

Abstract
Bottom-up proteomics (analyzing peptides that result from protein digestion) has demonstrated capability for broad proteome coverage and good throughput. However, due to incomplete sequence coverage, this approach is not ideally suited to the study of modified proteins. The modification complement of a protein can best be elucidated by analyzing the intact protein. Two-dimensional gel electrophoresis, typically coupled with the analysis of peptides that result from in-gel digestion, is the most frequently applied protein separation technique in MS-based proteomics. As an alternative, numerous column-based liquid phase techniques, which are generally more amenable to automation, are being investigated. In this work, the combination of size exclusion chromatography (SEC) fractionation with reversed-phase liquid chromatography (RPLC)-Fourier-transform ion cyclotron resonance (FTICR)-mass spectrometry (MS) is compared with the combination of RPLC fractionation with capillary isoelectric focusing (CIEF)-FTICR-MS for the analysis of the Shewanella oneidensis proteome. SEC-RPLC-FTICR-MS allowed the detection of 297 proteins, as opposed to 166 using RPLC-CIEF-FTICR-MS, indicating that approaches based on LC-MS provide better coverage. However, there were significant differences in the sets of proteins detected and both approaches provide a basis for accurately quantifying changes in protein and modified protein abundances.
Keywords: protein separations, FTICR-MS, proteomics, post-translational modifications, Shewanella oneidensis
Abbreviations: BCA, bicinchoninic acid; CA, carrier ampholyte; FTICR, Fourier-transform ion cyclotron resonance; MMA, mass measurement accuracy; TIC, total ion chromatogram; QIT, quadrupole ion trap
Introduction
Peptide mass fingerprinting [1–5] applied to proteins separated by two-dimensional gel electrophoresis (2-DE) is still the most commonly used technique in mass spectrometry (MS)-based proteomics [6]. 2-DE protein separation performance is impressive: the detection of over 10000 spots in a single gel has been demonstrated for large-gel 2-DE [7, 8]. In a recent study of the Arabidopsis thaliana proteome where samples from different tissues were fractionated during the extraction process, protein identifications were obtained for 2943 out of a total of 6000 excised spots [9]. However, this work exemplified many of the disadvantages commonly attributed to 2-DE: the majority of identified proteins were abundant, so-called, housekeeping proteins; there was a bias against membrane proteins due to solubility, and there was a bias against proteins with outlying masses and isoelectric points. Many methods exist to alleviate these problems, such as more extensive protein fractionation before 2-DE [10], but additional protein separation strategies are clearly necessary to extend proteome coverage.
One-dimensional protein separations cannot at present compete with the performance of 2-DE, but numerous alternative 2-D approaches have been proposed. (Multidimensional peak capacities are not directly equivalent to one-dimensional peak capacities [11], but additional separation dimensions can significantly improve overall performance.) True 2-D separations for MS, where the second dimension is fast enough to record a number of chromatograms over the width of a first-dimension peak and where the mass spectrometer is fast enough to record a number of mass spectra over the width of a second-dimension peak, are rare. For simple peptide mixtures, a reversed-phase liquid chromatography (RPLC)-capillary zone electrophoresis (CZE)-MS system that fulfills these criteria has been described by Lewis et al. [12]. Most reported methods would better be described as fractionations (frequently automated). For peptide mixtures, the combination of strong cation exchange (SCX) chromatography (either on- or off-line) with RPLC-MS [13, 14] has become standard, but no such consensus yet exists for protein separations.
The most frequently employed protein separation technique for direct coupling to MS is RPLC. The utility of common RPLC-MS operating parameters has recently been investigated by Wang et al. [15]. Capillary isoelectric focusing (CIEF), an equilibrium technique that can provide isoelectric point measurements useful for assigning identifications, is ideally suited to being the first separation dimension; in combination with RPLC-MS, excellent results have been reported for peptides [16–19] and proteins [20, 21]. CIEF has also been elegantly combined with transient capillary isotachophoresis-CZE; a peak capacity of 1600 was reported for peptides [22], and the detection of 1174 proteins was reported when interfaced to Fourier-transform ion cyclotron resonance (FTICR)-MS [23]. Chromatofocusing [24–27], a variant of ion exchange chromatography that provides a separation based on local protein surface charge, which approximates to a separation based on isoelectric point, provides an additional option for the first separation dimension. This method, in conjunction with RPLC-MS, using non-porous chromatographic support particles, has successfully been applied to the profiling of proteins from human breast cancer cell lines [28–32].
Electrophoretic fractionation methods have recently been reviewed by Righetti et al. [33]. Methods that use immobilized pH gradient gels to fractionate samples between liquid-filled chambers are of particular interest due to claimed ease of protein recovery and minimal fraction overlap [34–38]. Other chromatographic methods applied to protein fractionation prior to RPLC-MS include strong anion exchange chromatography [39, 40] and size exclusion chromatography (SEC) [41], but RPLC itself is an excellent fractionation method. Separations compatible with direct MS-interfacing that are orthogonal to RPLC include CIEF and CZE. CZE-MS protein separations are rare, but interesting CIEF-MS applications have been reported. For example, Jensen et al. [42] reported the detection of > 400 Escherichia coli proteins in a single CIEF-FTICR-MS analysis. Naturally, fractions may also be collected from the second dimension separation to allow off-line MS-analysis; for example, Meng et al. [43] used continuous-elution gel electrophoresis in combination with RPLC, using an acid-labile surfactant in the gel separation to simplify clean-up, to provide fractions for protein tandem mass spectrometry (MS/MS) experiments (top-down proteomics).
This work is focused on comparing SEC-RPLC-MS with RPLC-CIEF-MS for protein analysis. Fractionation methods were chosen for orthogonality with the on-line separation and to minimize sample handling losses (both fractionation methods allow fraction concentration by evaporation alone). Protein samples used in this work originated from Shewanella oneidensis, an organism that is of interest because of metal ion reduction capabilities that give it relevance to bioremediation efforts [44, 45]. In addition to direct protein analysis, an aliquot of each fraction was digested using trypsin and analyzed by RPLC-quadrupole ion trap (QIT)-MS/MS to obtain peptide identifications (a schematic representation of the entire experiment is given in Figure 1). Ideally, MS/MS spectra would also have been collected for the protein separations, but protein MS/MS is currently too slow to be effective for on-line separations. FTICR-MS was employed for protein mass measurements; good mass measurement accuracy (MMA) allowed tentative identifications to be proposed for some proteins [40, 46–48].
Figure 1.
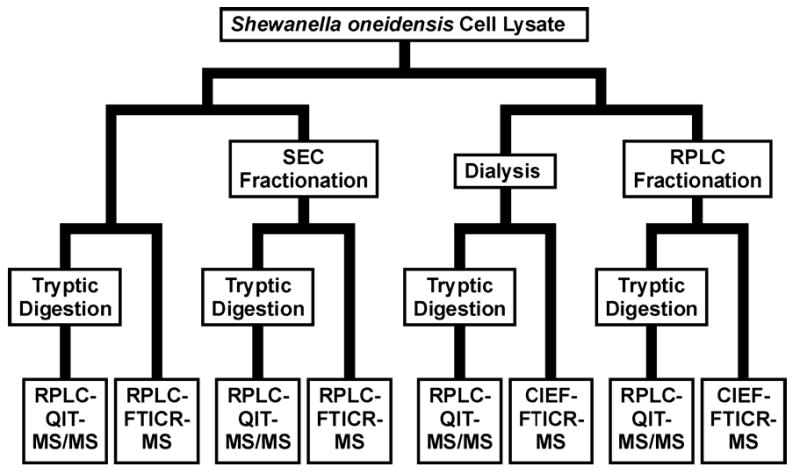
Schematic representation of the experimental pathway from S. oneidensis cell lysate to MS-analysis.
Methods
Sample Preparation
S. oneidensis MR-1 cells were cultured for high biomass in a defined medium using a BioFlo model 3000 fermentor (New Brunswick Scientific, Edison, NJ, USA) that was operated in continuous feed mode [49]; dissolved oxygen was maintained at 50%, while the carbon source, sodium DL-lactate, was present in the medium at 90 mM. Cells were harvested in the steady state growth phase by centrifugation and stored at −80°C until required. Cell disruption was achieved by bead-beating: pelletized cells were thawed and suspended in a minimum volume of 100 mM ammonium bicarbonate; bead-beating was performed using 0.1 mm zirconia/silica beads (BioSpec Products; Bartlesville, OK, USA); lysate was drawn off the beads by centrifugation. The bicinchoninic acid (BCA) assay [50], using a kit supplied by Pierce (Rockford, IL, USA), was used to determine protein concentration in the cell lysate.
Protein Fractionation
SEC and RPLC fractionations of the lysate were performed using a Shimadzu (Kyoto, Japan) HPLC system (DGU-14A degasser; 2 × LC-10AD pumps; FRC-10A fraction collector; SCL-10A controller); absorbance detection at 280 nm was achieved using a Thermo Separation Products (Rivera Beach, FL, USA) Spectra 100 absorbance detector. For SEC, a Phenomenex (Torrance, CA, USA) BioSep-SEC-S2000, 600 × 21.2 mm column, preceded by a 75 × 21.2 mm guard column, was used; the mobile phase was 100 mM ammonium bicarbonate (adjusted to pH 6.8) and the flow rate was 5 mL/min. For RPLC, a Phenomenex Jupiter (C4 stationary phase; 10 μm particles; 300 Å pore size) 250 × 10 mm column, preceded by a 50 × 10 mm guard column, was used; mobile phase A was 95/5 (% v/v) water/acetonitrile with 0.1% trifluoroacetic acid (TFA), mobile phase B was 10/90 (% v/v) water/acetonitrile with 0.1% TFA, and the flow rate was 4 mL/min. The gradient program was 100% A for 10 min, then a linear gradient to 30% B over 5 min, and then a linear gradient to 100% B over 45 min. Fractions were collected manually during the SEC run while fixed-duration (6 min) fractions were collected during the RPLC run. Protein concentrations for each fraction were determined using the BCA assay after evaporation to 100–200 μL; a comparison of these measurements with the initial protein concentration determined for the cell lysate was used to calculate fractionation recoveries; both chromatograms, with fraction boundaries indicated, are displayed as Figure 2. The amounts of protein injected were 22.5 mg for the SEC fractionation and 4 mg for the RPLC fractionation; the difference between the injected amounts reflects the sample requirements of the second separation dimension.
Figure 2.
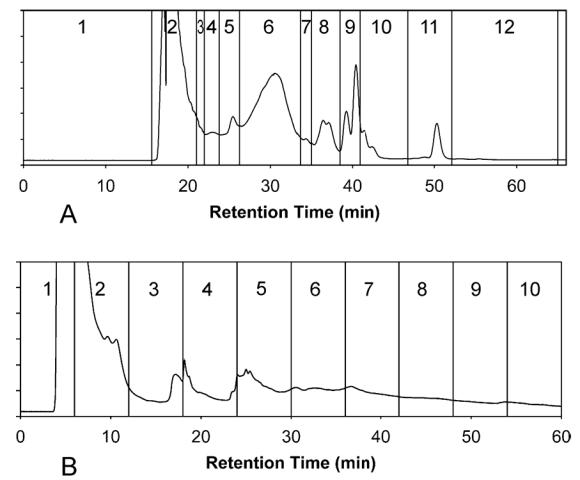
Chromatograms for (A) SEC fractionation and (B) RPLC fractionation of S. oneidensis cell lysate; absorbance detection was performed at 280 nm; vertical lines indicate boundaries between fractions.
Bottom-up Analyses
Aliquots of each fraction and of the cell lysate preparation, each containing 50 μg of protein, were digested using trypsin [49]. Salts present in the cell lysate preparation degrade CIEF separation performance, so a 50 μg aliquot of dialyzed cell lysate that was prepared for protein CIEF-MS was also subjected to digestion. Peptide samples were prepared for MS-analysis using Supelclean LC-18 (Supelco; Bellefonte, PA, USA) solid phase extraction cartridges [51]. De-salted peptide samples were evaporated to dryness and then reconstituted at 1 μg/μL in 25 mM ammonium bicarbonate for RPLC-QIT-MS/MS analysis (BCA assay used to guide concentration adjustment); 10 μg of sample was loaded to the column. RPLC was conducted on a 60 cm × 150 μm column packed in-house with Phenomenex Jupiter particles (C18 stationary phase; 5 μm particle diameter, 300 Å pore size). The separation was performed at 5000 psi using two ISCO (Lincoln, NE, USA) model 100 DM high-pressure syringe pumps. Mobile phase A consisted of 0.2% acetic acid and 0.05% TFA in water, while mobile phase B was 0.1% TFA, 9.9% water, and 90% acetonitrile; the initial (100% A) column flow rate was 2.0 μL/min. The gradient program began with a 20 min hold at 100% A, which was necessary to allow analytes to be loaded onto the column, followed by a nonlinear gradient to ~ 70% solvent B over 150 min. The gradient was produced using a stirred (2.5 mL) mixing chamber, initially filled with mobile phase A, to which mobile phase B was added at a known rate; the mixing chamber is located prior to a split, allowing a higher flow rate through the mixing chamber than is found through the column. The performance and design of the chromatographic system has been described by Shen et al. [52]. The capillary RPLC system was interfaced, using a pulled fused-silica needle and electrospray ionization (ESI), to a QIT mass spectrometer (Finnigan MAT LCQ; San Jose, CA, USA); the mass spectrometer cycled between single MS scans and three consecutive MS/MS scans. The minimum amount of peptide required for a good MS/MS spectrum, expressed as an amount of peptide loaded to the chromatographic column, was approximately 1 femtomole. Peptide identifications were derived from QIT-MS/MS data using SEQUEST (ThermoFinnigan; San Jose, CA, USA) [53]; no enzyme rules were applied. The S. oneidensis MR-1 protein sequence database that SEQUEST used has previously been described by Romine et al. [54]. SEQUEST peptide identifications were filtered using the rules suggested by Washburn et al. [55]; two or more constituent peptides were required to identify a protein.
On-line RPLC Separation
The liquid chromatography used for intact protein work was identical to that used for peptide RPLC-QIT-MS/MS. However, numerous operating parameters were altered. Protein RPLC was conducted on a 80 cm × 150 μm column packed in-house with Phenomenex Jupiter particles (C5 stationary phase; 5 μm particle diameter; 300 Å pore size); a guard column was used to protect the main column from clogging. Mobile phase A was changed to 0.1% TFA, 25% acetonitrile, 74.9% water, while mobile phase B remained as 0.1% TFA, 9.9% water, 90% acetonitrile; pump operating pressure was raised to 8000 psi, resulting in an initial (100% A) column flow rate of 2.8 μL/min. The non-linear gradient was allowed to run for 110 min, again resulting in a final mobile phase B concentration of ~ 70%. Prior to loading, protein samples were adjusted to 1.5 μg/μL in mobile phase A with TFA, acetonitrile and water; centrifugation was used after adjustment to remove precipitated proteins that could clog the chromatographic column (extensive precipitation was not observed). Approximately 75 μg protein was loaded to the column using a 50 μL sample loop. The detection limit for the RPLC-FTICR-MS system, expressed as an amount of protein loaded to the chromatographic column, was approximately 100 femtomoles.
On-line CIEF Separation
CIEF was performed using a Crystal model 300 capillary electrophoresis system (Prince Technologies, Emmen, The Netherlands). CIEF columns were constructed by coating 100 cm × 50 μm inside diameter (ID) × 190 μm outside diameter (OD) fused-silica capillaries (Polymicro Technologies, Phoenix, AZ, USA) with hydroxypropyl cellulose (HPC; average Mr = 88,000 Da) as described by Shen et al. [56]. Columns were trimmed to 80 cm in length before use. Sample preparation for CIEF simply required the addition of carrier ampholytes (CAs); Pharmalyte 3–10 (Amersham Pharmacia Biotech; Uppsala, Sweden) was added to the sample to give a final CA concentration of ~ 0.5% [57, 58]. Fractions originating from the RPLC method used in this work were free of salt, so no processing in addition to evaporation and adjustment to the desired CA and protein concentration was required. However, the lysate sample was subjected to dialysis to reduce salt concentrations (a membrane designed to retain molecules having molecular weight > 3500 Da was used to minimize protein loss). Column loading was achieved by filling the entire length of the column with the sample mixture of intact proteins and CAs. Samples were prepared at a protein concentration of ~ 0.3 mg/mL, resulting in ~ 0.5 μg protein being loaded to the column (the column volume was ~ 1.5 μL).
CIEF-MS Interfacing
Hyphenation was achieved using an approach similar to that described by Clarke and Naylor [59]. The catholyte was 1% aqueous ammonium hydroxide solution (pH ~ 10.7) while the anolyte was 1% aqueous acetic acid solution (pH ~ 2.5); on mobilization, the sheath-liquid was changed from catholyte to 2% acetic acid, 40% methanol, 58% water to generate an efficient electrospray. Swapping sheath-liquids resulted in chemical mobilization, but a small pressure of ~ 10 mbar was also applied to the inlet reservoir to reduce elution times and improve peak shapes. Sheath-liquids were delivered using a Harvard Apparatus (South Natick, MA, USA) model 22 syringe pump at a flow rate of 2 μL/min. A constant voltage of 25 kV was held across the column throughout the focusing and mobilization stages; the duration of focusing was 30 min.
FTICR-MS
Analyses were performed using an in-house developed mass spectrometer equipped with a 7.0 Tesla superconducting magnet [60]. The mass spectrometer incorporates an ESI ion source with an electrodynamic ion funnel [61], an octopole for collisional focusing, an ion pre-selection and external accumulation quadrupole, an ion guide, and an orthorhombic ICR cell operated at ~ 10−10 Torr. The ion accumulation time for protein analyses was 0.3 s, and one mass spectrum was recorded every 3.0 s. Additionally, to reduce contributions from CAs in CIEF-FTICR-MS experiments, peaks below m/z 600 were removed by tuning the radio frequency applied to one of the quadrupole ion guides. Raw FTICR-MS data were processed using an in-house software package, ICR-2LS. Subsequent to external calibration and mass transform, the THRASH algorithm [62] was used to deconvolute and deisotope individual ESI mass spectra. This resulted in lists of neutral, deisotoped masses that were then clustered across consecutive mass spectra. Filtered clusters were, using measured mass, compared between experiments to estimate overlap.
Results
The present study was not designed, primarily, to obtain the best possible proteome coverage. The principal purpose of this work was to compare the proteome coverage provided by the investigated analytical strategies. Filtered 2-D plots generated for the RPLC-FTICR-MS analysis of cell lysate and for the CIEF-FTICR-MS analyses of dialyzed cell lysate are shown as Figures 3A and 3C, respectively. As examples, Figure 3B shows the mass spectrum responsible for the indicated spot in Figure 3A, while Figure 3D shows the mass spectrum responsible for the indicated spot in Figure 3C. RPLC-QIT-MS/MS of digested cell lysate resulted in the identification of 150 proteins from 796 peptides; the number of proteins detected directly by RPLC-FTICR-MS was 142. For the dialyzed cell lysate that was required for CIEF, RPLC-QIT-MS/MS resulted in the identification of 84 proteins from 473 peptides; the number of proteins detected directly by CIEF-FTICR-MS was 58. Counts of MS/MS-identified peptides and proteins, and of proteins detected directly using RPLC- or CIEF-FTICR-MS, for all experiments, are given in Table 1. In addition, Supporting Data Table 1 lists MS/MS-identified proteins and the protein sequence coverage found for each experiment in which the protein in question was identified.
Figure 3.
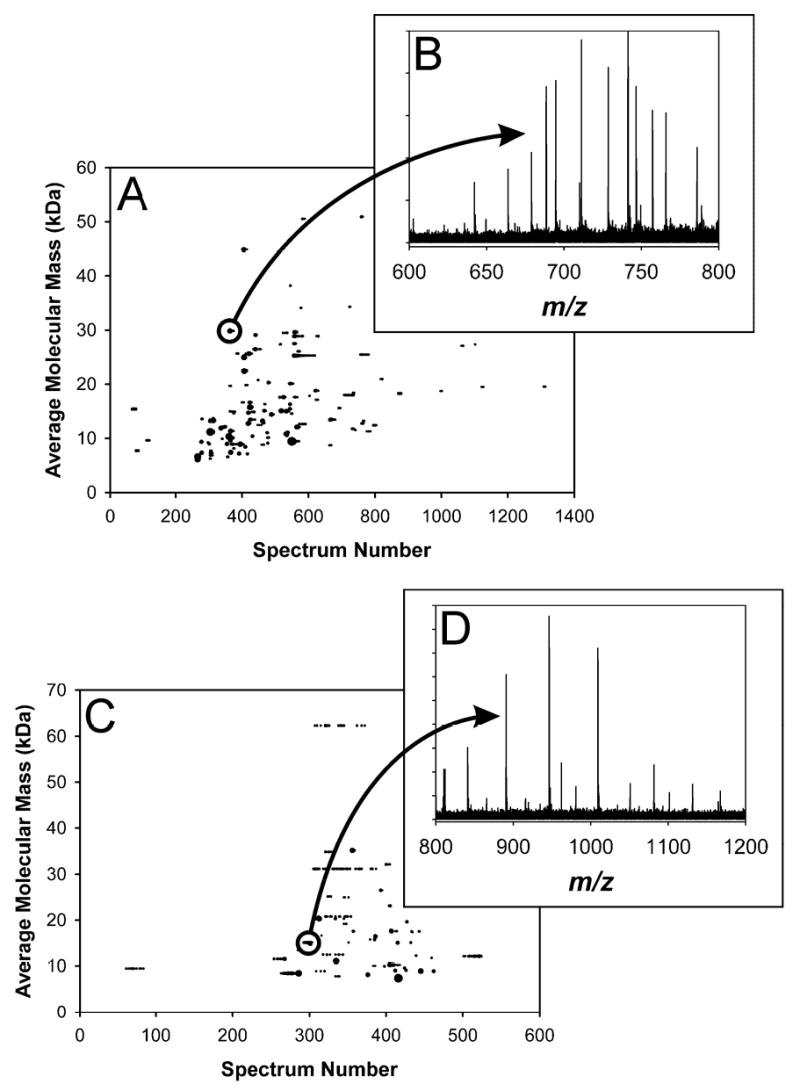
Comparison of (A) RPLC-FTICR-MS with (C) CIEF-FTICR-MS data, displayed as 2-D plots, for S. oneidensis cell lysate; CIEF sample preparation, in addition, required dialysis to reduce salt concentrations. 142 proteins were detected in the RPLC run, while 58 were detected in the CIEF run. (B) is the mass spectrum for the indicated spot in (A); the measured mass (average isotopic composition) for this spot was 29832.40 Da, which matches the mass of ribosomal protein L2 (SO0234) minus methionine at 9.6 ppm error. (D) is the mass spectrum for the indicated spot in (C); the measured mass (average isotopic composition) for this spot was 15127.80 Da, which matches the mass of hypothetical protein SO0691 at 0.3 ppm error.
Table 1.
S. oneidensis cell lysate was fractionated by SEC and by RPLC. Each fraction, along with cell lysate and dialyzed cell lysate, was split into aliquots: one aliquot was digested using trypsin, the resulting peptides being subjected to RPLC-QIT-MS/MS, while another was analyzed at the intact protein level (for the SEC fractions and cell lysate, RPLC-FTICR-MS was used; for the RPLC fractions and dialyzed cell lysate, CIEF-FTICR-MS was used). The first data column gives the number of peptides found by applying SEQUEST [53] to the RPLC-QIT-MS/MS data, using the rules given in Washburn et al. [55]; the second data column gives the number of proteins that were identified by the presence of two or more distinct constituent peptides; the third and fourth data columns contain the number of proteins that were detected at the intact protein level (the third column is for RPLC-FTICR-MS analyses while the fourth is for CIEF-FTICR-MS analyses).
| Experiment |
Peptide Measurements |
Protein Measurements |
||
|---|---|---|---|---|
| Number Peptides Identified by RPLC-QIT-MS/MS | Number Proteins Identified by RPLC-QIT-MS/MS | Number Proteins Detected by RPLC-FTICR-MS | Number Proteins Detected by CIEF-FTICR-MS | |
| Lysate | 796 | 150 | 142 | - |
| Dialyzed Lysate | 473 | 84 | - | 58 |
| SEC Fraction 01 | 64 | 17 | - | - |
| SEC Fraction 02 | 801 | 136 | 27 | - |
| SEC Fraction 03 | 552 | 83 | 62 | - |
| SEC Fraction 04 | 697 | 104 | 15 | - |
| SEC Fraction 05 | 480 | 78 | 164 | - |
| SEC Fraction 06 | 383 | 66 | 92 | - |
| SEC Fraction 07 | 49 | 9 | - | - |
| SEC Fraction 08 | 71 | 16 | - | - |
| SEC Fraction 09 | 3 | 0 | - | - |
| SEC Fraction 10 | 54 | 9 | - | - |
| SEC Fraction 11 | 43 | 7 | - | - |
| SEC Fraction 12 | 136 | 24 | - | - |
| All SEC Fractions | 2429 | 321 | 297 | - |
| RPLC Fraction 01 | 39 | 5 | - | - |
| RPLC Fraction 02 | 8 | 1 | - | - |
| RPLC Fraction 03 | 31 | 5 | - | - |
| RPLC Fraction 04 | 390 | 62 | - | 75 |
| RPLC Fraction 05 | 449 | 85 | - | 62 |
| RPLC Fraction 06 | 246 | 40 | - | 72 |
| RPLC Fraction 07 | 138 | 29 | - | 78 |
| RPLC Fraction 08 | 259 | 43 | - | 29 |
| RPLC Fraction 09 | 18 | 3 | - | 2 |
| RPLC Fraction 10 | 3 | 1 | - | - |
| All RPLC Fractions | 1135 | 149 | - | 166 |
| All Experiments | 3336 | 367 | 372 | 186 |
Figure 4A gives the RPLC-FTICR-MS total ion chromatogram (TIC) and Figure 4B gives the filtered 2-D plot obtained for SEC Fraction 5. Figure 4C gives the mass spectrum for the indicated spot in Figure 4B. Analysis of the tryptic digest of SEC Fraction 2 yielded 136 bottom-up protein identifications from 801 peptides, while FTICR-MS analysis of the undigested sample resulted in the detection of only 27 proteins. In contrast, SEC Fraction 5 gave 78 bottom-up protein identifications from 480 peptides, while FTICR-MS analysis resulted in the detection of 164 proteins. SEC Fraction 2 represents the highest molecular mass cell lysate components (based on bottom-up protein identifications, the average protein mass for SEC Fraction 2 is > 50 kDa while that for SEC Fraction 5 is ~ 23 kDa), so it is probable that the particularly wide discrepancy between the number of bottom-up identifications and the number of detected proteins was due simply to the prevalence of high molecular mass proteins in this sample. SEC Fraction 5 exemplifies a set of proteins more accessible to FTICR-MS, as evidenced by the fact that the number of directly detected proteins is greater than the number of bottom-up identifications (from a single analysis). For the RPLC-FTICR-MS intact protein analyses, of the 297 proteins detected in the SEC fractions, 67 were also detected in the cell lysate sample. Considering the bottom-up peptide analyses, of the 321 proteins identified from the SEC fractions, 134 were also identified in the cell lysate sample.
Figure 4.
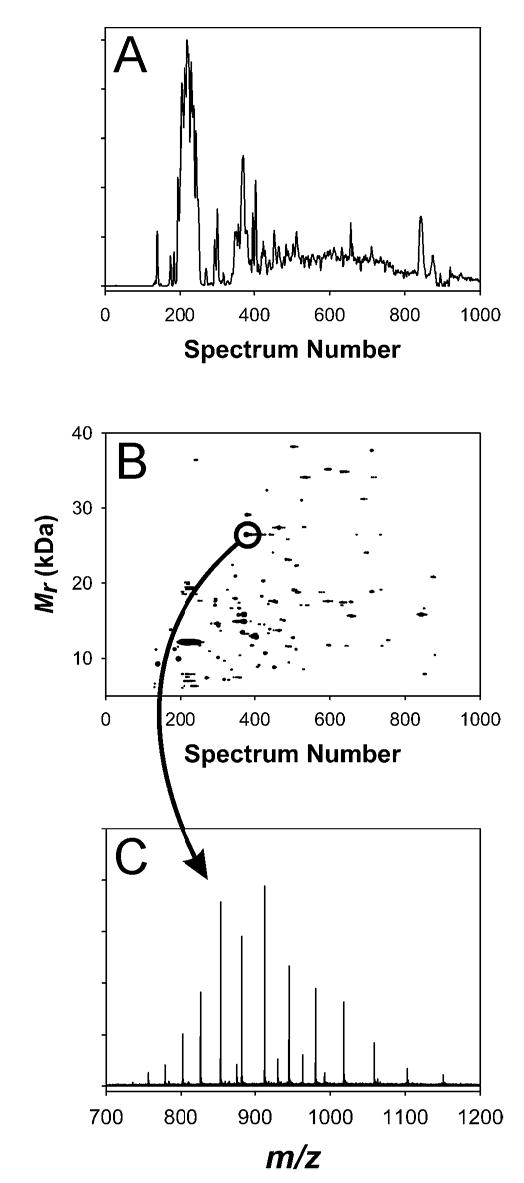
(A) TIC and (B) 2-D plot for SEC Fraction 5. (C) is the mass spectrum for the indicated spot in (B); the measured mass (average isotopic composition) for this spot was 26445.11 Da, which matches the mass of conserved hypothetical protein SO4719 minus a predicted signal peptide at 6.6 ppm error.
Figure 5A gives the CIEF-FTICR-MS TIC and Figure 5B gives the filtered 2-D plot obtained for RPLC Fraction 4. Figure 5C gives the mass spectrum for the indicated spot in Figure 5B, while Figure 5D illustrates the isotopic resolution available even for a relatively low signal-to-noise peak. RPLC Fraction 4 yielded 62 bottom-up protein identifications from 390 peptides, while CIEF-FTICR-MS direct protein analysis resulted in the detection of 75 proteins. For the RPLC fractions that were subjected to CIEF-FTICR-MS intact protein analysis a total of 166 proteins were detected; 38 of these were also detected in the dialyzed cell lysate sample. Similarly, considering only bottom-up peptide analyses, of the 149 proteins identified from the RPLC fractions, 51 were also identified in the dialyzed cell lysate sample.
Figure 5.
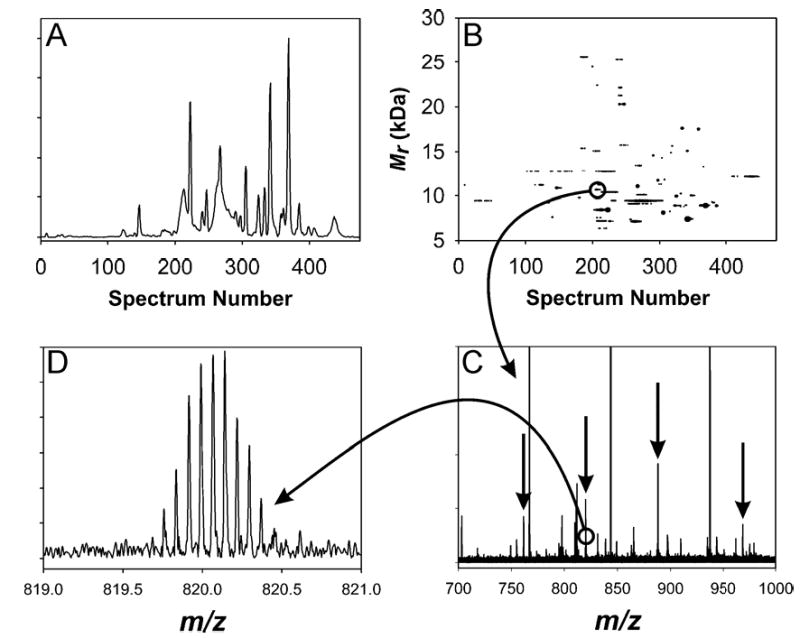
(A) TIC and (B) 2-D plot for RPLC Fraction 4. (C) is the mass spectrum for the indicated spot in (B); arrows highlight the peaks that constitute the indicated spot; the measured mass (average isotopic composition) for this spot was 10648.27 Da, which matches the mass of integration host factor beta subunit (SO2401) at 4.6 ppm error. (D) is an expansion of the 13+ peak that exemplifies the isotopic resolution achievable even for less intense peaks.
Discussion
For the fractions where data was recorded at both the peptide and the protein levels, counts of proteins identified from RPLC-QIT-MS/MS data are comparable to the number of proteins detected directly using FTICR-MS. This reflects the generally better coverage for the bottom-up approach in combination with the ability of the FTICR-MS method to detect proteins in more than one modification state. In addition to allowing the comparison of the described 2-D protein separation approaches, this data provides information about the extent of modification in the S. oneidensis proteome.
Protein recovery for SEC fractionation was 25% while that for RPLC fractionation was 13% (initial measurements were made on freshly prepared lysate; final measurements were made after evaporation to reduce fraction volume); the bottom-up (peptide-level) measurements also support the superiority of the SEC fractionation. Lower RPLC fractionation recovery was probably due to precipitation losses in acidic RPLC mobile phases; in contrast, the SEC mobile phase was very similar to the cell disruption buffer in which the initial measurements were made. TFA is present in the RPLC mobile phases to improve separation quality, but it may have been better to accept wider peaks in exchange for improved proteome coverage. Wider peaks would, however, result in greater overlap between fractions, in opposition to the chief purpose of fractionation, namely, to reduce signal overlap by splitting a complicated sample into simpler parts. Overall performance for the 2-D approaches could have been similar because SEC fractions were acidified for RPLC-FTICR-MS analysis subsequent to the BCA protein concentration assay. RPLC fractions are prepared for CIEF-FTICR-MS simply by the addition of CAs, a procedure that would not be expected to significantly impact protein solubility. Short guard column lifetimes for the fractionations indicate that the centrifugation used to clarify cell lysate was not sufficient. Therefore, much of the reported losses, for both fractionation methods, could be due to aggregated proteins and proteins bound to cellular debris being caught at the top of the guard column. Such proteins are probably at least partly available to trypsin, indicating that (unsurprisingly) proteome coverage will be maximized by performing the digestion as early as possible in the sample preparation process. Finally, for the SEC fractionation, over half of recovered protein mass was found in Fraction 2, while for the RPLC fractionation, over 80% of recovered protein mass was found in Fractions 4–8. Naturally, a more equal distribution between the fractions would have been desirable.
PSORTb [63, 64] was used to examine protein localization for proteins identified using the bottom-up approach (i.e., proteins identified using SEQUEST peptide identifications). Predictably, since no special efforts were made, an over-representation of cytoplasmic and periplasmic, and an under-representation of inner and outer membrane proteins was found. Surfactants and chaotropic reagents are not usually suitable for use on-line with MS, but organic solvents can safely be used to expand proteome coverage to hydrophobic proteins [65]. A bias towards proteins with acidic isoelectric points was observed for the bottom-up identifications, but not against proteins with outlying isoelectric points. As membrane proteins are expected to have predominantly basic isoelectric points [66], this bias is not surprising. No significant protein mass bias was observed for the bottom-up approach, but, as has been mentioned above, there was significant bias against high mass proteins for the intact protein mass measurements.
When attempting to compare the performance of RPLC-FTICR-MS with CIEF-FTICR-MS, which would appear possible using the cell lysate samples, it should be remembered that the cell lysate for CIEF-FTICR-MS analysis was subjected to dialysis with a protein recovery of only ~ 30%. In contrast, dialysis was not required for the RPLC fractions that were subsequently analyzed by CIEF-FTICR-MS, allowing the combination of SEC fractionation with RPLC-FTICR-MS to be compared with the combination of RPLC fractionation with CIEF-FTICR-MS. Since SEC and MS cannot be regarded as highly orthogonal separations, it might be expected that the combination of RPLC fractionation with CIEF-FTICR-MS would provide more detections. However, the combination of SEC fractionation with RPLC-FTICR-MS seems to be the better of the two, although the difference may not be significant. The relatively poorer performance of RPLC fractionation followed by CIEF-FTICR-MS might be attributable to protein precipitation as proteins are brought to their isoelectric point, to low sample loading amounts that reduce protein concentrations below detection limits, and also to proteins binding to the capillary wall during the CIEF process. However, the lower recovery found for the RPLC fractionation is just as likely to be responsible for the overall difference in performance (if this lower recovery is not misleading as suggested above). Nevertheless, the fact that the system based on RPLC fractionation and CIEF-FTICR-MS produces comparable results from a much smaller amount of protein (i.e., ~ 0.5 μg versus ~ 75 μg, when considering the amount of protein loaded to the on-line separation) is certainly of note in the sample-limited situations that are often encountered in multidimensional separations.
While protein modifications in prokaryotes are generally considered to be much less common than in eukaryotes, numerous modifications were expected to be found in a study of the S. oneidensis proteome. N-terminal modifications are perhaps most common, with many proteins suffering the loss of N-terminal methionines [67] or signal peptides [68, 69]; N-terminal acetylation and N-terminal formylmethionine are also common. Other common modifications include oxidation [70], acetylation (at points other than the N-terminus), and histidine phosphorylation (instability in acidic solutions means that this modification is not accessible by the methods described here) [71].
Considering only SEQUEST output for the bottom-up data obtained from digested samples (no enzyme rules were applied), evidence of N-terminal methionine loss (in the form of partially-tryptic peptides) was found for 53 of the 367 identified proteins (14%). Signal peptide deletions were predicted by the SignalP version 3.0 neural network method [68, 69] for 78 of the 367 identified proteins (21%), but only 25 were supported both by the presence of partially-tryptic peptides beginning at the predicted break and by no sequence coverage being found for the signal peptide region. Coverage was found within the signal peptide region for 8 of the remaining predictions, while no evidence, either for or against the deletion, was found for the remaining 45 predictions. Protein oxidation was also investigated, but this required an additional SEQUEST search for singly and doubly oxidized methionines (the most likely site of protein oxidation [70]). 6% of peptides and 45% of proteins showed evidence of oxidation in the form of at least one modified constituent peptide. From this information, collected for a limited set of modifications, most proteins must be expected to be modified in some manner.
Additional investigation would no doubt find evidence of further modifications. However, without complete sequence coverage, determining whether the complete modification complement had been discovered would not be possible. In such cases, and in cases where numerous protein forms with different modification complements were suspected to be present, intact protein mass measurements might be expected to settle the matter.
The concept of matching measured intact protein masses to hypothetical protein masses to which a number of possible modifications have been applied [40, 46–48] was investigated for the proteins detected in this work. Proteins identified from digested samples using the bottom-up approach were used to provide lists of expected masses for each fraction; to these masses were added or subtracted masses for each protein having undergone mass changes equivalent to formylation of the N-terminal methionine, loss of the N-terminal methionine, up to three oxidations, and acetylation. Proteins for which a signal peptide deletion was predicted [68, 69] were included in both the nascent and processed state. All combinations of the listed modifications were permitted (except for the combination of loss and formylation of the N-terminal methionine). The maximum accepted difference between a hypothetical and a measured mass was 50 ppm and tentative identifications were only assigned if there was one single hypothetical mass within 50 ppm of the measured mass (these are listed in Supporting Data Table 2). A total of 117 out of 472 detected proteins (~ 25%) could be assigned tentative identifications; of these proteins, 20 were unmodified, 15 had just lost the N-terminal methionine, and 12 had just been singly oxidized; the remaining 70 were spread between many combinations of the permitted modifications. Evidence of signal peptide deletion was found for 27 detected proteins, which, due to the detection of protein isoforms, represented 18 open reading frames; 9 out of 18 were supported by the bottom-up data. From this and from the bottom-up approach, it seems clear that the S. oneidensis proteome is extensively modified.
The MMA achieved for higher mass proteins is inferior to that routinely achieved for short peptides. Therefore, identifications made on the basis of measured mass alone must be viewed as tentative (for example, measurements of quite exceptional accuracy would be required to distinguish between acetylation and tri-methylation of a high-mass protein). However, even incremental improvements in MMA would be expected to result in significantly enhanced utility; higher field magnets and the use of automatic gain control [72] would be expected to be beneficial. The application of high-throughput proteomics to direct protein measurements could be very useful in comparative proteomics; that is, in quantifying changes in the abundance of particular protein isoforms. Such techniques could rapidly be applied to investigations where either isotopic labeling [73, 74] or direct ion abundance comparisons are used to determine abundance changes in response to a perturbation. Species of interest (e.g., differentially abundant proteins) could then be characterized using targeted MS/MS [75], employing a variety of ion dissociation schemes including electron capture dissociation (ECD) [76], which has proved to be advantageous for the characterization of post-translational modifications.
The reported SEC-RPLC method has been shown to be more effective than the reported RPLC-CIEF method for 2-D protein separations; nearly twice as many proteins were detected using the former in comparison with the latter. Bottom-up measurements, which reflect fractionation performance, indicate bias against hydrophobic proteins, but no bias against proteins with outlying masses or isoelectric points; it would probably be easier to modify the SEC-RPLC method for improved compatibility with hydrophobic proteins. Both methods, however, without alteration, would be expected to be valuable in extending proteome coverage. Given the large difference in sample consumption, CIEF-FTICR-MS intact protein analysis was shown to be remarkably effective when compared with RPLC-FTICR-MS intact protein analysis. Improvements in mass spectrometer sensitivity would be expected to result in a more significant improvement to the CIEF- than to the RPLC-based technique because of the lower sample loading amounts found in the former. The careful choice of separation techniques to simplify the transfer of sample from the first to the second dimension successfully minimized losses at this stage, but significant protein loss still occurred at entry to the first dimension.
Acknowledgments
Particular thanks go to Ken Auberry, Navdeep Jaitly, Sam Purvine and Nikola Tolic for computational assistance, to Margaret Romine for protein localization and signal peptide databases, to Rui Zhao for assistance with high-pressure chromatographic instrumentation, and to Dwayne Elias for growing the cells. We thank the U.S. Department of Energy (DOE) Office of Biological and Environmental Research and the NIH National Center for Research Resources (Grant RR018522) for supporting portions of this research. We also thank the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by DOE and located at Pacific Northwest National Laboratory (PNNL), for use of instrumentation. PNNL is operated by Battelle for the DOE under Contract No. DE AC05 76RLO 1830.
References
- 1.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.James P, Quadroni M, Carafoli E, Gonnet G. Biochem Biophys Res Comm. 1993;195:58–64. doi: 10.1006/bbrc.1993.2009. [DOI] [PubMed] [Google Scholar]
- 3.Mann M, Hojrup P, Roepstorf P. Biol Mass Spectrom. 1993;22:338–345. doi: 10.1002/bms.1200220605. [DOI] [PubMed] [Google Scholar]
- 4.Pappin DJC, Hojrup P, Bleasby AJ. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 5.Yates JR, 3rd, Speicher S, Griffin PR, Hunkapiller T. Anal Biochem. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- 6.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 7.Klose J, Kobalz U. Electrophoresis. 1995;16:1034–1059. doi: 10.1002/elps.11501601175. [DOI] [PubMed] [Google Scholar]
- 8.Klose J, Link AJ, editors. 2-D Proteome Analysis Protocols. Humana Press; Totowa, NJ, USA: 1999. pp. 147–172. [Google Scholar]
- 9.Giavalisco P, Nordhoff E, Kreitler T, Klöppel KD, Lehrach H, Klose J, Gobom J. Proteomics. 2005;5:1902–1913. doi: 10.1002/pmic.200401062. [DOI] [PubMed] [Google Scholar]
- 10.Lescuyer P, Hochstrasser DF, Sanchez JC. Electrophoresis. 2004;25:1125–1135. doi: 10.1002/elps.200305792. [DOI] [PubMed] [Google Scholar]
- 11.Davis JM. Anal Chem. 1991;63:2141–2152. [Google Scholar]
- 12.Lewis KC, Opiteck GJ, Jorgenson JW, Sheeley DM. J Am Soc Mass Spectrom. 1997;8:495–500. [Google Scholar]
- 13.Opiteck GJ, Lewis KC, Jorgenson JW, Anderegg RJ. Anal Chem. 1997;69:1518–1524. doi: 10.1021/ac961155l. [DOI] [PubMed] [Google Scholar]
- 14.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Balgley BM, Rudnick PA, Lee CS. J Chromatogr A. 2005;1073:35–41. doi: 10.1016/j.chroma.2004.08.140. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Lee CS, Shen Y, Smith RD, Baehrecke EH. Electrophoresis. 2002;23:3143–3148. doi: 10.1002/1522-2683(200209)23:18<3143::AID-ELPS3143>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Balgley BM, DeVoe DL, Lee CS. Anal Chem. 2003;75:3145–3152. doi: 10.1021/ac034014+. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Balgley BM, Rudnick PA, Evans EL, DeVoe DL, Lee CS. J Proteome Res. 2005;4:36–42. doi: 10.1021/pr049876l. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Rudnick PA, Evans EL, Li J, Zhuang Z, Devoe DL, Lee CS, Balgley BM. Anal Chem. 2005;77:6549–6556. doi: 10.1021/ac050491b. [DOI] [PubMed] [Google Scholar]
- 20.Zhou F, Johnston MV. Anal Chem. 2004;76:2734–2740. doi: 10.1021/ac035446n. [DOI] [PubMed] [Google Scholar]
- 21.Zhou F, Johnston MV. Electrophoresis. 2005;26:1383–1388. doi: 10.1002/elps.200410125. [DOI] [PubMed] [Google Scholar]
- 22.Mohan D, Lee CS. Electrophoresis. 2002;23:3160–3167. doi: 10.1002/1522-2683(200209)23:18<3160::AID-ELPS3160>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 23.Mohan D, Pasa-Tolic L, Masselon CD, Tolic N, Bogdanov B, Hixson KK, Smith RD, Lee CS. Anal Chem. 2003;75:4432–4440. doi: 10.1021/ac0342572. [DOI] [PubMed] [Google Scholar]
- 24.Sluyterman LAA, Elgersma O. J Chromatogr. 1978;150:17–30. [Google Scholar]
- 25.Sluyterman LAA, Wijdenes J. J Chromatogr. 1978;150:31–44. [Google Scholar]
- 26.Sluyterman LAA, Wijdenes J. J Chromatogr. 1981;206:429–440. [Google Scholar]
- 27.Sluyterman LAA, Wijdenes J. J Chromatogr. 1981;206:441–447. [Google Scholar]
- 28.Chong BE, Yan F, Lubman DM, Miller FR. Rapid Commun Mass Spectrom. 2001;15:291–296. doi: 10.1002/rcm.227. [DOI] [PubMed] [Google Scholar]
- 29.Kreunin P, Urquidi V, Lubman DM, Goodison S. Proteomics. 2004;4:2754–2765. doi: 10.1002/pmic.200300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu K, Kachman MT, Miller FR, Lubman DM, Zand R. J Chromatogr A. 2004;1053:133–142. [PubMed] [Google Scholar]
- 31.Zhu K, Miller FR, Barder TJ, Lubman DM. J Mass Spectrom. 2004;39:770–780. doi: 10.1002/jms.650. [DOI] [PubMed] [Google Scholar]
- 32.Zhu K, Zhao J, Lubman DM, Miller FR, Barder TJ. Anal Chem. 2005;77:2745–2755. doi: 10.1021/ac048494w. [DOI] [PubMed] [Google Scholar]
- 33.Righetti PG, Castagna A, Herbert B, Candiano G. Biosci Rep. 2005;25:3–17. doi: 10.1007/s10540-005-2844-2. [DOI] [PubMed] [Google Scholar]
- 34.Zuo X, Speicher DW. Anal Biochem. 2000;284:266–278. doi: 10.1006/abio.2000.4714. [DOI] [PubMed] [Google Scholar]
- 35.Zuo X, Echan L, Hembach P, Tang HY, Speicher KD, Santoli D, Speicher DW. Electrophoresis. 2001;22:1603–1615. doi: 10.1002/1522-2683(200105)22:9<1603::AID-ELPS1603>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 36.Zuo X, Hembach P, Echan L, Speicher DW. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;782:253–265. doi: 10.1016/s1570-0232(02)00567-6. [DOI] [PubMed] [Google Scholar]
- 37.Zuo X, Speicher DW. Proteomics. 2002;2:58–68. [PubMed] [Google Scholar]
- 38.Heller M, Michel PE, Morier P, Crettaz D, Wenz C, Tissot JD, Reymond F, Rossier JS. Electrophoresis. 2005;26:1174–1188. doi: 10.1002/elps.200410106. [DOI] [PubMed] [Google Scholar]
- 39.Millea KM, Kass IJ, Cohen SA, Krull IS, Gebler JC, Berger SJ. J Chromatogr A. 2005;1079:287–298. doi: 10.1016/j.chroma.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 40.Millea KM, Krull IS, Cohen SA, Gebler JC, Berger SJ. J Proteome Res. 2006 doi: 10.1021/pr050278w. published on the Internet. [DOI] [PubMed] [Google Scholar]
- 41.Opiteck GJ, Ramirez SM, Jorgenson JW, Moseley MA., 3rd Anal Biochem. 1998;258:349–361. doi: 10.1006/abio.1998.2588. [DOI] [PubMed] [Google Scholar]
- 42.Jensen PK, Pasa-Tolic L, Anderson GA, Horner JA, Lipton MS, Bruce JE, Smith RD. Anal Chem. 1999;71:2076–2084. doi: 10.1021/ac990196p. [DOI] [PubMed] [Google Scholar]
- 43.Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. Anal Chem. 2002;74:2923–2929. doi: 10.1021/ac020049i. [DOI] [PubMed] [Google Scholar]
- 44.Lovley DR, Phillips EJP, Gorby YA, Landa ER. Nature. 1991;350:413–416. [Google Scholar]
- 45.Fredrickson JK, Romine MF. Curr Opin Biotechnol. 2005;16:269–274. doi: 10.1016/j.copbio.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Lee SW, Berger SJ, Martinovic S, Pasa-Tolic L, Anderson GA, Shen Y, Zhao R, Smith RD. Proc Natl Acad Sci USA. 2002;99:5942–5947. doi: 10.1073/pnas.082119899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.VerBerkmoes NC, Bundy JL, Hauser L, Asano KG, Razumovskaya J, Larimer F, Hettich RL, Stephenson JL., Jr J Proteome Res. 2002;1:239–252. doi: 10.1021/pr025508a. [DOI] [PubMed] [Google Scholar]
- 48.Strader MB, Verberkmoes NC, Tabb DL, Connelly HM, Barton JW, Bruce BD, Pelletier DA, Davison BH, Hettich RL, Larimer FW, Hurst GB. J Proteome Res. 2004;3:965–978. doi: 10.1021/pr049940z. [DOI] [PubMed] [Google Scholar]
- 49.Elias DA, Monroe ME, Marshall MJ, Romine MF, Belieav AS, Fredrickson JK, Anderson GA, Smith RD, Lipton MS. Proteomics. 2005;5:3120–3130. doi: 10.1002/pmic.200401140. [DOI] [PubMed] [Google Scholar]
- 50.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 51.Adkins JN, Monroe ME, Auberry KJ, Shen Y, Jacobs JM, Camp DG, 2nd, Vitzthum F, Rodland KD, Zangar RC, Smith RD, Pounds JG. Proteomics. 2005;5:3454–3466. doi: 10.1002/pmic.200401333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen Y, Zhao R, Belov ME, Conrads TP, Anderson GA, Tang K, Pasa-Tolic L, Veenstra TD, Lipton MS, Udseth HR, Smith RD. Anal Chem. 2001;73:1766–1775. doi: 10.1021/ac0011336. [DOI] [PubMed] [Google Scholar]
- 53.Eng JK, McCormack AL, Yates JR., 3rd J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 54.Romine MF, Elias DA, Monroe ME, Auberry K, Fang R, Fredrickson JK, Anderson GA, Smith RD, Lipton MS. Omics. 2004;8:239–254. doi: 10.1089/omi.2004.8.239. [DOI] [PubMed] [Google Scholar]
- 55.Washburn MP, Wolters D, Yates JR., 3rd Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 56.Shen Y, Smith RD. J Microcol Sep. 2000;12:135–141. [Google Scholar]
- 57.Tang Q, Harrata AK, Lee CS. Anal Chem. 1995;67:3515–3519. [Google Scholar]
- 58.Tang Q, Harrata AK, Lee CS. Anal Chem. 1996;68:2482–2487. doi: 10.1021/ac960169o. [DOI] [PubMed] [Google Scholar]
- 59.Clarke NJ, Naylor S. Biomed Chromatogr. 2002;16:287–297. doi: 10.1002/bmc.159. [DOI] [PubMed] [Google Scholar]
- 60.Vilkov AN, Bogdanov B, Pasa-Tolic L, Prior DC, Anderson GA, Masselon CD, Moore RJ, Smith RD. Rapid Commun Mass Spectrom. 2004;18:2682–2690. doi: 10.1002/rcm.1664. [DOI] [PubMed] [Google Scholar]
- 61.Shaffer SA, Tang KQ, Anderson GA, Prior DC, Udseth HR, Smith RD. Rapid Commun Mass Spectrom. 1997;11:1813–1817. [Google Scholar]
- 62.Horn DM, Zubarev RA, McLafferty FW. J Am Soc Mass Spectrom. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 63.Gardy JL, Spencer C, Wang K, Ester M, Tusnady GE, Simon I, Hua S, deFays K, Lambert C, Nakai K, Brinkman FS. Nucleic Acids Res. 2003;31:3613–3617. doi: 10.1093/nar/gkg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- 65.Blonder J, Goshe MB, Moore RJ, Pasa-Tolic L, Masselon CD, Lipton MS, Smith RD. J Proteome Res. 2002;1:351–360. doi: 10.1021/pr0255248. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz R, Ting CS, King J. Genome Res. 2001;11:703–709. doi: 10.1101/gr.gr-1587r. [DOI] [PubMed] [Google Scholar]
- 67.Giglione C, Meinnel T. Trends Plant Sci. 2001;6:566–572. doi: 10.1016/s1360-1385(01)02151-3. [DOI] [PubMed] [Google Scholar]
- 68.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 69.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 70.Berlett BS, Stadtman ER. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 71.Klumpp S, Krieglstein J. Eur J Biochem. 2002;269:1067–1071. doi: 10.1046/j.1432-1033.2002.02755.x. [DOI] [PubMed] [Google Scholar]
- 72.Page JS, Bogdanov B, Vilkov AN, Prior DC, Buschbach MA, Tang K, Smith RD. J Am Soc Mass Spectrom. 2005;16:244–253. doi: 10.1016/j.jasms.2004.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pasa-Tolic L, Jensen PK, Anderson GA, Lipton MS, Peden KK, Martinovic S, Tolic N, Bruce JE, Smith RD. J Am Chem Soc. 1999;121:7949–7950. [Google Scholar]
- 74.Jensen PK, Pasa-Tolic L, Peden KK, Martinovic S, Lipton MS, Anderson GA, Tolic N, Wong KK, Smith RD. Electrophoresis. 2000;21:1372–1380. doi: 10.1002/(SICI)1522-2683(20000401)21:7<1372::AID-ELPS1372>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 75.Masselon C, Pasa-Tolic L, Tolic N, Anderson GA, Bogdanov B, Vilkov AN, Shen Y, Zhao R, Qian WJ, Lipton MS, Camp DG, 2nd, Smith RD. Anal Chem. 2005;77:400–406. doi: 10.1021/ac049043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zubarev RA, Kelleher NL, McLafferty FW. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]