytokine/production in these tissues relative to WT controls. The results also suggest IFN-γ levels in the TG and brain stem of WT mice parallel CXCL9 and CXCL10 levels. In contrast, the decrease in HSV-1 recovered in the cornea of GIFN mice was not associated with a decrease in chemokine expression compared to WT animals (Fig. 3).
FIGURE 3.
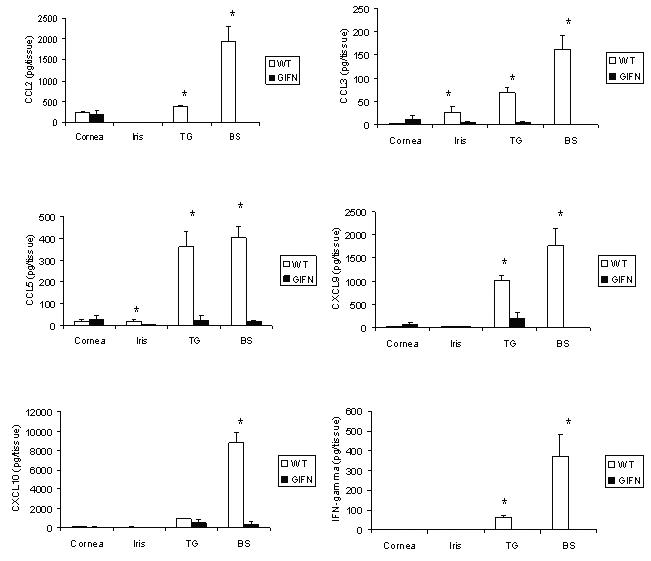
Chemokine levels are reduced in GIFN mice following ocular HSV-1 infection. Wild type (WT) and GIFN transgenic mice (n=6-7 mice) were infected with HSV-1 (1,500 pfu/eye) and anesthetized and perfused seven days post infection. The corneas, irises, trigeminal ganglia, and brain stems were removed and homogenized. The clarified homogenate was assayed for the quantity of chemokine or IFN-γ by ELISA. The results expressed as mean ± SEM are the summary of two experiments with 3-4 mice/group/experiment. *p<.05 comparing the WT to GIFN mice for each chemokine/cytokine.
DISCUSSION
Previously, we have reported a significant rise in TNF-α and CCL5 mRNA expression in the nervous system of mice in response to an increase in viral burden (8). We interpreted these results to suggest the antigenic load elicits the inflammatory response and drives the expression of soluble mediators in the HSV-1-infected host. However, in the original work there was no direct link between virus load and the incidence of encephalitis. More recent findings using mice deficient in toll-like receptor (TLR) 2 also concluded virus loads within the brain do not predict mortality (14). The present findings do show an increase in virus recovered in the nervous system and iris correlated with chemokine expression. In contrast, the results suggest a disconnect between lower infectious virus in the cornea and yet, similar chemokine levels comparing the resistant GIFN mice to the WT animals. We interpret the results to suggest an alternative or unique mechanism stimulates chemokine production in the cornea relative to other sites of infection. Although there may be multiple mechanisms, one possible pathway involves virus DNA.
HSV-1 DNA persists in the cornea associated with lesions long after the virus is cleared (19). These results are consistent with the development of herpetic keratitis reported in mice infected with an HSV-1 mutant unable to undergo anterograde transport from the ganglia (21). The basis for HSV DNA induction of inflammatory mediators involves the high content of cytidylate-phosphate-deoxyguanylate (CpG) motifs that are recognized by the pattern recognition receptor, TLR9 (12, 17, 28). TLR9 stimulation induces intracellular signals leading to the activation of NFκB and induction of cytokine secretion (7). We investigated the role of TLR9 in the induction of cytokines and chemokines in response to ocular HSV-1 infection using mice deficient in TLR9. We found TLR9 expression was necessary for up-regulation of the chemokines CXCL9 and CXCL10 but not other chemokines including CXCL1, CCL3, CCL5 and the cytokines IL-1β and IL-6 in the cornea in response to HSV-1 (25). CCL2 levels were also reduces although the variability precluded significant differences. It was also found the absence of TLR9 greatly diminished the early recruitment of PMNs but not macrophages into the cornea following infection. Taken together, TLR9 does appear to play a role in the expression of selective chemokines that impact on the early recruitment of leukocytes into the infected eye. However, other mechanisms must also be operational in the regulation of chemokines within the cornea. One likely alternative is another pattern recognition receptor, the mannose receptor that is induced by HSV-1 resulting in the production of IFN-α by dendritic cells (18). Whether dendritic cells residing in the cornea (11) have this capacity is currently unknown.
The critical role of type I IFNs in blocking HSV-1 replication and spread is demonstrated in the present findings using GIFN mice as well as previously through the application of type I IFN transgenes onto the cornea of HSV-1-infected animals (2). Further evidence for a key role of type I IFNs in controlling HSV-1 replication is typified by the number of proteins such as ICP34.5 (15), ICP0 (10, 16), and vhs (26) that target various levels of the type I IFN signaling cascade. In the absence of these proteins, viral replication is substantially reduced. In contrast, over-expression or re-introduction of these genes allows for successful replication. While type I IFN transgene treatment of the cornea has been found to reduce the number of infiltrating cells during acute HSV-1 infection (20), in light of the present findings in which chemokine levels in the cornea were similar between the highly resistant GIFN transgenic mice and wild type controls, type I IFN may ultimately have little impact on the severity or incidence of corneal pathology and the development of herpes keratitis. Preservation of the visual axis is balanced by the need to clear the pathogen. Therefore, a combinational therapy that includes anti-viral and anti-inflammatory compounds is predicted to be necessary to maximize a positive outcome for the host.
ACKNOWLEDGMENTS
This work was supported by USPHS grants, MH62331 to ILC, and EY015566 and core grant EY12190 as well as a Jules and Doris Stein Research to Prevent Blindness research professorship to DJJC.
REFERENCES
- 1.Akwa Y, Hassett DE, Eloranta M-L, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J. Immunol. 1998;161:5016. [PubMed] [Google Scholar]
- 2.Austin BA, James C, Silverman RH, Carr DJJ. Critical role for the oligoadenylate synthetase/RNase L pathway in response to IFN-β during acute ocular herpes simplex virus type 1 infection. J. Immunol. 2006;175:1100. doi: 10.4049/jimmunol.175.2.1100. [DOI] [PubMed] [Google Scholar]
- 3.Biswas PS, Banerjee K, Kim B, Rouse BT. Mice transgenic for IL-1 receptor antagonist protein are resistant to herpetic stromal keratitis: possible role for IL-1 in herpetic stromal keratitis pathogenesis. J. Immunol. 2004;172:3736. doi: 10.4049/jimmunol.172.6.3736. [DOI] [PubMed] [Google Scholar]
- 4.Carr DJJ, Chodosh J, Ash J, Lane TE. Effect of anti-CXCL10 monoclonal antibody on herpes simplex virus type 1 keratitis and retinal infection. J. Virol. 2003;77:10037. doi: 10.1128/JVI.77.18.10037-10046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carr DJJ, Veress LA, Noisakran S, Campbell IL. Astrocyte-targeted expression of IFN-α1 protects mice from acute ocular herpes simplex virus type 1 infection. J. Immunol. 1998;161:4859. [PubMed] [Google Scholar]
- 6.Cook ML, Stevens JG. Pathogenesis of herpetic neuritis and ganglionitis in mice: evidence for intra-axonal transport infection. Infect. Immun. 1973;7:272. doi: 10.1128/iai.7.2.272-288.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hacker, Vabulas HRM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J. Exp. Med. 192:595. doi: 10.1084/jem.192.4.595. 20000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halford WP, Gebhardt BM, Carr DJJ. Persistent cytokine expression in the trigeminal ganglion latently infected with herpes simplex virus type 1. J. Immunol. 1996;157:3542. [PubMed] [Google Scholar]
- 9.Halford WP, Gebhardt BM, Carr DJJ. Acyclovir blocks cytokine gene expression in trigeminal ganglia latently infected with herpes simplex virus type 1. Virology. 1997;238:53. doi: 10.1006/viro.1997.8806. [DOI] [PubMed] [Google Scholar]
- 10.Halford WP, Weisend C, Grace J, Soboleski M, Carr DJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virology J. 2006 doi: 10.1186/1743-422X-3-44. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamrah P, Huq SO, Liu Y, Zhang Q, Dana MR. Corneal immunity is mediated by heterogenous population of antigen-presenting cells. J. Leukoc. Biol. 74:172. doi: 10.1189/jlb.1102544. [DOI] [PubMed] [Google Scholar]
- 12.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002;20:709. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 13.Kumaraguru U, David I, Rouse BT. Chemokine and ocular pathology caused by corneal infection with herpes simplex virus. J. Neurovirol. 1999;5:42. doi: 10.3109/13550289909029744. [DOI] [PubMed] [Google Scholar]
- 14.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA. 2004;101:1315. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leib DA, Machalek MA, Williams BRG, Silverman RH, Virgin HW. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA. 2000;97:6097. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004;78:1675. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundberg P, Welander P, Han X, Cantin E. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J. Virol. 2003;77:11158. doi: 10.1128/JVI.77.20.11158-11169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milone MC, Fitzgerald-Bocarsly P. The mannose receptor mediates induction of IFN-α in peripheral blood dendritic cells by enveloped RNA and DNA viruses. J. Immunol. 1998;161:2391. [PubMed] [Google Scholar]
- 19.Mitchell WJ, Gressens P, Martin JR, DeSanto R. Herpes simplex virus type 1 DNA persistence, progressive disease and transgenic immediate early gene promoter activity in chronic corneal infections in mice. J. Gen. Virol. 1994;75:1201. doi: 10.1099/0022-1317-75-6-1201. [DOI] [PubMed] [Google Scholar]
- 20.Noisakran S, Carr DJJ. Plasmid DNA encoding IFN-α1 antagonizes herpes simplex virus type 1 ocular infection through CD4+ and CD8+ T lymphocytes. J. Immunol. 2000;164:6435. doi: 10.4049/jimmunol.164.12.6435. [DOI] [PubMed] [Google Scholar]
- 21.Polcicova K, Biswas PS, Banerjee K, Wisner TW, Rouse BT, Johnson DC. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA. 2005;102:11462. doi: 10.1073/pnas.0503230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimeld C, Whiteland JL, Nicholls SM, Grinfeld E, Easty DL, Gao H, Hill TJ. Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J. Neuroimmunol. 1995;61:7. doi: 10.1016/0165-5728(95)00068-d. [DOI] [PubMed] [Google Scholar]
- 23.Streilein JW, Dana MR, Ksander BR. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol. 1997;18:443. doi: 10.1016/s0167-5699(97)01114-6. Today. [DOI] [PubMed] [Google Scholar]
- 24.Tumpey TM, Cheng H, Cook DN, Smithies O, Oakes JE, Lausch RN. Absence of macrophage inflammatory protein-1α prevents the development of blinding herpes stromal keratitis. J. Virol. 1998;72:3705. doi: 10.1128/jvi.72.5.3705-3710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wuest T, Austin BA, Uematsu S, Thapa M, Akira S, Carr DJJ. Intact TLR 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J. Neuroimmunol. 2006 doi: 10.1016/j.jneuroim.2006.06.020. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokota S.-i., Yokosawa N, Okabayashi T, Suzutani T, Miura S, Jimbow K, Fujii N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 2004;78:6282. doi: 10.1128/JVI.78.12.6282-6286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 2001;75:9828. doi: 10.1128/JVI.75.20.9828-9835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng M, Klinman DM, Gierynska M, Rouse BT. DNA containing CpG motifs induces angiogenesis. Proc. Natl. Acad. Sci. USA. 2002;99:8944. doi: 10.1073/pnas.132605599. [DOI] [PMC free article] [PubMed] [Google Scholar]