

Abstract
Phenobarbital is a lipophilic molecule used as a sedative and antiepileptic drug that elicits a multitude of effects in the liver, including gross liver enlargement, hepatocyte hypertrophy, and induced expression of drug-metabolizing enzymes and other liver-specific genes. The constitutive androstane receptor (CAR; NR1I3) and to a lesser extent the pregnane X receptor (PXR; NR1I2) are responsible for mediating induction of many phenobarbital-responsive genes. However, CAR-mediated transcriptional control of some genes is critically dependent on hepatocyte nuclear factor 4 alpha (HNF-4α; NR2A1), which itself regulates multiple liver-specific genes involved in hepatic growth, metabolism, and differentiation. We studied the effects of phenobarbital on HNF-4α expression in hepatocytes and provide evidence that HNF-4α nuclear expression is regulated in response to phenobarbital. Real-time polymerase chain reaction analyses revealed that HNF-4α mRNA is modestly up-regulated by phenobarbital. In addition, nuclear expression of HNF-4α protein is significantly elevated 3 hours after the administration of phenobarbital in wild-type, CAR−/−, and CAR−/−/PXR−/− mice. In vitro analysis revealed that phenobarbital-induced HNF-4α expression is both time- and dose dependent. In addition, the phosphatase inhibitor okadaic acid and the Ca2−/calmodulin-dependent protein kinase II inhibitor KN62 block nuclear induction of HNF-4α by phenobarbital. Furthermore, HNF-4α nuclear expression is enhanced by inhibition of cyclic AMP– dependent protein kinase A. In conclusion, induced nuclear expression of HNF-4α and CAR is an integral part of the phenobarbital response, aimed at coordinated regulation of genes involved in drug metabolism and detoxification as well as maintenance of liver function.
Abbreviations: CAR, constitutive androstane receptor; PXR, pregnane X receptor; HNF-4α, hepatocyte nuclear factor 4 alpha; PB, phenobarbital; CYP, cytochrome P450; PCR, polymerase chain reaction
Phenobarbital (PB) is an antiepileptic drug and is the prototype of a large family of lipophilic PB-like compounds that have profound effects in the liver. Its effects on liver physiology are typified by hepatic hypertrophy, hyperproliferation of the smooth endoplasmic reticulum, and induction or repression of numerous genes, especially the genes of cytochrome P450 enzymes.1–3 Hepatic hypertrophy induced by PB is mediated by a moderate increase in hepatocyte DNA synthesis and dramatic enlargement of individual hepatocytes.4,5 Hepatic enlargement subsides after PB withdrawal and this decrease in liver size is mediated by hepatocyte apoptosis.6 Liver regeneration in response to partial hepatectomy is markedly diminished in PB-treated rat and mouse livers.7,8 PB is also one of the most effective liver tumor promoters in both rats and mice. The mechanism of liver tumor promotion by PB is not understood; however, there is evidence that PB affects EGF signaling.9. Furthermore, TGFβ1 receptors I, II, and III are specifically down-regulated in early neoplastic lesions following treatment with PB.10,11 It has been hypothesized that the inhibition of DNA synthesis in normal hepatocytes gives a proliferative advantage to initiated cells.
The complex effects of PB on hepatocytes are also seen in primary cultures. Typically, hepatocytes in primary culture lose characteristic gene expression markers. Treatment of the cultures with PB results in stabilization of the differentiated hepatocyte phenotype, as assessed by many differentiated functions including expression of albumin and cytochrome P450 family members and metabolic activities.12 PB treatment in culture also is associated with decreased response to hepatocyte mitogens, including TGFα and HGF.13
The mechanisms by which PB exerts all these complex effects in hepatocytes are not clear. The same pathways involved in changes in PB-induced gene expression may mediate these diverse effects. PB induces changes in expression of several cytochrome P450s and conjugation enzymes. Regulation of these genes is part of an integrated cellular defense response aimed at detoxification of potentially harmful endo- and xenobiotics.14,15 The constitutive androstane receptor (CAR) is responsible for phenobarbital-induced expression of many cytochrome P450s and other genes involved in detoxification such as glucuronosyl transferases.16 CAR is a liver-enriched nuclear receptor that, in the absence of signaling, is retained in the cytoplasm in an inactive state, complexed with HSP90. PB treatment results in translocation of CAR to the nucleus through a phosphorylation-dependent mechanism and subsequent activation, leading to transcriptional regulation of PB-responsive genes.17
Interestingly, hepatocyte nuclear factor 4 (NR2A1; HNF-4α) also has a role in the regulation of many P450 genes that have HNF-4α-binding sites.18,19 HNF-4α is an orphan nuclear hormone receptor expressed in liver, kidney, intestine, and pancreas and regulates expression of numerous liver enzymes involved in glucose, cholesterol, and fatty acid metabolism, as well as in apolipoprotein and urea biosynthesis. Recently it has been shown that HNF-4α is critical to CAR- and pregnane X receptor (PXR)-mediated induction of CYP3A4 and CYP2C9.20,21 Furthermore, HNF-4 regulates expression of UGT1A9, which is involved in catabolism and elimination of endo-and xenobiotics.22 From these and other studies, it is evident that HNF-4α plays a crucial role in regulating expression of many hepatic genes; however, little is known about if and how HNF-4α expression is regulated in response to phenobarbital and other drugs. In the present study, we showed that PB treatment results in increased nuclear expression of HNF-4α protein in hepatocytes. This induction is independent of the xenobiotic-sensing CAR and PXR nuclear receptors and occurs at both the mRNA and protein levels. In vitro analysis of cellular phosphatase and kinase pathways involved in PB signaling revealed their roles in regulating HNF-4α expression. These data suggest that regulation of HNF-4α protein expression is a distinct part of a coordinated mechanism mediating the hepatic response to PB.
Materials and Methods
Animals
Male Fisher 344 rats were used for rat hepatocyte isolation. Male wild-type C57 black, PXR-null, and CAR-null mice were used for in vivo studies. All animals received humane care and were treated according to protocols approved by the University of Pittsburgh’s institutional animal care and use committee (IACUC).
Isolation and Culture of Hepatocytes
Rat hepatocytes were isolated by two-step collagenase perfusion, as previously described23 and then seeded at 3 × 106 cells per 100 mm of a collagen-coated dish in minimal essential media containing insulin. After 1 hour, the medium was changed to serum-free hepatocyte growth medium containing ITS and dexamethasone as described elsewhere.24 After 18–20 hours, cells were treated with PB (2 mmol/L unless otherwise indicated) and harvested at various times thereafter. When indicated, okadaic acid (10 nmol/L), KN62 (5 μmol/L), and RP isomer of 8-bromo-cyclic adenosine monophosphate (RP-8Br-cAMP; 10 μmol/L) were added 30 minutes prior to administration of PB.
RNA Isolation and Real-Time PCR Analysis
Total RNA was isolated using RNA-B solution (Tel-Test, Friendswood, TX), then treated with Turbo DNA-Free (Ambion, Austin, TX) according to the manufacturer’s instructions. Two micrograms of total RNA was reverse-transcribed with SuperScript II (Invitrogen, Carlsbad, CA). Forty nanograms of the resulting cDNA was used for Sybr Green real-time polymerase chain reaction (PCR) analysis (Applied Biosystems, Foster City, CA), along with mouse HNF-4α (SuperArray Biosciences, Frederick, MD) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers.25 Real-time PCR analysis was performed using an ABI Prism 7000 sequence detection system. Results were compiled from two independent experiments with assays performed three times with each sample in triplicate. Data presented are based on the comparative CT method and reflect relative quantitation to GAPDH as the internal control. Statistical significance was determined using the Student t test.
Immunofluorescence Staining
Hepatocytes grown on collagen-coated coverslips were washed with (PBS), fixed with cold 2% paraformaldehyde in PBS for 15 minutes, permeabilized with 0.1% TritonX-100 in PBS for 15 minutes, washed five times in 0.5% BSA solution (PBS containing 20 mmol/L glycine and 0.5% BSA), then blocked for 1 hour with 2% BSA solution. Following five washes with 0.5% BSA solution, coverslips were incubated overnight with goat anti-HNF-4α, 1:500 (Santa Cruz Biotechnology, Santa Cruz CA), in 0.5% BSA solution. Cells were washed five times with 0.5% BSA solution, then incubated for 1 hour with donkey antigoat CY3 in 0.5% BSA solution. Following five washes in 0.5% BSA solution and then PBS, coverslips were incubated with Hoescht stain (30–45 s), washed with PBS, and mounted on slides. Fluorescence was visualized using an Olympus Provis fluorescence microscope at 60× magnification.
Protein Isolation
Cells from one 100-mm plate or 100 mg of liver tissue was lysed by sonication in RIPA buffer containing protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO), then incubated on ice for 30 minutes and centrifuged at 14,000g for 30 minutes. Supernatants were saved as total cell lysate.
For nuclear protein isolation, cells from 3–5 plates were washed and harvested in 40 mmol/L Tris (pH 7.6), 14 mmol/L NaCl, and 1 mmol/L EDTA, then pooled and centrifuged (5 min, 100g). Cell pellets were suspended in 2 mL of hypotonic buffer [10 mmol/L Hepes (pH 7.9), 10 mmol/L NaH2PO4, 1.5 mmol/L MgCl2, 1 mmol/L DTT, 0.5 mmol/L spermidine, and 1 mol/L NaF with protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO)]. Following 10 minutes of incubation on ice, samples were homogenized in a Dounce homogenizer and then centrifuged (5 min, 800g). Cell lysis was monitored with trypan blue stain. Supernatants were saved as cytoplasmic extracts. The nuclei pellets were washed two additional times in the same buffer. Mouse liver tissue (0.2–0.3 g) was directly homogenized in 5 mL of hypotonic buffer and then centrifuged (5 min, 100g). The pellets were resuspended in 2 mL of hypotonic buffer and processed as above.
Nuclear proteins were extracted in 50–100 μL of hypertonic buffer [30 mmol/L Hepes, (pH 7.9), 25% glycerol, 450 mmol/L NaCl, 12 mmol/L MgCl2, 1 mmol/L DTT, and 0.1 mmol/L EDTA with protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO)] for 45 minutes at 4°C with continuous agitation. Extracts were centrifuged at 30,000g, and the supernatants were collected and dialyzed for 2 hours against the same solution but containing 150 mmol/L NaCl. Protein concentration was determined by the Bicinchoninic Acid assay (Sigma, St. Louis, MO).
Western Blot Analysis
Nuclear proteins (20 μg), cytoplasmic extract (50 μg), or total cell lysate (100 μg) were separated by SDS-PAGE in 4%–12% NuPage Bis-Tris gels with 1× MOPS Buffer (Invitrogen, Carlsbad, CA), then transferred to Immobilon-P membranes (Millipore, Bedford, MA) in NuPAGE transfer buffer containing 0.005% SDS and 10% methanol. Membranes were stained with Ponceau S to verify loading and transfer efficiency. Membranes were probed with antibodies in TBST containing 5% nonfat milk, then processed with SuperSignal West Pico chemiluminescence substrate (Pierce, Rockford, IL) and exposed to blue X-ray film (Lab Product Sales, Rochester, NY). The primary antibodies used were: rabbit anti-HNF-4α N1.14 (F. Sladek, UC Riverside), 1:5,000; goat anti-HNF-4α C-19, rabbit anti-CAR H-150, and goat anti-PXR R14 (Santa Cruz Biotechnology, Santa Cruz CA), 1:500; and mouse anti-actin MAB150 (Chemicon, Temecula, CA) 1:2000. HRP-conjugated secondary antibodies were used at a 1:80,000 ratio (Chemicon, Temecula, CA). Protein expression was normalized to β-actin for total cell lysate or total protein per lane from Ponceau S stain of membrane for nuclear extracts.
Results
Phenobarbital Induces HNF-4α Protein Expression in the Nucleus
Our laboratory has previously shown that PB induces or maintains differentiation and high levels of HNF-4α DNA-binding activity as well as cytochrome P450s in cultured rat hepatocytes.12 To determine whether HNF-4α induction was physiologically relevant in vivo, we analyzed its expression in mouse liver following a single injection of PB (100 mg/kg, i.p.). Western blot analysis of liver nuclear protein extracts revealed a dramatic elevation in HNF-4α protein expression 2–4 hours post-PB in wild-type mice (Fig. 1A). Peak HNF-4 nuclear protein expression occurred after 3 hours and was, on average, at least threefold higher than untreated liver. This pattern of induced expression paralleled that of CAR, the primary mediator of PB-induced transcription and was consistent with previous reports of peak CAR induction by PB occurring after 3 hours.26 Levels of both the HNF-4α and CAR nuclear proteins remained elevated compared with those in untreated liver through 6 hours post-PB (data not shown). However, PXR protein levels increased slightly from 2 to 4 hours and then fell to normal levels. These results reveal the temporal regulation of HNF-4α expression by PB; however, they do not rule out CAR or PXR as potential mediators of this regulation.
Fig. 1.
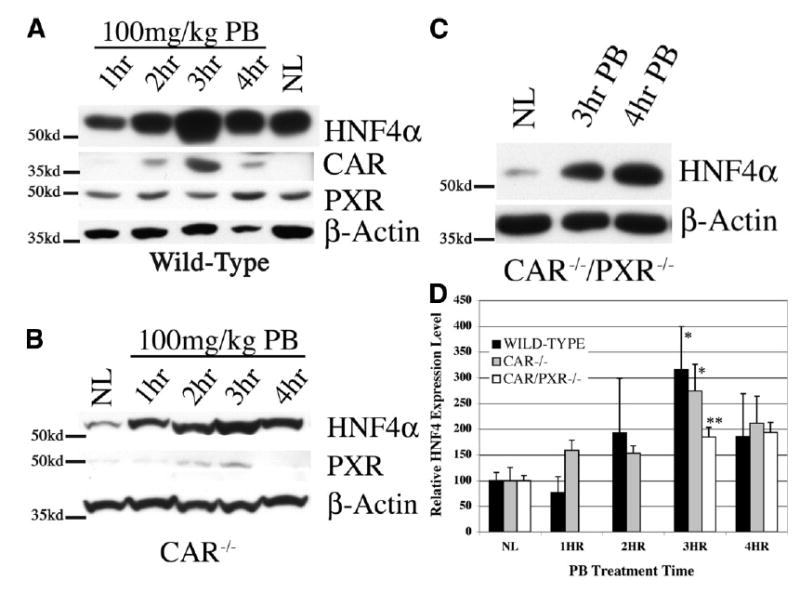
Effect of PB on nuclear expression of HNF-4α, CAR, and PXR in vivo. Representative Western blot analyses of liver nuclear protein from (A) wild-type, (B) CAR−/−, and (C) CAR−/−/PXR−/− mice given a single intraperitoneal injection (100 mg/kg) of PB (NL, normal liver [saline injection]), Locations of molecular-weight standards are indicated on the left; HNF-4α (~54 kd); CAR (~40 kd); PXR (~49 kd). Note the significant increase in HNF-4α expression after 3 hours compared to that of normal liver in each mouse strain. β-Actin is shown for approximation of sample loading. (D) Graphical representation of HNF-4αexpression following PB treatment. Expression levels were normalized to total protein/lane, and the combined results from three independent experiments are presented as a mean ± SEM. Statistical significance was determined using the Student t test. (*P < .05; **P < .01.)
Phenobarbital-Induced HNF-4α Nuclear Expression Is Independent of CAR and PXR
To examine whether PB-induced HNF-4α protein expression is dependent on CAR and/or PXR, we analyzed the response in knockout mice. Western blot analysis of nuclear extracts from CAR−/− mice treated with PB revealed a marked induction of HNF-4α protein between 2 and 4 hours, reaching a threefold increase after 3 hours (Fig. 1B). This pattern of expression is very similar to that seen in wild-type mice; however, because PXR expression was slightly increased in CAR−/−mice after 2–3 hours, and to rule out the possibility that PXR may compensate for the absence of CAR in mediating HNF-4α induction, we examined the response to PB in double-knockout mice (Fig. 1C). Again, HNF-4α nuclear protein expression was significantly induced in CAR−/−/PXR−/− mice 3–4 hours after the administration of PB, confirming HNF-4α nuclear protein induction by PB is independent of CAR and PXR.
To determine whether PB-induced HNF-4αexpression is regulated at the mRNA level, we performed real-time PCR analysis on RNA isolated from control and PB treated livers of wild-type and CAR−/− mice (Fig. 2). Real-time PCR analysis revealed that HNF-4α mRNA expression was consistently induced up to 1.6-fold at 2 hours post-PB. The mRNA change was modest but statistically significant (P < .01) and levels remained slightly elevated through 4 hours post PB treatment, consistent with higher levels of HNF-4α protein seen from 3 to 6 hours. To verify that HNF-4α mRNA induction is independent of CAR, we performed an analogous experiment in CAR−/− animals (Fig. 2). Again HNF-4α mRNA levels were modestly but significantly increased at 2 hours post-PB (P < .05) and remained slightly elevated through 4 hours. This result parallels that of the wild type mice, thus indicating that HNF-4α mRNA induction by PB occurs through a CAR-independent mechanism.
Fig. 2.
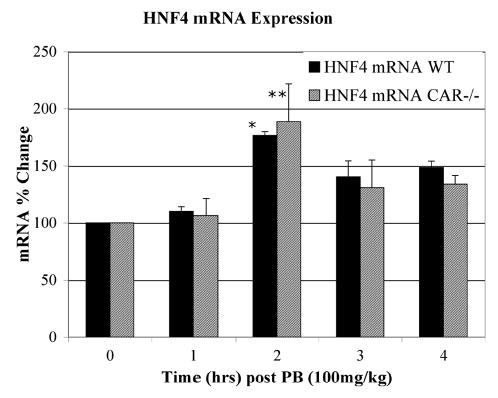
Semiquantitative real-time PCR analysis of HNF-4α expression in wild-type and CAR−/− mice. Real-time PCR analysis was performed by the Sybr Green method in an ABI Prism 7000 sequence detection system, using mouse HNF-4α and GAPDH primers. Results were compiled from two independent experiments with assays performed three times with each sample in triplicate. Data presented are based on the comparative CT method and reflect relative quantitation to GAPDH as the internal control. Statistical significance was determined using the Student t test. (*P < .01; **P < .05)
HNF-4α Nuclear Protein Induction Is Dose- and Time Dependent
To further characterize PB-induced HNF-4α protein expression, we studied the response in vitro using primary rat hepatocytes cultured in the presence of PB. Analysis of nuclear protein extracts from PB-treated hepatocyte cultures showed a temporally regulated induction of HNF-4α expression similar to that seen in vivo (Fig. 3A). HNF-4α nuclear protein levels had increased by 3 hours and remained elevated through 6 hours.
Fig. 3.
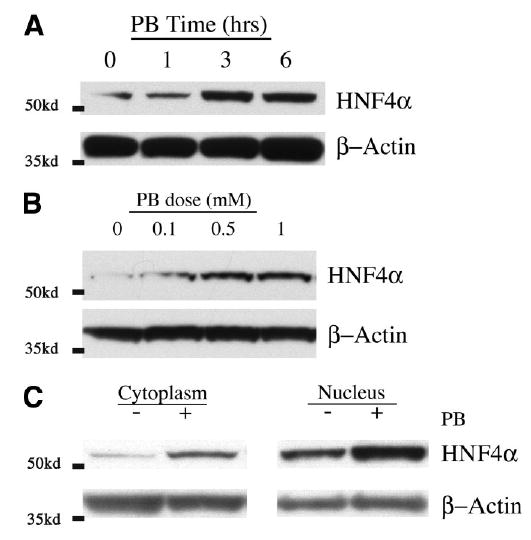
Characterization of HNF-4α protein induction by PB in vitro. Rat hepatocytes cultured for 20 hours were treated with PB and subjected to Western blot analysis as described in the Materials and Methods section. (A) Time course of HNF-4α expression in nuclear proteins from 2 mmol/L PB-treated hepatocytes. (B) Dose response of HNF-4α expression in nuclear proteins from hepatocytes treated with PB (0.1–1 mmol/L) for 3 hours. (C) Expression profile of HNF-4α protein in cytoplasm and nucleus with and without PB treatment. β-Actin is shown for approximation of sample loading. Representative blots of at three independent experiments are depicted, and the locations of molecular-weight standards are indicated on the left.
We next treated hepatocytes with increasing amounts of PB to determine the dose dependence of this response. Western blot analysis revealed that HNF-4α nuclear protein expression was induced in a dose-dependent manner, increasing two- to fivefold with doses from 0.1 to 1 mmol/L PB (Fig. 3B). Together these results reveal a direct association between PB signaling and induced expression of HNF-4α and validate this model system as appropriate for studying the hepatic response to PB.
Cellular HNF-4α protein is primarily in the nucleus; however, a small portion may also be in the cytoplasm, as seen by immunofluorescence, discussed later. Therefore, we sought to determine whether PB-induced expression of HNF-4α nuclear protein was a result of an overall increase in HNF-4α protein resulting in part from increased mRNA expression, or possibly from secondary modifications as a consequence of increased localization in the nucleus, comparable to that seen with CAR. We isolated cytoplasmic and nuclear extracts from PB-treated hepatocytes and performed Western blot analysis. Figure 3C shows that both the cytoplasmic and nuclear fractions of cultured hepatocytes contain detectable amounts of HNF-4α protein, which increased proportionally three-to fivefold in response to PB treatment. Hence, PB signaling results in an increase in total cellular levels of HNF-4α protein, which is primarily in the nucleus to begin with, and not an increase in nuclear translocation. We were unable to detect HNF-4 protein expression in cytosolic fractions from whole liver, and the levels observed in cytoplasmic extracts of hepatocyte cultures were mainly a result of contaminating nuclear protein from the protein isolation procedure.
Signaling Pathways Regulating HNF-4α Nuclear Expression in Response to PB
It is well documented that PB signaling causes nuclear translocation of CAR via a phosphorylation-sensitive event that can be blocked by treatment with the phosphatase inhibitor okadaic acid. We treated primary hepatocyte cultures with 10 nmol/L okadaic acid and then stimulated them with PB for 3 hours to see if HNF-4α protein induction was affected. Western blot analyses of total cell lysates and nuclear protein extracts revealed detectable levels of HNF-4α protein, which increased approximately twofold after 3 hours of PB treatment, much like in vivo (Fig. 4). However, okadaic acid treatment alone resulted in a dramatic 80% reduction in nuclear HNF-4α expression while only minimally changing total HNF-4α protein levels. Furthermore, okadaic acid treatment blocked induction by PB of HNF-4α protein expression. These Western blot data were also confirmed by HNF-4α immunofluorescence (Fig. 5). Untreated hepatocytes showed predominantly nuclear HNF-4α immunoreactivity with minor cytoplasmic staining. Following PB treatment HNF-4α protein level increased significantly in the nuclei compared with that in untreated cells, whereas cytoplasmic staining was still weak. On the other hand, okadaic acid treatment caused a dramatic reduction in nuclear HNF-4α immunoreactivity, whereas the cytoplasmic level increased slightly with a punctate staining pattern. PB treatment was unable to induce HNF-4α expression, nuclear or otherwise, in the presence of okadaic acid. Taken together, these data indicate that HNF-4α nuclear expression and PB inducibility require a phosphatase activity that is inhibited by okadaic acid.
Fig. 4.
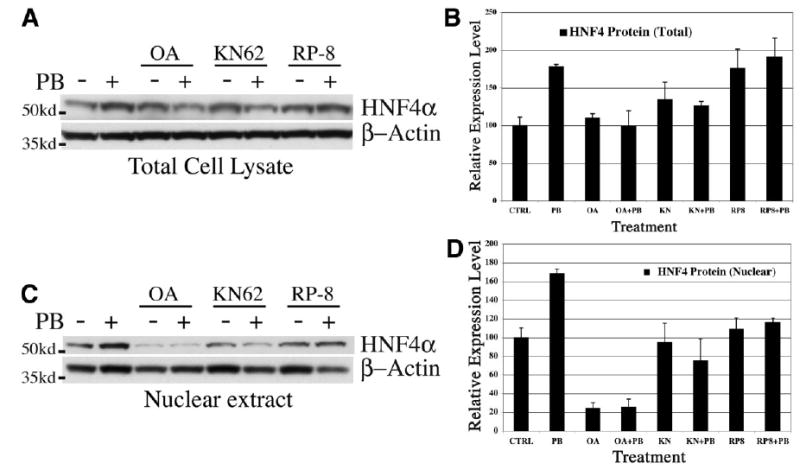
Effects of phosphatase and kinase pathways on PB induction of HNF-4α. Rat hepatocytes were cultured for 20 hours, then treated with 2 mmol/L PB for 3 hours with and without phosphatase or kinase inhibitors and subjected to Western blot analyses for HNF-4α. (A) Representative Western blot analysis of HNF-4α expression in total cell lysates from PB-treated hepatocytes. (B) Graphical representation of total HNF-4α expression normalized to β-actin with combined results from three independent experiments presented as a mean ± SEM. (C) Representative Western blot analysis of HNF-4α expression in nuclear extracts from PB-treated hepatocytes. (D) Graphical representation of HNF-4α nuclear expression normalized to total protein/lane with combined results from three independent experiments presented as a mean ± SEM. Where indicated, the inhibitors okadaic acid (10 nmol/L), KN62 (5 μmol/L), or Rp-8Br-cAMP (10 μmol/L) were added 30 minutes prior to PB. The locations of molecular-weight standards are indicated on the left.
Fig. 5.
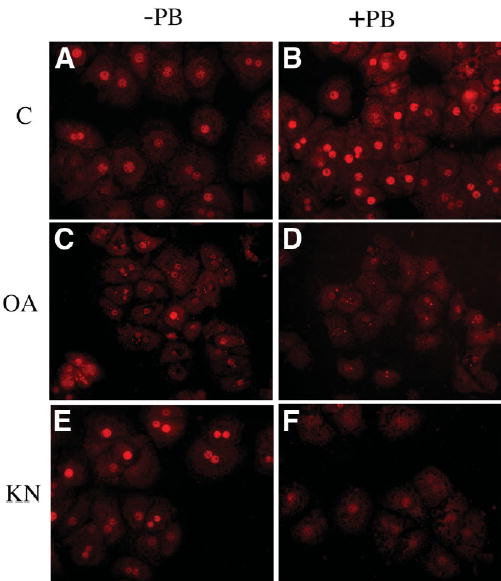
PB induction of HNF-4α protein in vitro. Rat hepatocytes cultured on coverslips and treated with 2 mmol/L PB for 3 hours were analyzed by immunofluorescent staining with HNF-4α antibody as described in the Materials and Methods section. HNF-4α is visualized with CY3 fluorescence (red). (A and B) PB treatment resulted in increased nuclear expression of HNF-4α (compare A and B). (C and D) Okadaic acid treatment alone caused marked loss of nuclear HNF-4α and inhibited nuclear induction by PB. (E and F) KN62 treatment caused a modest increase in overall HNF-4α expression, but nuclear levels were repressed when combined with PB treatment.
To further characterize the signaling pathways governing HNF-4α protein induction by PB, we analyzed inhibitors of protein kinases known to be involved in PB signaling. Other studies have shown that Ca2−/calmod-ulin-dependent protein kinase (CaM-PK) and cAMP-dependent protein kinase A (PKA) are involved in PB-induced cytochrome P450 expression in mouse and rat hepatocytes.27,28 We investigated the potential role of these kinases in regulation of PB-induced HNF-4α expression by treating cultured hepatocytes with the kinase-specific inhibitors KN62 and RP-8Br-cAMP. As seen in Fig. 4, inhibition of CaM-PK with KN62 alone caused a modest increase in total but not in nuclear HNF-4α protein. This cytoplasmic increase was negated by PB treatment, and nuclear levels were sharply reduced. These data were confirmed by immunofluorescent staining, which showed that blockage of CaM-PK with KN62 in conjunction with PB treatment resulted in significant reduction in nuclear HNF-4α immunoreactivity (Fig. 5). Together, these data show that CaM-PK activity is necessary for nuclear induction of HNF-4α by PB.
Inhibition of PKA with the inhibitor RP-8Br-cAMP resulted in a modest increase in total and nuclear HNF-4α protein, which was not significantly changed by PB treatment, indicating that PKA activity negatively regulates endogenous expression of HNF-4α; however, its role in PB induction is not clear.
Discussion
The present study provides insight into molecular events mediating the pleiotropic response of liver to phenobarbital. PB is commonly used in clinical medicine and causes liver enlargement in humans and rodents. PB is also the prototype of liver tumor promoters, dramatically increasing tumor numbers when chronically administered after initial genotoxic carcinogen treatment. Because of the dramatic effects of PB signaling in liver, elucidating the mechanisms involved has been the subject of numerous studies, leading to the discovery of CAR as a major mediator of PB-induced gene expression. Several recent studies using HNF-4α −/− mice and antisense-HNF-4α adenovirus also found that many CAR- and PXR-regulated genes require HNF-4α for efficient induction by PB and other drugs or xenobiotics. However, regulation of HNF-4α in response to PB has not been previously demonstrated. To our knowledge, we are the first to show that increased nuclear expression of HNF-4α is a distinct part of the PB response in liver and a likely mediator of PB-induced gene regulation in concert with CAR.
The present study showed the temporal pattern of induced HNF-4α expression to be similar to but independent of CAR, indicating they may be part of a coordinated mechanism of PB signaling. Specifically, HNF-4α and CAR expression in the nucleus are dramatically increased 3 hours post-PB administration. The kinetics of induction observed are consistent with those of others who observed peak CAR nuclear localization after 3 hours and subsequent PB-induced gene transcription.26 This study also has shown that HNF-4α protein induction occurs in both mouse and rat hepatocytes and that it is time- and dose dependent, indicating HNF-4α induction by PB is a specific and direct result of PB signaling. Preliminary findings with human hepatocytes also indicate that PB induces HNF-4α protein expression, although only a modest 1.5-fold induction was observed (A. Bell, unpublished observation). Furthermore, HNF-4α induction by PB is also independent of PXR because it occurred in the CAR−/−/PXR−/− mice and PXR nuclear expression was not significantly affected after 3 hours in wild-type animals treated with PB. These findings concur with those of Squires et. al., who showed that PXR nuclear translocation is not significant in PB-treated mice after 6 hours of treatment.29 However, we did see minor increases in PXR nuclear expression after 4 hours of treatment (Fig. 1A), possibly a result of increased HNF-4α, which has been shown to regulate expression of PXR in fetal hepatocytes, and an HNF-4α-binding site has been identified in the PXR promoter.30 In addition, HNF-4α induction by PB in CAR−/−/PXR−/− mice was diminished, although a statistically significant nearly twofold induction does occur in these animals. These findings suggest that PXR may augment PB induction of HNF-4α expression and vice versa, but neither PXR nor CAR is a necessary component of HNF-4α induction by PB.
The increase in HNF-4α protein expression is regulated, at least in part, at the mRNA level. We showed that in vivo, PB modestly induced HNF-4α mRNA expression 1.7-fold after 2 hours of treatment in wild-type and CAR−/− mice, which preceded the induction of significant protein expression observed after 3 hours and beyond. This indicates a likely increase in HNF-4α transcription; however, we cannot rule out mRNA stabilization accounting for the increase. Ueda et. al. showed through microarray analysis using CAR−/− mice that PB induces expression of some genes independently of CAR, although HNF-4α was not reported as such.31 Our results suggest that one possible explanation for this is that HNF-4α mRNA induction is time sensitive and occurs within 2 hours of treatment, then falls to slightly elevated levels. The microarray analysis was done after 12 hours of PB treatment, a time when the HNF-4α mRNA increase would not have been seen. It is possible that some of the CAR-independent gene induction seen in the microarray analysis may be mediated in part by HNF-4α. Nevertheless, our observed mRNA increase is consistent with the subsequent increase in protein expression observed.
The mRNA increase was modest and may not account for the threefold increase in protein expression; therefore, posttranscriptional modifications that stabilize HNF-4α protein expression may be involved. HNF-4α is known to be phosphorylated, and its DNA-binding and transactivation potential are tightly governed by its state of phosphorylation and acetylation.32–34 However, it is not known whether these modifications actually stabilize nuclear levels of HNF-4α protein. In the present study we found that inhibition of cellular phosphatases with okadaic acid greatly diminished endogenous and PB-induced nuclear levels of HNF-4α protein. Clues to the identity of the phosphatase activity governing HNF-4α nuclear localization and PB induction may come from investigations of CAR activation. Interestingly, PB signaling results in recruitment of protein phosphatase 2A to the CAR-Hsp90 complex in the cytoplasm leading to CAR nuclear translocation, which can be blocked by okadaic acid.35 It is likely that PP2A or another phosphatase is required for PB induction of HNF-4α expression because okadaic acid blocks the increase in total HNF-4α protein.
HNF-4α induction by PB is likely regulated by kinases as well. One phosphorylation pathway implicated in PB signaling involves the CaM-PK. Several studies have shown that inhibition of CaM-PK resulted in inhibition of PB-induced CYP expression.27,28 Yamamoto et. al. also showed that inhibition of CaM-PK with KN-62 can block TCPOBOP-induced CYP expression, but CAR nuclear translocation still occurs. Thus, other factors or events requiring CaM-PK activity are necessary for induced CYP expression by PB-like inducers. Our results show that inhibition of CaM-PK with KN62 blocks PB induction of HNF-4α nuclear expression. Therefore, CaM-PK activity is required for PB-induced HNF-4α nuclear expression, which may play a complementary role in induction of CYP gene expression.
The PKA phosphorylation pathway has also been implicated in PB signaling. Our results demonstrate that PKA activity negatively regulates endogenous HNF-4α nuclear expression. With regard to PB signaling, our findings also indicate that inhibition of PKA results in only minor inhibition of PB-induced HNF-4α expression and nuclear accumulation. Interestingly, the DNA-binding domain of HNF-4α is a direct target of PKA serine-phosphorylation, and the phosphorylation of this domain results in decreased HNF-4α DNA-binding affinity.36 PKA activation is known to block CYP3A induction by PB, whereas PKA inhibitors enhance induction.27 It is reasonable to think that increased CAR in the nucleus and increased HNF-4α DNA-binding affinity resulting from decreased PKA activity leads to coordinated induction of CYP 3A and other target genes.
Although we show no direct evidence of induction of a CAR- and HNF4-dependent target gene in response to PB, the circumstantial evidence of increased nuclear HNF-4α accumulation coincident with CAR and of similarities in regulation by phosphatase and kinase pathways is compelling. This is further supported by findings that HNF-4α is required for full inducibility of CAR- and PXR-regulated genes, many of which have HNF-4α-binding sites like CYP2C9 and CYP3A4.19–22 In conclusion, it is apparent that regulation of HNF-4α protein expression by PB is a distinct part of the PB response that occurs via a complex phosphorylation-sensitive mechanism.
We propose that induction of HNF-4α is part of a coordinated response to PB-like inducers and possibly other xenobiotics, acting as a key regulator of hepatic gene expression separately or in concert with CAR and PXR. It is likely that hepatocytes maintain CAR-independent mechanisms of gene regulation to confer adaptive responses to PB and other xenobiotics. Identifying CAR-independent mechanisms by which PB and PB-like molecules regulate gene expression will strengthen our understanding of how hepatocytes and other cell types respond to such agents and give insight into the fundamental mechanism mediating the dramatic whole-tissue response of liver to PB.
Acknowledgments
The authors thank Dr. Wen Xie (University of Pittsburgh) for the CAR- and PXR-null mice and Dr. Frances Sladek (University of California) for the HNF-4α antibody. We also thank William Bowen for technical support and Zahida Khan for critical review of this manuscript.
Footnotes
Potential conflict of interest: Nothing to report.
Supported by NIH grants CA35373 and CA103958 (to G.K.M.).
References
- 1.Orrenius S, Ericsson JL, Ernster L. Phenobarbital-induced synthesis of the microsomal drug-metabolizing enzyme system and its relationship to the proliferation of endoplasmic membranes. A morphological and biochemical study. J Cell Biol. 1965;25:627–639. doi: 10.1083/jcb.25.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez FJ. The molecular biology of cytochrome P450s. Pharmacol Rev. 1988;40:243–288. [PubMed] [Google Scholar]
- 3.Garcia-Allan C, Lord PG, Loughlin JM, Orton TC, Sidaway JE. Identification of phenobarbitone-modulated genes in mouse liver by differential display. J Biochem Mol Toxicol. 2000;14:65–72. doi: 10.1002/(sici)1099-0461(2000)14:2<65::aid-jbt1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 4.Kolaja KL, Stevenson DE, Johnson JT, Walborg EF, Jr, Klaunig JE. Subchronic effects of dieldrin and phenobarbital on hepatic DNA synthesis in mice and rats. Fundam Appl Toxicol. 1996;29:219–228. doi: 10.1006/faat.1996.0025. [DOI] [PubMed] [Google Scholar]
- 5.Carthew P, Edwards RE, Nolan BM. The quantitative distinction of hyperplasia from hypertrophy in hepatomegaly induced in the rat liver by phenobarbital. Toxicol Sci. 1998;44:46–51. doi: 10.1006/toxs.1998.2473. [DOI] [PubMed] [Google Scholar]
- 6.Bursch W, Lauer B, Timmermann-Trosiener I, Barthel G, Schuppler J, Schulte-Hermann R. Controlled death (apoptosis) of normal and putative preneoplastic cells in rat liver following withdrawal of tumor promoters. Carcinogenesis. 1984;5:453–458. doi: 10.1093/carcin/5.4.453. [DOI] [PubMed] [Google Scholar]
- 7.Aletti MG, Presta M, Ragnotti G. Liver growth after partial hepatectomy: influence of phenobarbital administration. Exp Mol Pathol. 1981;34:216–225. doi: 10.1016/0014-4800(81)90078-2. [DOI] [PubMed] [Google Scholar]
- 8.Burki K, Schindler R, Pfenninger M. Studies on liver regeneration. II. Effects of phenobarbital on the onset and pattern of rat liver regeneration following partial hepatectomy. Cell Tissue Kinet. 1971;4:529–537. doi: 10.1111/j.1365-2184.1971.tb01560.x. [DOI] [PubMed] [Google Scholar]
- 9.Jirtle RL, Meyer SA. Liver tumor promotion: effect of phenobarbital on EGF and protein kinase C signal transduction and transforming growth factor-beta 1 expression. Dig Dis Sci. 1991;36:659–668. doi: 10.1007/BF01297035. [DOI] [PubMed] [Google Scholar]
- 10.Reisenbichler H, Chari RS, Boyer IJ, Jirtle RL. Transforming growth factor-beta receptors type I, II and III in phenobarbital-promoted rat liver tumors. Carcinogenesis. 1994;15:2763–2767. doi: 10.1093/carcin/15.12.2763. [DOI] [PubMed] [Google Scholar]
- 11.Mansbach JM, Mills JJ, Boyer IJ, De Souza AT, Hankins GR, Jirtle RL. Phenobarbital selectively promotes initiated cells with reduced TGF beta receptor levels. Carcinogenesis. 1996;17:171–174. doi: 10.1093/carcin/17.1.171. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki M, Mars WM, Runge D, Kim TH, Bowen WC, Michalopoulos GK. Phenobarbital suppresses growth and accelerates restoration of differentiation markers of primary culture rat hepatocytes in the chemically defined hepatocyte growth medium containing hepatocyte growth factor and epidermal growth factor. Exp Cell Res. 1998;241:445–457. doi: 10.1006/excr.1998.4085. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki M, Mars WM, Michalopoulos GK, Namba M. Dose-dependent biphasic effects of phenobarbital on growth and differentiation of primary culture rat hepatocytes. J Gastroenterol Hepatol. 1998;13 (Suppl):S78–S82. doi: 10.1111/jgh.1998.13.s1.78. [DOI] [PubMed] [Google Scholar]
- 14.Okey AB. Enzyme induction in the cytochrome P-450 system. Pharmacol Ther. 1990;45:241–298. doi: 10.1016/0163-7258(90)90030-6. [DOI] [PubMed] [Google Scholar]
- 15.Omiecinski CJ, Remmel RP, Hosagrahara VP. Concise review of the cytochrome P450s and their roles in toxicology. Toxicol Sci. 1999;48:151–156. doi: 10.1093/toxsci/48.2.151. [DOI] [PubMed] [Google Scholar]
- 16.Zelko I, Negishi M. Phenobarbital-elicited activation of nuclear receptor CAR in induction of cytochrome P450 genes. Biochem Biophys Res Commun. 2000;277:1–6. doi: 10.1006/bbrc.2000.3557. [DOI] [PubMed] [Google Scholar]
- 17.Swales K, Negishi M. CAR, driving into the future. Mol Endocrinol. 2004;18:1589–1598. doi: 10.1210/me.2003-0397. [DOI] [PubMed] [Google Scholar]
- 18.Jover R, Bort R, Gomez-Lechon MJ, Castell JV. Cytochrome P450 regulation by hepatocyte nuclear factor 4 in human hepatocytes: a study using adenovirus-mediated antisense targeting. Hepatology. 2001;33:668–675. doi: 10.1053/jhep.2001.22176. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson SS, Chen Y, LeCluyse EL, Negishi M, Goldstein JA. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol Pharmacol. 2005;68:747–757. doi: 10.1124/mol.105.013169. [DOI] [PubMed] [Google Scholar]
- 20.Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, Parviz F, et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med. 2003;9:220–224. doi: 10.1038/nm815. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Kissling G, Negishi M, Goldstein JA. The Nuclear Receptors Constitutive Androstane Receptor and Pregnane X Receptor Cross-Talk with Hepatic Nuclear Factor 4{alpha} to Synergistically Activate the Human CYP2C9 Promoter. J Pharmacol Exp Ther. 2005;314:1125–1133. doi: 10.1124/jpet.105.087072. [DOI] [PubMed] [Google Scholar]
- 22.Barbier O, Girard H, Inoue Y, Duez H, Villeneuve L, Kamiya A, et al. Hepatic expression of the UGT1A9 gene is governed by hepatocyte nuclear factor 4alpha. Mol Pharmacol. 2005;67:241–249. doi: 10.1124/mol.104.003863. [DOI] [PubMed] [Google Scholar]
- 23.Kost DP, Michalopoulos GK. Effect of 2% dimethyl sulfoxide on the mitogenic properties of epidermal growth factor and hepatocyte growth factor in primary hepatocyte culture. J Cell Physiol. 1991;147:274–280. doi: 10.1002/jcp.1041470212. [DOI] [PubMed] [Google Scholar]
- 24.Block GD, Locker J, Bowen WC, Petersen BE, Katyal S, Strom SC, et al. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol. 1996;132:1133–1149. doi: 10.1083/jcb.132.6.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L, Melenhorst JJ, Hennighausen L. Loss of interleukin 6 results in delayed mammary gland involution: a possible role for mitogen-activated protein kinase and not signal transducer and activator of transcription 3. Mol Endocrinol. 2002;16:2902–2912. doi: 10.1210/me.2001-0330. [DOI] [PubMed] [Google Scholar]
- 26.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galisteo M, Marc N, Fautrel A, Guillouzo A, Corcos L, Lagadic-Gossmann D. Involvement of cyclic nucleotide- and calcium-regulated pathways in phenobarbital-induced cytochrome P-450 3A expression in mouse primary hepatocytes. J Pharmacol Exp Ther. 1999;290:1270–1277. [PubMed] [Google Scholar]
- 28.Joannard F, Galisteo M, Corcos L, Guillouzo A, Lagadic-Gossmann D. Regulation of phenobarbital-induction of CYP2B and CYP3A genes in rat cultured hepatocytes: involvement of several serine/threonine protein kinases and phosphatases. Cell Biol Toxicol. 2000;16:325–337. doi: 10.1023/a:1026702615125. [DOI] [PubMed] [Google Scholar]
- 29.Squires EJ, Sueyoshi T, Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J Biol Chem. 2004;279:49307–49314. doi: 10.1074/jbc.M407281200. [DOI] [PubMed] [Google Scholar]
- 30.Kamiya A, Inoue Y, Gonzalez FJ. Role of the hepatocyte nuclear factor 4alpha in control of the pregnane X receptor during fetal liver development. Hepatology. 2003;37:1375–1384. doi: 10.1053/jhep.2003.50212. [DOI] [PubMed] [Google Scholar]
- 31.Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, et al. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Jiang G, Nepomuceno L, Yang Q, Sladek FM. Serine/threonine phosphorylation of orphan receptor hepatocyte nuclear factor 4. Arch Biochem Biophys. 1997;340:1–9. doi: 10.1006/abbi.1997.9914. [DOI] [PubMed] [Google Scholar]
- 33.Guo H, Wei J, Inoue Y, Gonzalez FJ, Kuo PC. Serine/threonine phosphorylation regulates HNF-4alpha-dependent redox-mediated iNOS expression in hepatocytes. Am J Physiol Cell Physiol. 2003;284:C1090–C1099. doi: 10.1152/ajpcell.00394.2002. [DOI] [PubMed] [Google Scholar]
- 34.Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell. 2000;5:745–751. doi: 10.1016/s1097-2765(00)80253-1. [DOI] [PubMed] [Google Scholar]
- 35.Yoshinari K, Kobayashi K, Moore R, Kawamoto T, Negishi M. Identification of the nuclear receptor CAR:HSP90 complex in mouse liver and recruitment of protein phosphatase 2A in response to phenobarbital. FEBS Lett. 2003;548:17–20. doi: 10.1016/s0014-5793(03)00720-8. [DOI] [PubMed] [Google Scholar]
- 36.Viollet B, Kahn A, Raymondjean M. Protein kinase A-dependent phosphorylation modulates DNA-binding activity of hepatocyte nuclear factor 4. Mol Cell Biol. 1997;17:4208–4219. doi: 10.1128/mcb.17.8.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]