

Abstract
The Evi5 oncogene has recently been shown to regulate the stability and accumulation of critical G1 cell cycle factors including Emi1, an inhibitor of the anaphase-promoting complex/cyclosome, and cyclin A. Sequence analysis of the amino terminus of Evi5 reveals a Tre-2, Bub2, Cdc16 domain, which has been shown to be a binding partner and GTPase-activating protein domain for the Rab family of small Ras-like GTPases. Here we describe the identification of Evi5 as a candidate binding protein for Rab11, a GTPase that regulates intracellular transport and has specific roles in endosome recycling and cytokinesis. By yeast two-hybrid analysis, immunoprecipitation, and Biacore analysis, we demonstrate that Evi5 binds Rab11a and Rab11b in a GTP-dependent manner. However, Evi5 displays no activation of Rab11 GTPase activity in vitro. Evi5 colocalizes with Rab11 in vivo, and overexpression of Rab11 perturbs the localization of Evi5, redistributing it into Rab11-positive recycling endosomes. Interestingly, in vitro binding studies show that Rab11 effector proteins including FIP3 compete with Evi5 for binding to Rab11, suggesting a partitioning between Rab11–Evi5 and Rab11 effector complexes. Indeed, ablation of Evi5 by RNA interference causes a mislocalization of FIP3 at the abscission site during cytokinesis. These data demonstrate that Evi5 is a Rab11 binding protein and that Evi5 may cooperate with Rab11 to coordinate vesicular trafficking, cytokinesis, and cell cycle control independent of GTPase-activating protein function.
Keywords: cytokinesis, GTPase-activating protein, recycling endosome
Various forms of cellular signaling have been linked to the control of the cell division cycle. Notably, cyclin accumulation and its destruction in mitosis by ubiquitin-dependent proteolysis is a central mechanism controlling cell division (1). Cyclin destruction in mitosis is directed by the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase that targets a number of critical substrates with cell cycle functions (2). During S and G2 phases, the APC/C must be kept inactive by its pseudosubstrate inhibitor Emi1 (3). As cells progress through G1 into S phase, E2F-dependent transcription of Emi1 mRNA in late G1 results in accumulation of Emi1 protein, thereby inhibiting the APC/C and allowing reaccumulation of APC/C substrates including cyclin A, which drives S phase entry (4). Later in mitosis, APC/C activation requires Emi1 destruction (5–8). Recent work showed that Evi5 binds and stabilizes Emi1 and functions by blocking both its phosphorylation by the Polo-like kinase Plk and its association with the SCFβTrCP ubiquitin ligase (9). Accumulation of Evi5 occurs in early G1, 4 h after mitotic exit, whereas the accumulation of Emi1 and cyclin A occurs over the next several hours leading up to the G1–S transition. The details of how Evi5 ultimately contributes to Emi1 accumulation may represent a previously undescribed link from mitotic exit to the eventual passage to S phase.
The Rab GTPases make up the largest subfamily of monomeric Ras-like GTPases, with >60 members in the human genome, and have been shown to regulate multiple aspects of vesicle transport, budding, and fusion through the binding of specific effector proteins (10–13). Like other GTPases, Rabs cycle between an inactive (GDP-bound) and an active (GTP-bound) state capable of binding effector proteins. Rabs generally have a low intrinsic rate of GTP hydrolysis, requiring GTPase-activating proteins (GAPs) to stimulate GTP hydrolysis as well as guanine nucleotide exchange factors to catalyze GDP release and reloading of GTP. A variety of factors regulate targeting of active Rabs to specific membranes in the cell, including interactions with Rab escort protein, guanine-nucleotide dissociation inhibitor (GDI), and GDI displacement factors (14). Several aspects of Rab-RabGAP function remain poorly understood: notably, differences in the intrinsic GTPase activity of various Rabs, the extent of GTPase stimulation by specific GAPs and their effect on Rab membrane association, and the role of a diverse family of Rab effector proteins. Several laboratories have identified candidate GAPs for mammalian Rabs, including Rab3, Rab5, Rab6, and Rab7 (15–18).
Sequence analysis of the Evi5 protein revealed a Tre-2, Bub2, Cdc16 (TBC) domain, which has been generally shown to act as a specific binding domain and GAP domain for the Rab family of small Ras-like GTPases (16, 17, 19–21). A homologous protein, GAPCenA, possesses a similar domain structure and has been shown to be a centrosomally localized GAP for Rab6 (16). Like GAPCenA, Evi5 localizes to the centrosome (9, 22). However, a critical role for the Evi5 GAP domain, the identity of a Rab capable of binding Evi5, and how Rab function might link to the cell cycle, remained unknown.
In an effort to identify a Rab protein that binds Evi5, we screened a panel of Rab GTPases for their ability to bind Evi5 and demonstrated that Evi5 binds specifically to Rab11, which is localized to vesicles in the pericentriolar region of cells and is required for endocytic recycling of cell surface proteins, including transferrin (23, 24), IgA (25), and CXCR2 chemokine receptors (26). Both Rab11a and Rab11b bind Evi5 in a GTP-dependent manner, but no stimulation of Rab11 GTPase activity was detected. Indeed, Rab11 colocalizes with Evi5 and redistributes Evi5 when overexpressed. Evi5 ablation by RNA interference causes cytoplasmic defects similar to those seen after Rab11 depletion (27). We hypothesize that association of Evi5 with Rab11 may be important for allowing targeting of Evi5 to the centrosome and that degradation of Evi5 in early mitosis may free Rab11 to participate in late mitotic events, such as cleavage furrow formation.
Results
Identification of a Rab Binding Partner for Evi5.
To identify a potential Evi5–Rab interaction a yeast two-hybrid screen was performed by using the amino-terminal TBC domain of Evi5 [Evi5N (9)] to test interactions against a panel of GTPase-deficient Rab GTPases. Mutation of the conserved glutamine residue required for catalysis to leucine yields a constitutively active Rab that is predicted to stably interact with GAP domains. Likewise, the catalytic arginine within Evi5N′s arginine finger (Arg-208) was mutated to alanine, an accommodating mutation that has recently been proposed to compensate sterically for the introduction of the bulky hydrophobic residue in the Rab (18). Rab11a–Q70L was the sole GTPase observed to interact with Evi5N after quadruple selection [supporting information (SI) Fig. 5]. We next sought to confirm the specific binding seen by yeast two-hybrid through in vitro methods using recombinant proteins expressed and purified from Escherichia coli. A maltase binding protein (MBP)-tagged version of Evi5N bound to GST–Rab11a, but not GST alone, immobilized on glutathione-Sepharose resin (Fig. 1A). Importantly, the Evi5–Rab11a interaction was enhanced in the presence of guanosine 5′-[β,δ-imido] triphosphate (GMP-PNP), a nonhydrolyzable analogue of GTP, a feature characteristic of Rab effector proteins (13).
Fig. 1.

Evi5 binds Rab11a and Rab11b in a GTP-dependent manner. (A) Recombinant GST-tagged Rab11 and MBP-tagged Evi5N proteins bind in vitro. GST or GST–Rab11a was immobilized on glutathione-Sepharose and then tested for its ability to bind MBP–Evi5N in the presence of either GDP or GMP-PNP (nonhydrolyzable GTP analog). Captured proteins were detected by immunoblotting with antibodies against Evi5 and GST. (B) Evi5 specifically binds to the active forms of Rab11a and Rab11b by surface plasmon resonance. MBP–Evi5N was immobilized on a Biacore chip and tested for binding to various Rab and ARF GTPases. GTPases were preloaded with either GDP or GMP-PNP to assay the nucleotide dependency of the interaction. (C) Recombinant Evi5 binds activated Rab11 expressed in mammalian cells. GST–Rab fusion proteins were transiently expressed in 293T cells and captured on glutathione-Sepharose beads before addition of MBP–Evi5N. Bead-bound proteins were detected by immunoblotting with antibodies against Evi5 and GST as in A. (D) Evi5 does not display significant GAP activity toward Rab11 in vitro. Bacterially expressed GST–Rab proteins were preloaded with [α-32P]GTP and incubated with MBP or MBP–Evi5N protein at 30°C. Rab GTPase activity is expressed as the percentage of total nucleotide converted to GDP after incubation with Evi5 or control protein.
To further test the specificity and GTP dependence of the Evi5–Rab11 interaction, we next sought to study the binding between recombinant Rab11 and Evi5 more rigorously using surface plasmon resonance analysis. Bacterially expressed GTPases were subjected to an exchange reaction to preload them with either GDP or GMP-PNP and tested for interaction with MBP–Evi5N immobilized on a Biacore chip (Fig. 1B). Again Rab11a–GMP–PNP bound Evi5 efficiently, with a dissociation constant of 55 nM. Rab11a–GDP showed a >10-fold weaker binding to Evi5 (650 nM), arguing that in vivo association of Evi5 with Rab11 is likely to be GTP-dependent. Rab11b, which shares 90% sequence identity with Rab11a, displayed similar Evi5 binding characteristics as Rab11a, with a dissociation constant of 50 nM. As a control for binding specificity, Evi5 was shown to be unable to bind GMP-PNP-loaded versions of Rab1 and Rab3b, as well as ARF6, another GTPase implicated in vesicular trafficking (28).
Because these results suggested that Evi5 has greater binding affinity for the GTP-bound form of recombinant Rab11 than the GDP-bound form, we next asked whether Evi5 showed similar GTP-dependent binding specificity for Rab11 expressed in mammalian cells. Expression in eukaryotic cells allows prenylation of the Rab11 protein, and previous studies have shown that geranyl-geranylation of Rab GTPases at their carboxyl termini may be important for the ability of RabGAPs to stimulate Rab GTPase activity (16). Consistent with the above data, we found that Evi5N bound both wild-type Rab11a and Rab11a–Q70L (Fig. 1C Left). Conversely, Rab11a–S25N, a dominant negative version of Rab11a that is preferentially GDP-bound, showed no binding to Evi5 over that seen with the negative control, suggesting that prior loading of Rab11 with GTP is required for association of Evi5 with Rab11 in vivo. Although Evi5 shares significant sequence homology to the Evi5 family member and Rab6–GAP GAPCenA (25% amino acid similarity), Evi5 was unable to bind Rab6b (Fig. 1C and SI Fig. 5). These data show that the high degree of in vivo binding specificity of Rab GTPases for their partner proteins is maintained in vitro. The third member of the Rab11 subfamily of GTPases, Rab25, is expressed exclusively in epithelial tissues (29, 30), where it is proposed to regulate vesicle trafficking events specific to epithelial cells. Rab25 shares binding affinity with a subset of the family of Rab11-intereacting proteins (FIPs), which collectively mediate a variety of Rab11 functions, including regulation of vesicle trafficking, protein sorting, and cytokinesis (10, 13). Evi5, however, showed no binding affinity for immobilized GST–Rab25 expressed and purified from mammalian cells (Fig. 1C Right), again demonstrating the specificity of the Evi5–Rab11 interaction.
Because Evi5 contains a conserved TBC domain (SI Fig. 6) and binds Rab11 in a GTP-specific manner, we next asked whether we could detect stimulation of Rab11 GTPase activity by Evi5 in vitro. Bacterially expressed, GST-tagged Rab11, as well as various control Rabs, were loaded with GTP and incubated with MBP or MBP–Evi5N, and GTP hydrolysis was determined by measuring either [α-32P]GTP conversion to GDP via thin-layer chromatography or inorganic phosphate release by indirect fluorescence detection. In both cases Evi5 caused no significant increase in intrinsic Rab11 GTPase activity, even at late time points (up to 2 h) and using high concentrations of Evi5 (Fig. 1D and SI Fig. 7).
In Vivo Colocalization and Interaction of Rab11 with Evi5.
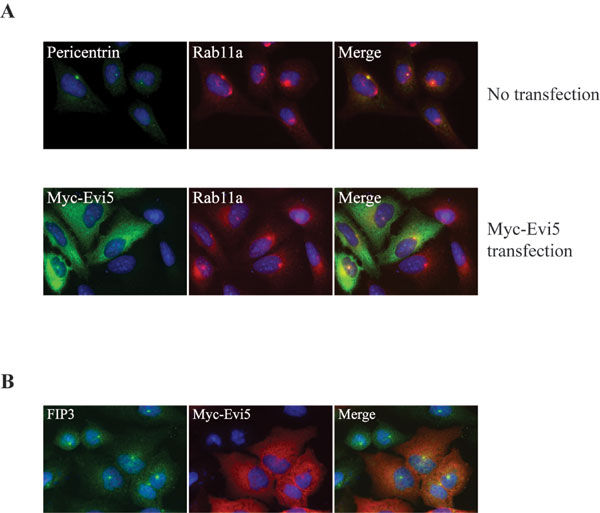
Recent studies have shown that Evi5 protein localizes to the cytoplasm, with focal staining at the centrosome (9, 22). Because Rab11a localizes to the pericentriolar recycling endosome compartment (27, 31), we next asked whether endogenous Evi5 and Rab11a colocalize in cells. In agreement with prior reports, we found endogenous Rab11a strongly localized in a punctate pattern surrounding the centrosome, with more diffuse staining in puncta throughout the cytoplasm (27, 31). Costaining with antibodies specific for Evi5 and Rab11a confirmed that these proteins colocalize in the pericentriolar region of U2OS cells (Fig. 2A). The absence of a suitable antibody specific for Rab11b precluded attempts to colocalize Evi5 with Rab11b. In addition, Evi5 was immunoprecipitated from these cells by a polyclonal antibody that specifically recognizes Rab11a (Fig. 2B), demonstrating that a pool of Evi5 and Rab11a exists in a complex in vivo.
Fig. 2.

Evi5 and Rab11 bind in vivo. (A) Endogenous Evi5 and Rab11a proteins colocalize in the pericentriolar region of the cell. U2OS cells were processed for immunofluorescence analysis by using affinity-purified anti-Evi5 and anti-Rab11 antibodies, as well as Hoechst to mark DNA. (B) Endogenous Evi5 and Rab11a proteins form a complex in vivo. Equal amounts of 293T cell lysate were subjected to immunoprecipitation by using either rabbit IgG or antibody against Rab11a. Captured immune complexes were eluted with sample buffer and subjected to SDS/PAGE and immunoblotting with anti-Evi5 antibody.
Activated Forms of Rab11 Target Evi5 to Vesicular Compartments.
To determine whether the interaction between Rab11 and Evi5 is functionally relevant in vivo, we next asked whether expression of Rab11 caused a redistribution of Evi5. As previously reported, transiently expressed Evi5 shows centrosome localization, as well as some diffuse cytoplasmic staining, especially when conditions are not optimized to maintain centrosome localization (SI Fig. 8A) (9, 22). Whereas endogenous Rab11 shows localization predominantly to pericentriolar recycling endosomes, overexpressed GFP–Rab11a and GFP–Rab11b have been shown to localize to both recycling endosomes and additional cytoplasmic puncta indicative of vesicular localization (SI Fig. 8A) (27, –33). Rab11a–Q70L localizes similar to wild-type Rab11a, whereas the S25N mutant causes dispersal of the vesicles (SI Fig. 8A) (34, 35). Consistent with our in vitro binding data, Evi5 partially colocalized with Rab11a-positive vesicles (Fig. 3 and SI Fig. 8B), suggesting that Rab11 may be responsible in part for targeting Evi5 within the cell. The inactive Rab11a mutant (S25N) did not affect Evi5 localization, consistent with its inability to bind Evi5 in vitro. Expression of Rab11b, which binds Evi5 in vitro (Fig. 1B), redistributed Evi5 to vesicles, whereas overexpression of Rab5, which localizes to the early endosome compartment of the cell (SI Fig. 8A) (36), was completely unable to alter the localization of Evi5-positive structures, further demonstrating the specificity of the Rab11 effect (SI Fig. 8B). In the reciprocal experiment we found that overexpression of Evi5 does not significantly alter the targeting of endogenous Rab11 to recycling endosomes (SI Fig. 9A), nor does it affect the localization of Rab11 effectors that bind Rab11–GTP, including FIP3, RCP/FIP1, and RIP11/FIP5 (SI Fig. 9B and data not shown). The ability of Rab11 to target Evi5 to specific populations of recycling endosomes argues for a functional interaction of Rab11 with Evi5.
Fig. 3.

Expressed wild type and constitutively active Rab11 recruits endogenous Evi5 to Rab11-positive endosomes. U2OS cells were transfected with GFP–Rab11 wild type or inactive S25N mutant, fixed with 4% PFA, and stained with anti-Evi5 antibody as well as Hoechst to mark the DNA. White arrows highlight sample regions of colocalization, which appear yellow in the merged panel.
FIP3 Competes with Evi5 for Binding to Rab11.
Examination of recent crystal structures of Rab11 in complex with its effectors FIP2 (37) or FIP3 (38, 39) and Rab33 in complex with the budding yeast RabGAP Gyp1 (21) suggested that the binding surfaces on Rabs for their GAPs and effectors likely overlap (SI Fig. 10). We thus predicted that a Rab11 effector might compete with the TBC domain of Evi5 for interaction with Rab11. As previously demonstrated, Evi5 efficiently bound to Rab11 protein immobilized on beads (Fig. 4A). Addition of increasing amounts of the Rab11 effector FIP3, however, strongly reduced binding of Evi5 to Rab11. To ensure that this effect was not a unique feature of a single effector protein, we also tested the ability of RIP11/FIP5, an effector of Rab11 important in regulation of vesicle trafficking (40), to compete for Rab11 binding. The carboxyl-terminal fragment of RIP11/FIP5, which contains its Rab binding domain and is both necessary and sufficient for its association with Rab11 (41), was able to compete very effectively with Evi5 for binding to Rab11 (SI Fig. 11). A point mutant of RIP11/FIP5 that is unable to bind Rab11 (42) shows no competition with Evi5 for Rab11. We suspect that the enhanced competition seen with the RIP11/FIP5 fragment compared with FIP3 is due to its stronger binding because the dissociation constant of FIP3 for Rab11 binding is comparable to that of the Evi5N protein [≈360 nM (38)], whereas the binding affinity of the RIP11/FIP5 fragment for Rab11 is ≈10-fold stronger (J.R.J., unpublished data). Finally, to demonstrate that this competition is not an artifact arising from our use of the amino-terminal fragment of Evi5, we also found that FIP3 is able to compete with in vitro-translated, full-length Evi5 for binding to Rab11 GMP-PNP (data not shown).
Fig. 4.

Evi5 regulates Rab11 effector function in vitro and in vivo. (A) FIP3 competes with Evi5 for binding to Rab11a. Glutathione-Sepharose beads were incubated with GST–Rab11a GMP-PNP and then washed. The Rab11a beads were then incubated with a fixed amount of MBP–Evi5N protein and an increasing molar ratio of FIP3 protein. After washing the beads, bound proteins were eluted with SDS sample buffer and analyzed by SDS/PAGE followed by silver staining. (B) Evi5 depletion causes mislocalization of FIP3 during late cytokinesis. HeLa cells stably expressing GFP–FIP3 were transfected with control or Evi5 siRNAs for 48 h and then processed for immunofluorescence analysis by using antibodies against MKLP1 to mark the midbody and Hoechst to mark the DNA. In each panel, the midbody of the dividing cell (outlined in white) is magnified and shown in Right for additional clarity.
Ablation of Evi5 Alters FIP3 Localization During Cytokinesis.
Having established that FIP3 and Evi5 compete for the same binding surface on Rab11, we were interested in the functional importance of this competition in vivo. Previous work from Cowell and colleagues (43) suggested that Evi5 was important for the accurate completion of cytokinesis because treatment of cells with siRNAs against Evi5 caused a multinucleation phenotype. Furthermore, FIP3 and Rab11 have also been shown to have a critical role in cytokinesis (27). Upon depleting Evi5 by RNA interference, we found that the localization of FIP3 was perturbed at the abscission structure in ≈100% (10 of 10) of randomly chosen cells undergoing late stages of cytokinesis (Fig. 4B). A substantial fraction of FIP3 localized outside of the midbody in Evi5-depleted cells, whereas control cells displayed midbody localization of all visible FIP3. Notably, we saw asymmetric recruitment of GFP–FIP3 to the midbody, as well as what appeared to be an enlarged midbody by bright-field microscopy analysis (data not shown), consistent with a role for Evi5 in regulation of abscission.
Discussion
The Rab family of Ras-like GTPases is known to be critically important in a variety of vesicle trafficking events, and Rab11 has been specifically implicated in regulation of pericentriolar recycling endosome function. More recently, studies have demonstrated a role for Rab11 and Rab11 effector proteins in the accurate completion of cytokinesis (27, 44). Herein we describe an interaction of Rab11 with the candidate oncogene Evi5, which has been shown to be a critical regulator of the cell cycle through its ability to stabilize Emi1 and cyclin protein in interphase. We show that Evi5 binds Rab11a and Rab11b in a GTP-dependent manner, both in vitro and in vivo, and colocalizes with Rab11 in recycling endosomes. Furthermore, Rab11 expression is capable of redistributing Evi5 by recruiting a substantial fraction of Evi5 to Rab11-positive vesicles. Rab11 effector proteins compete with Evi5 for binding to Rab11, suggesting a possible role for Evi5 in controlling downstream events gated by the Rab11 GTPase. In support of this idea, ablation of Evi5 by RNA interference causes mislocalization of FIP3 at sites of abscission during late stages of cytokinesis.
The TBC domain is the best-known RabGAP domain. We suspect that Evi5's amino-terminal TBC domain is quite likely to be the functional determinant responsible for binding to Rab11 because the domain is conserved throughout candidate Evi5 orthologs (SI Fig. 6), the amino terminus of Evi5 is sufficient for Rab11 binding, and we have identified no other conserved domains, such as Rab binding domains, in this fragment. It was recently reported that Evi5 promotes Rab11 GTPase activity in vitro (45). We have tested Evi5 extensively for GAP activity toward Rab11, however, and thus far we have been unable to demonstrate a significant enhancement of intrinsic Rab11 GTPase activity (Fig. 1D). Although we cannot explain this discrepancy in the in vitro GAP activity assays, we note that overexpression of Evi5 in vivo does not significantly alter the targeting of endogenous Rab11 to pericentriolar recycling endosomes, nor does it affect centrosomal localization of FIP3 (SI Fig. 9B) or localization of other Rab11 effectors that bind Rab11–GTP. These data stand in contrast to results published for RabGAP5, which show that its overexpression strongly disrupts GTP-dependent binding of EEA1 to Rab5, as well as Rab5's localization to early endosomes (18). Thus, we suspect that Evi5's ability to bind Rab11 regulates Rab11 function independent of GTP hydrolysis. Importantly, we have found, as have others, that Rab11 displays a higher amount of intrinsic GTPase activity on its own in comparison to some other Rabs (Fig. 1D) (17), suggesting that perhaps stimulation of Rab11 GTPase activity may not be as important to Rab11 function as tight control of Rab11 localization and/or interaction with specific effector proteins (see below). We cannot exclude the possibility, however, that weak GAP activity toward Rab11 may be sufficient to modulate Rab11 function in vivo, or that we are missing a critical cofactor required for GAP activity in our in vitro assays.
We find that Rab11 overexpression redistributes Evi5 to Rab11-containing endosomes (Fig. 3 and SI Fig. 8B), presumably by virtue of the in vivo association of the two proteins. It is tempting to speculate that Rab11 may be required for the localization of Evi5 to the pericentriolar space as well. Although our previous work has shown that the carboxyl-terminal coiled-coil domain of Evi5 is capable of localizing to the centrosome when overexpressed (9), it is possible that vesicle trafficking, perhaps mediated in part by Rab11, is important for localization of endogenous Evi5 protein to the centrosome in interphase. Indeed, we find that acute nocodazole treatment to disrupt the microtubule cytoskeleton results in a loss of the majority of Evi5 from the centrosome (A. Loktev, A.G.E., and P.K.J., unpublished results), arguing that microtubule-guided vesicles may drive the initial targeting of Evi5 to the pericentriolar space, thereby allowing Evi5's coiled coil to tether Evi5 at this site.
Our data showing that Evi5 competes with Rab11 effectors for binding to Rab11 suggest a model for control of Rab11-directed events by Evi5. Rab11, in conjunction with FIP3 and FIP4, are important for targeting recycling endosomes to the cleavage furrow during cytokinesis, where they provide the physical membrane required for cellularization as well as factors that promote and regulate furrow formation (27, 44). Localization of a pool of Evi5 at interphase centrosomes or recycling endosomes may limit the extent or duration of association of FIP3 with Rab11 during interphase, effectively inhibiting FIP3's activity in promoting cytokinesis. In this model, ubiquitin-dependent degradation of Evi5 in early mitosis, induced by Evi5's phosphorylation by the Polo-like kinase Plk1 (9), would shift the balance of Rab11 complexes away from Rab11–Evi5 (toward Rab11–FIP3), which would in turn help to initiate downstream events that require Rab11 activity, such as cytokinesis. In this manner, Evi5 could play a role in regulating cytokinesis even though the bulk pool of Evi5 is no longer present when cytokinesis actually occurs. Consistent with this model, we note that Evi5-depleted cells show a highly penetrant abscission defect with mislocalized FIP3 (Fig. 4B). Moreover, Cowell and colleagues (43) have reported that Evi5 siRNA treatment results in a multinucleation phenotype, which has been shown to frequently reflect a failed cytokinesis event (46). In addition, we have found that Evi5 ablation causes a substantial increase in centrosome number in HeLa cells (9), which could be due to failure in the previous cytokinesis. It is possible that this phenotype arises from a defect in recycling endosome trafficking, and we are further investigating the involvement of Evi5 in these and other Rab11-dependent trafficking events.
The clearest role for the Evi5 protein is in cell cycle control, in which Evi5 is required to allow the accumulation of the APC/C inhibitor Emi1 in late G1 (9). In the absence of Evi5, Emi1 degradation is precociously activated in a Plk- and βTrCP-dependent manner, resulting in a failure to inactivate the APC/C, which in turn leads to a failure in S phase entry. It is possible that Evi5 and Rab11 cooperate in a pathway upstream of Emi1 accumulation at the G1–S transition, in which Evi5 may regulate the transmission of signals from growth factor receptors or other receptors at the cell surface, through the Rab11-positive recycling endosome compartment, and into the nucleus.
Materials and Methods
Reagents, Plasmids, and Transfections.
Antibodies used were rabbit polyclonal anti-Rab11a (Invitrogen, Carlsbad, CA), mouse monoclonal Rab11a (Genetex, San Antonio, TX), rabbit anti-FIP3 (27), mouse anti-Myc and rabbit anti-GST (both generated at Genentech), and rabbit anti-MKLP1 (M. Glotzer, University of Chicago, Chicago, IL). cDNAs encoding Rab11a, Rab11b, Rab6b, Rab8a, and Rab25 in the pENTR vector, Gateway system donor (pDONR), and destination (pDEST15, pDEST17, pDEST26, pDEST27, and pDEST53) vectors were from Invitrogen. Inserts were subcloned into destination vectors by using Clonase II LR using standard methods. Plasmids containing Evi5 (9) and FIP3 (27) have been previously described.
Cell Culture and Transfections.
U2OS, HeLa, and 293T cells were grown as per American Type Culture Collection guidelines. Plasmid transfections used FuGENE 6 (Roche, Indianapolis, IN), and siRNA transfections used either Lipofectamine 2000 or oligofectamine (Invitrogen), according to the manufacturers' instructions.
Protein Purification.
Evi5 proteins were purified as described (9). Rabs were generated from bacterial systems using standard methods (see SI Materials and Methods). To isolate prenylated Rabs, GST and His6 fusions were expressed in 293T cells and purified as above with the exception of lysis conditions, which were as generally described (47). His-FIP3 was generated by using the baculovirus system (H. Matern, personal communication), and RIP11/FIP5 fragments were purified as described (42).
In Vitro Binding Assays.
Briefly, 25 μg of GST–Rab protein was immobilized on glutathione-Sepharose beads (Sigma, St. Louis, MO), and nucleotide was removed by washing with exchange buffer (1× PBS, pH 7.4/10 mM EDTA/0.1% Triton X-100) and replaced by incubation in fixation buffer (1× PBS, pH 7.4/5 mM MgCl2/0.1% Triton X-100 with 300 μM GDP or GMP-PNP). GST–Rab beads were then incubated with 10 μg of MBP–Evi5N in 500 μl of binding buffer (1× PBS, pH 7.4/5 mM MgCl2/1% Nonidet P-40/300 μM nucleotide) at 4°C for 1 h. Beads were washed with 1× PBS (pH 7.4)/5 mM MgCl2 and eluted with sample buffer.
For competition experiments, glutathione-Sepharose beads were incubated with 1 μg of GST–Rab11a protein (preloaded with GMP-PNP) and washed with 20 mM Hepes (pH 7.4)/100 mM NaCl/0.1% Triton X-100. Beads were incubated with 5 μg of MBP–Evi5N in the presence of 2 mg/ml BSA and either His-FIP3 or RIP11/FIP5 fragments as indicated. Beads were washed extensively and eluted with sample buffer.
Endogenous Coimmunoprecipitation.
293T cells were lysed in 20 mM Hepes (pH 7.4)/150 mM NaCl/5 mM MgCl2/0.1% Triton X-100/1 mM DTT/Complete protease inhibitors (Roche). Lysate (500 μg) was incubated for 1 h with 20 μl of protein A Affiprep beads coupled to either rabbit IgG or affinity-purified Evi5 antibodies. Beads were washed extensively in lysis buffer, and proteins were eluted with sample buffer.
Surface Plasmon Resonance Analysis.
Binding affinities were measured on a BIAcore 3000 as described previously (48) except as follows. Evi5N (5–10 pmol) was coupled to the chip, and sensograms were recorded by injecting Rab solutions (8 nM to 5 μM) in the presence of 0.2 mM GMP-PNP or GDP. Three hundred seconds were allowed for association, and 600 seconds were allowed for dissociation, at a flow rate of 30 μl/min in 50 mM Hepes (pH 7.2)/100 mM NaCl/2.5 mM MgCl2/0.05% Tween 20. Evi5N-coated surfaces were regenerated with 10 μl of 10 mM HCl followed by reequilibration in 50 μl of dissociation buffer. Dissociation constants were derived by fitting the data into a 1:1 Langmuir binding model by using BIAevaluation 3.2 software.
Immunofluorescence.
Cells were grown on polyl-lysine-coated coverslips (BD Biosciences, Bedford, MA), washed with PBS (pH 7.4), and fixed with 4% PFA in PBS according to standard protocols. Cells were either permeabilized with 25 μg/ml digitonin for 10 min at 25°C or maintained throughout staining in 1× PBS (pH 7.4)/0.2% saponin/1% BSA. Coverslips were incubated with primary antibodies in PBS plus 1% BSA, washed, and detected with Alexa Fluor-conjugated secondary antibodies and Hoechst 33342 (Invitrogen). Slides were imaged by using a Zeiss Axiovert 200 microscope running AxioVision 3.1 software.
GAP Assays.
For GTP-hydrolysis determinations, 0.5 μM GST–Rab was loaded with 50 nM [α-32P]GTP in reaction buffer (20 mM Tris, pH 7.5/2 mM EDTA/5 μM ATP/1 mM DTT/10 mM reduced glutathione) for 30 min at 25°C. GTP binding was determined by using the rapid filtration method (49). GAP assays were initiated by addition of 10 mM MgCl2 and either MBP or MBP–Evi5N and incubated at 30°C for various times. Reactions were stopped by addition of stop buffer (0.2% SDS/25 mM EDTA/1 mM GTP/1 mM GDP), followed by heating to 65°C for 2 min. Samples were spotted onto PEI Cellulose F plates (EMD Chemicals, Gibbstown, NJ) and run in a glass chamber equilibrated with 0.75 M NaH2PO4. Plates were exposed to phosphorimager cassettes and scanned on a Typhoon 8600 variable mode imager.
Supplementary Material
Acknowledgments
We are grateful to Hugo Matern (Exelixis, South San Francisco, CA) for full-length His-tagged FIP3 protein, Michael Glotzer for MKLP1 antibody, and the DNA sequencing and oligo synthesis facilities at Genentech for their assistance. We thank Maxence Nachury for helpful advice concerning GTPase assays. The major part of this work was done at Genentech. This work was partly supported by National Institutes of Health Grant R01 GM073023 (to P.K.J.) and National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Grant DK064380 (to R.P.).
Abbreviations
- APC/C
anaphase-promoting complex/cyclosome
- GAP
GTPase-activating protein
- TBC
Tre-2, Bub2, Cdc16
- MBP
maltose-binding protein
- GMP-PNP
guanosine 5′-[B,δ-imido]triphosphate.
Footnotes
Conflict of interest statement: C.J.W., J.R.J., M.P., R.H.S., P.K.J., and A.G.E. are employees of Genentech.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610500104/DC1.
References
- 1.Murray AW. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 2.Harper JW, Burton JL, Solomon MJ. Genes Dev. 2002;16:2179–2206. doi: 10.1101/gad.1013102. [DOI] [PubMed] [Google Scholar]
- 3.Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, Jackson PK. Genes Dev. 2006;20:2410–2420. doi: 10.1101/gad.1454006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu JY, Reimann JD, Sorensen CS, Lukas J, Jackson PK. Nat Cell Biol. 2002;4:358–366. doi: 10.1038/ncb785. [DOI] [PubMed] [Google Scholar]
- 5.Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Cell. 2001;105:645–655. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- 6.Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 7.Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, Jackson PK. Dev Cell. 2003;4:813–826. doi: 10.1016/s1534-5807(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 8.Hansen DV, Loktev AV, Ban KH, Jackson PK. Mol Biol Cell. 2004;15:5623–5634. doi: 10.1091/mbc.E04-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eldridge AG, Loktev AV, Hansen DV, Verschuren EW, Reimann JD, Jackson PK. Cell. 2006;124:367–380. doi: 10.1016/j.cell.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 10.Prekeris R. ScientificWorldJournal. 2003;3:870–880. doi: 10.1100/tsw.2003.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deneka M, Neeft M, van der Sluijs P. Crit Rev Biochem Mol Biol. 2003;38:121–142. doi: 10.1080/713609214. [DOI] [PubMed] [Google Scholar]
- 12.Wennerberg K, Rossman KL, Der CJ. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 13.Grosshans BL, Ortiz D, Novick P. Proc Natl Acad Sci USA. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seabra MC, Wasmeier C. Curr Opin Cell Biol. 2004;16:451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 15.Fukui K, Sasaki T, Imazumi K, Matsuura Y, Nakanishi H, Takai Y. J Biol Chem. 1997;272:4655–4658. doi: 10.1074/jbc.272.8.4655. [DOI] [PubMed] [Google Scholar]
- 16.Cuif MH, Possmayer F, Zander H, Bordes N, Jollivet F, Couedel-Courteille A, Janoueix-Lerosey I, Langsley G, Bornens M, Goud B. EMBO J. 1999;18:1772–1782. doi: 10.1093/emboj/18.7.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XM, Walsh B, Mitchell CA, Rowe T. Biochem Biophys Res Commun. 2005;335:154–161. doi: 10.1016/j.bbrc.2005.07.070. [DOI] [PubMed] [Google Scholar]
- 18.Haas AK, Fuchs E, Kopajtich R, Barr FA. Nat Cell Biol. 2005;7:887–893. doi: 10.1038/ncb1290. [DOI] [PubMed] [Google Scholar]
- 19.Albert S, Will E, Gallwitz D. EMBO J. 1999;18:5216–5225. doi: 10.1093/emboj/18.19.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Will E, Gallwitz D. J Biol Chem. 2001;276:12135–12139. doi: 10.1074/jbc.M011451200. [DOI] [PubMed] [Google Scholar]
- 21.Pan X, Eathiraj S, Munson M, Lambright DG. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 22.Faitar SL, Dabbeekeh JT, Ranalli TA, Cowell JK. Genomics. 2005;86:594–605. doi: 10.1016/j.ygeno.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green EG, Ramm E, Riley NM, Spiro DJ, Goldenring JR, Wessling-Resnick M. Biochem Biophys Res Commun. 1997;239:612–616. doi: 10.1006/bbrc.1997.7520. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Kumar R, Navarre J, Casanova JE, Goldenring JR. J Biol Chem. 2000;275:29138–29146. doi: 10.1074/jbc.M004410200. [DOI] [PubMed] [Google Scholar]
- 26.Fan GH, Lapierre LA, Goldenring JR, Sai J, Richmond A. Mol Biol Cell. 2004;15:2456–2469. doi: 10.1091/mbc.E03-09-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson GM, Fielding AB, Simon GC, Yu X, Andrews PD, Hames RS, Frey AM, Peden AA, Gould GW, Prekeris R. Mol Biol Cell. 2005;16:849–860. doi: 10.1091/mbc.E04-10-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Souza-Schorey C, van Donselaar E, Hsu VW, Yang C, Stahl PD, Peters PJ. J Cell Biol. 1998;140:603–616. doi: 10.1083/jcb.140.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldenring JR, Shen KR, Vaughan HD, Modlin IM. J Biol Chem. 1993;268:18419–18422. [PubMed] [Google Scholar]
- 30.Goldenring JR, Aron LM, Lapierre LA, Navarre J, Casanova JE. Methods Enzymol. 2001;329:225–234. doi: 10.1016/s0076-6879(01)29082-x. [DOI] [PubMed] [Google Scholar]
- 31.Horgan CP, Walsh M, Zurawski TH, McCaffrey MW. Biochem Biophys Res Commun. 2004;319:83–94. doi: 10.1016/j.bbrc.2004.04.157. [DOI] [PubMed] [Google Scholar]
- 32.Schlierf B, Fey GH, Hauber J, Hocke GM, Rosorius O. Exp Cell Res. 2000;259:257–265. doi: 10.1006/excr.2000.4947. [DOI] [PubMed] [Google Scholar]
- 33.Lapierre LA, Dorn MC, Zimmerman CF, Navarre J, Burnette JO, Goldenring JR. Exp Cell Res. 2003;290:322–331. doi: 10.1016/s0014-4827(03)00340-9. [DOI] [PubMed] [Google Scholar]
- 34.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. Proc Natl Acad Sci USA. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. J Cell Biol. 2000;151:1207–1220. doi: 10.1083/jcb.151.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- 37.Jagoe WN, Lindsay AJ, Read RJ, McCoy AJ, McCaffrey MW, Khan AR. Structure (London) 2006;14:1273–1283. doi: 10.1016/j.str.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 38.Eathiraj S, Mishra A, Prekeris R, Lambright DG. J Mol Biol. 2006;364:121–135. doi: 10.1016/j.jmb.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 39.Shiba T, Koga H, Shin HW, Kawasaki M, Kato R, Nakayama K, Wakatsuki S. Proc Natl Acad Sci USA. 2006;103:15416–15421. doi: 10.1073/pnas.0605357103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prekeris R, Klumperman J, Scheller RH. Mol Cell. 2000;6:1437–1448. doi: 10.1016/s1097-2765(00)00140-4. [DOI] [PubMed] [Google Scholar]
- 41.Prekeris R, Davies JM, Scheller RH. J Biol Chem. 2001;276:38966–38970. doi: 10.1074/jbc.M106133200. [DOI] [PubMed] [Google Scholar]
- 42.Junutula JR, Schonteich E, Wilson GM, Peden AA, Scheller RH, Prekeris R. J Biol Chem. 2004;279:33430–33437. doi: 10.1074/jbc.M404633200. [DOI] [PubMed] [Google Scholar]
- 43.Faitar SL, Sossey-Alaoui K, Ranalli TA, Cowell JK. Exp Cell Res. 2006;312:2325–2335. doi: 10.1016/j.yexcr.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 44.Fielding AB, Schonteich E, Matheson J, Wilson G, Yu X, Hickson GR, Srivastava S, Baldwin SA, Prekeris R, Gould GW. EMBO J. 2005;24:3389–3399. doi: 10.1038/sj.emboj.7600803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dabbeekeh JT, Faitar SL, Dufresne CP, Cowell JK. Oncogene. 2006 doi: 10.1038/sj.onc.1210081. in press. [DOI] [PubMed] [Google Scholar]
- 46.Shi Q, King RW. Nature. 2005;437:1038–1042. doi: 10.1038/nature03958. [DOI] [PubMed] [Google Scholar]
- 47.Soldati T, Shapiro AD, Pfeffer SR. Methods Enzymol. 1995;257:253–259. doi: 10.1016/s0076-6879(95)57030-6. [DOI] [PubMed] [Google Scholar]
- 48.Jin R, Junutula JR, Matern HT, Ervin KE, Scheller RH, Brunger AT. EMBO J. 2005;24:2064–2074. doi: 10.1038/sj.emboj.7600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walworth NC, Brennwald P, Kabcenell AK, Garrett M, Novick P. Mol Cell Biol. 1992;12:2017–2028. doi: 10.1128/mcb.12.5.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}