

Abstract
We present the first demonstration that fluorescence resonance energy transfer can be used to track the motion of a single molecule undergoing conformational changes. As a model system, the conformational changes of individual three-helix junction RNA molecules induced by the binding of ribosomal protein S15 or Mg2+ ions were studied by changes in single-molecule fluorescence. The transition from an open to a folded configuration was monitored by the change of fluorescence resonance energy transfer between two different dye molecules attached to the ends of two helices in the RNA junction. Averaged behavior of RNA molecules closely resembles that of unlabeled molecules in solution determined by other bulk assays, proving that this approach is viable and suggesting new opportunities for studying protein–nucleic acids interactions. Surprisingly, we observed an anomalously broad distribution of RNA conformations at intermediate ion concentrations that may be attributed to foldability differences among RNA molecules. In addition, an experimental scheme was developed where the real-time response of single molecules can be followed under changing environments. As a demonstration, we repeatedly changed Mg2+ concentration in the buffer while monitoring single RNA molecules and showed that individual RNA molecules can measure the instantaneous Mg2+ concentration with 20-ms time resolution, making it the world’s smallest Mg2+ meter.
The study of individual molecules allows one to look beyond ensemble-averaged properties. For example, one can measure the distributions of physical properties and observe the detailed time trajectories of molecular conformations. The study of single molecules has also led to the discovery that identical DNA molecules under identical conditions follow a multitude of paths as they extend in elongational flows (1). This “molecular individualism” was shown to be a consequence of thermal variations in the initial random polymer coil before extension and of nonequilibrium statistical mechanics (2). It is possible that complex processes such as protein folding and enzyme activity will also show a rich set of kinetic paths that can be fully characterized only at the single-molecule level. Thus, it is important to develop techniques that extend our ability to observe such molecular processes at this level of sensitivity.
Fluorescence resonance energy transfer (FRET) (3, 4) provides a powerful way of observing the behavior of molecules. Donor and acceptor dyes attached to two sites of a biological molecule can be used to measure the distance between the two dyes. Donor fluorescence emission is strongly quenched in a distance-dependent manner by the acceptor, whereas the acceptor emission increases because of the energy transfer. Thus, measurement of a change in fluorescence from the two dyes can be used as an indicator of a change in the conformation of the host molecule. Because the two fluorophores are on different parts of the molecule, intramolecular motion can be measured in the molecular center of mass. The FRET technique has recently been extended to the single-molecule domain (5–8), assisted by rapid developments in single-molecule fluorescence spectroscopy (9–12) and their subsequent application to biological systems (13–16). After its first demonstration on DNA conjugates in dry conditions (5), single-molecule FRET was recently extended to a protein-conjugated system under physiological conditions (7). Although many types of interesting fluctuations in energy transfer were seen, including those arising from photophysical events and orientational and distance changes, their relation to protein-conformational fluctuations was unknown. Although it has been proposed that FRET should reflect such motions (5, 12), its feasibility, especially in the presence of many dye- and surface-related fluctuations, was unknown. FRET was also used to identify subpopulations among diffusing DNA molecules (8), but the very nature of this geometry precluded the possibility of studying the motion of single molecules.
We report here that the shape and motion of specifically immobilized single RNA molecules, induced by protein binding or ion binding, can be measured by using FRET. Furthermore, the observed changes in FRET signal caused by the RNA motion are in quantitative agreement with the predicted conformational change.
We studied the motion of an RNA three-way junction containing the binding site for the ribosomal protein S15 (Fig. 1a). The 30S ribosomal subunit, which consists of a 1,540-nt 16S rRNA and 21 ribosomal proteins, is assembled in a cascade of RNA conformational changes induced by a sequence of protein-binding events (17). Binding of S15 to the 16S rRNA three-way junction nucleates the assembly of the central domain of the 30S ribosomal subunit (18–21) by folding the RNA so that distant sites are brought together, allowing the binding of subsequent proteins in the assembly cascade.
Figure 1.

(a) RNA junction with fluorescein, Cy3 and biotin labeling. Functionally important bases are marked by uppercase letters. (b) Cartoon of RNA conformational change on protein/Mg binding. Interhelix angles are taken from ref. 20. (c) Two-color overlay image of protein-bound and free RNA molecules (128 ×128 pixels, integration time for each pixel ≈5 ms). RNA molecules were preincubated with 3nM S15 protein solution before immobilization. The donor image (500 nm < λ < 540 nm) was pseudo-colored in green and the acceptor image (570 nm < λ < 610 nm) was pseudo-colored in red, and the two images were overlaid. The protein-bound RNA adopts a folded conformation, bringing the two dyes together, leading to significant donor quenching (red spots). The free RNA adopts an open conformation that has strong donor emission (green-yellow spots). Conformational state identification can be made quantitative by calculating the average intensity of two images over a rectangle (≈16 pixels) surrounding each molecule (if the dye photobleached during the scan, only the portion before the photobleaching was used). Donor average intensity and acceptor average intensity of ≈200 molecules from five images including c are represented as a two-dimensional histogram in d in gray scale. Each image is taken from a fresh area to avoid photobleaching caused by repeated exposure to the laser excitation. The peak at low donor intensity is caused by protein-bound RNA molecules and directly corresponds to the red spots in c, and the peak at high donor intensity corresponds to free RNA molecules or green spots in c. Distributions obtained from RNA molecules labeled with donor only and acceptor only are superimposed by using green and red, respectively.
The free RNA junction has been shown to be nearly planar, with ≈120° angles between each pair of helices, by using gel mobility shift and transient electric birefringence experiments (18, 20). Binding of S15 protein or Mg2+ causes Helix 1 to rotate by 60°, becoming collinear with Helix 3 as shown in Fig. 1b (20). A fluorescein dye [donor (D)] was attached at the end of Helix 1 and a Cy3 dye [acceptor (A)] was attached at the end of Helix 2 to detect the resulting distance change. A biotin tag was attached to Helix 3 to bind the RNA junction to a streptavidin-coated surface (Fig. 1 a and b). Three additional RNA junctions were also constructed for control studies, where one of the three tags was omitted.
MATERIALS AND METHODS
RNA Junction Synthesis.
The RNA oligonucleotides (with 5′-fluorescein, 5′-amino, and 5′-biotin modifications for donor, acceptor, and biotin strands, respectively; Fig. 1a) were synthesized by using 5′-o-silyl-2′-o-orthoester protecting groups (Dharmacon Research, Boulder, CO). After deprotection, the RNA oligos were purified by polyacrylamide gel electrophoresis and were eluted from the gel by soaking in 0.5 M NH4OAc, pH 5.2, and 1 mM EDTA. The eluant was loaded onto a SepPak C18 purification cartridge (Waters) and eluted with 50% acetonitrile. After lyophilization, 10–20 nmol of the amine-labeled RNA was resuspended in 500 μl of 300 mM NaHCO3, pH ≈ 8.3. This volume was added to one vial of Cy3 dye (monofunctional succinimidyl ester from Amersham Pharmacia), placed in foil, and incubated for 1 hr at 37°C with brief vortexing every 15 min, overnight at room temperature, and finally for 1 hr at 37°C. Free dyes were removed by using 10 ml Econo-Pac 10 DG columns (6,000 MW exclusion limit, Bio-Rad) equilibrated with 5 mM Na-borate buffer, pH 8. RNAs were all further purified by HPLC on an analytical C18 column (Vydac, Hesperia, CA) by using the solvent system described by Smith et al. (22) to resolve labeled from unlabeled RNA. The eluted fractions were pooled, lyophilized, and resuspended in 10 mM sodium phosphate buffer, pH 7.2. The concentrations of the RNAs were determined after base hydrolysis to circumvent effects of hypochromicity due to base stacking. The three RNA strands comprising the junction were annealed by combining them at 0.5 μM in 10 mM sodium phosphate, pH 7.2, and 100 mM NaCl (2-fold excess of acceptor strand was used). The sample was heated to 95°C for 2 min, then cooled by −1°C every 30 s until the temperature reached 35°C.
RNA Junction Immobilization.
We applied the following incubation–wash cycles to a microscope cover glass: 10-min incubation with 1 mg/ml biotinylated BSA (Sigma) in buffer A (Tris 10 mM, NaCl 50 mM, pH 8) → wash with buffer A → 10-min incubation with 0.2 mg/ml streptavidin (Molecular Probes) in buffer A → wash with buffer A → 10-min incubation with 20pM RNA or RNA/S15 complex → wash with buffer A. The specificity of the immobilization was better than 500:1, as determined by control experiments where the streptavidin step was skipped or nonbiotinylated RNA was used. Buffer A was also used as a base buffer for the experiments. β-Mercaptoethanol (1%) was used for the [Mg2+] dependence study to enhance the photostability of fluorescein.
Inter-Dye Distance and FRET Efficiency.
The acceptor arm is 15 bp long, and the donor arm is 16 bp long. Assuming an angular separation of 120° and 60° between helices 1 and 2 for open and folded RNA junctions, respectively (20), the inter-dye distance changes from 8.5 nm to 5 nm on protein binding. Energy transfer distance R0 was determined to be 5.3 nm by using R0 = (8.79·10−5n−4·φD·J·κ2)1/6 where n is the index of refraction of the medium, φD is the donor quantum yield, J is the spectral overlap of donor emission and acceptor absorption in [nm4M−1cm−1] units, and κ2 is the relative orientation factor of the two dipoles (3). We assumed free and rapid rotation of the dyes around their linkers, which gives κ2 = 2/3 (the polarization anisotropy is 0.17 for fluorescein and 0.22 for Cy3, so this is only an approximation). J and φD were experimentally determined from the RNA junction. Energy transfer efficiency E, the amount of donor quenching caused by the energy transfer, is estimated by using E = 1/(1+R6/R06) and is 4% for the open RNA and 60% for the folded one. The agreement with the single-molecule experiment (donor signal quenched by 70% on RNA folding) is good, considering the fact that there are uncertainties because of κ2 and the length of the dye linkers. Donor average intensity for the RNA molecules was indistinguishable within the experimental uncertainty (10%) from the donor intensity for the RNA molecules with donor only, indicating that RNA spends less than 10% of the time, if at all, in the folded conformation in the absence of the S15 protein or Mg2+.
Experimental Setup.
Individual RNA molecules were excited by 0.5 μW of focused laser light (488 nm, linearly polarized) by using an epiillumination configuration in a home-built confocal scanning microscope. Collected fluorescence emission was split by a dichroic beam splitter (550-nm long pass) onto donor and acceptor detectors (Silicon avalanche photodiodes, EG&G Canada, Vaudreuil, Quebec, Canada). Additional band pass filters (518-nm band pass for donor and 590-nm band pass for acceptor) were used to reduce the cross talk between the two detectors. The acceptor signal is composed of (i) donor fluorescence photons that are detected in the acceptor channel (RNA labeled with donor-only showed 1/3 intensity at the acceptor channel compared with the donor channel); (ii) the direct excitation of the acceptor molecule; and (iii) energy transfer from donor to acceptor. The change in acceptor signal on junction folding is small because the energy transfer increase is accompanied by a decrease in donor leakage. All filters were purchased from Chroma Technology. Throughout the experiment, room temperature was maintained at 18°C.
RESULTS AND DISCUSSION
Fluorescence from the donor and acceptor on the same RNA junction was measured simultaneously by using two detectors and a confocal scanning optical microscope of our own construction. When RNA molecules are preincubated with 3 nM S15 protein solution, we observe two clearly distinguishable populations from such measurements (Fig. 1 c and d). In one population, the folded form of the junction caused by protein binding, the average donor fluorescence is reduced by ≈70%, and the average acceptor fluorescence is increased by 20% compared with the other population, the open conformation of the junction (see the two-dimensional histogram of the average intensity at the donor and acceptor detector in Fig. 1d). The change in energy transfer caused by an RNA conformational change is clearly larger than measurement “noise” because of effects such as dye “blinking” (23, 24), spectral diffusion (25), and rotational dynamics (26). For comparison, the distributions obtained from single labeled (“donor only” or “acceptor only”) RNAs are shown in the same histogram in green and red, respectively. The absence of a donor-only peak and an acceptor-only peak for doubly labeled RNA molecules shows that most of the RNA molecules have both donor and acceptor dyes.
Polarization experiments demonstrated that the dye molecules were not attached directly to the surface but were freely rotating and indirectly immobilized via the RNA junction. On circularly polarized laser excitation of RNA molecules labeled donor only or acceptor only, the emission was split by a broadband polarizing beam splitting cube (CVI Lasers, Albuquerque, NM) and was measured by two detectors. The ratio between the two detector signals was the same among molecules and also as a function of time for a single molecule, indicating that the dyes are free to rotate rapidly in the laboratory frame.
Protein or Mg2+ binding on donor-only and acceptor-only RNA induced less than a 10% change in either donor or acceptor fluorescence channels. Furthermore, the observed donor quenching caused by the energy transfer is consistent with the distance change estimated from the published RNA conformations (20) and dye properties (see Materials and Methods). Therefore, we conclude that the observed fluorescence change from a single RNA molecule is caused by a protein-induced conformational change. The fluorescence change is large enough to allow easy discrimination of the two RNA conformations from the false-color overlay image (Fig. 1c).
Protein-binding kinetics and thermodynamic information can also be obtained from the single-molecule experiments. For this purpose, a 20-pM RNA solution was preincubated with S15 protein at various concentrations before being immobilized on the surface. For the FRET signal from each molecule, we define a proximity factor P = IA/(IA+ID), where IA and ID are the single-molecule fluorescence intensities measured at the acceptor detector and the donor detector, respectively. A larger value of P corresponds to higher proximity between the two dyes, if all other factors are equal. P is related, but not identical, to the energy transfer efficiency. The initial fraction of RNA molecules bound to S15 was determined from a histogram of the single-molecule proximity factors as a function of protein concentration (Fig. 2a). The histogram for the free RNA shows a single peak at P ≈ 0.45, characteristic of the open form of the junction (P is nonzero because of direct excitation of the acceptor and leakage of the donor emission into the acceptor detector spectral range). At intermediate protein concentrations, there are two populations, the open configuration at P ≈ 0.45 and the other at P ≈ 0.75 for the protein-bound junction. The fraction of protein-bound molecules estimated from the relative area under the P ≈ 0.75 peak as a function of protein concentration fits well to a simple bimolecular equilibrium association with a Kd ≈ 3.5 nM. At very high protein concentrations (≥2.5 μM), the donor signal was completely quenched (which was not seen on the donor-only sample), consistent with further folding of the RNA junction, likely because of multiple protein binding. Indeed, earlier studies showed evidence of nonspecific multiple protein binding at protein concentrations above 400 nM (18).
Figure 2.

(a) Initial proximity factor histograms after incubation with S15 protein solution of various concentration (0 M, 1 nM, 3 nM, 10 nM, 200 nM). Data (200 nM) were obtained by incubating preimmobilized RNA with the protein solution. Each histogram is fit with two Gaussian functions to estimate the protein-bound fraction. (b) Time sequence of single-molecule proximity factor histograms for RNA molecules preincubated with 3 nM S15 protein. Protein-bound fraction under P ≈ 0.75 peak decreases in time because of protein dissociation.
The rate of protein dissociation from the RNA can also be measured by monitoring the fraction of protein bound as a function of time. The histogram of proximity factors for a population of molecules in the presence of 3 nM S15 protein is shown in Fig. 2b. The protein-bound fraction decreases with a dissociation rate constant koff ≈ 10−4 s−1. In a bimolecular association reaction, KD = koff/kon, where kon is the association rate constant. Using the measured values for KD and koff, we obtain a value of kon ≈ 3 × 104 M−1 s−1. The values of KD, koff, and kon measured by single-molecule fluorescence are consistent with the values determined from gel electrophoresis mobility assays by using RNAs without the dye labels (18, 21). Thus neither the dye labeling nor the immobilization on the glass substrate has a significant effect on the biological activity of the RNA junction. The two distinct populations of RNA molecules shown in Fig. 2 are a result of the slow dissociation rate of the protein from the RNA, and we do not detect the RNA in transition.
Divalent ions such as Mg2+ and Ca2+ have also been shown to fold the RNA junction in a similar manner to the S15 protein (20, 21). The effect of Mg2+ ions on the RNA junction conformational distribution was investigated as a function of [Mg2+], as shown in Fig. 3. At very low [Mg2+], there is a peak at P ≈ 0.45, indistinguishable from the free RNA. At high [Mg2+], there is a peak at P ≈ 0.75, which is in the same position as the protein-bound peak shown in Fig. 2. We found no evidence of Mg2+ binding kinetics slower than the integration time (5 ms) in our single molecule study. This Mg2+- induced conformational change is consistent with the transient electric birefringence measurements that showed RNA conformation is the same for both S15 and Mg2+ binding (20). In the following analysis, we assume a two-state model for Mg2+ binding as in the case of S15 binding (21).
Figure 3.

Proximity factor histograms of single RNA molecules for [Mg2+] = 1 μM, 100 μM, 170 μM, 300 μM, 1 mM, and 3 mM. Dashed lines are the expected distribution assuming only the dye variations and instrumental noise and do not match the broad distribution observed for intermediate [Mg2+]. Solid lines show the fits with the variability of KD, as described in the text.
In contrast to the results obtained for S15 binding, we observed a gradual shift of the RNA conformational distribution between the two extremes at intermediate [Mg2+] (Fig. 3). The gradual change of the proximity factor as a function of [Mg2+] is likely to be caused by the fast dissociation rate of Mg2+ from the RNA, resulting in time averaging of the rapid motion of the RNA between open and folded states. Conventional stop-flow experiment with millisecond resolution could not resolve the transient response.
The change in the average proximity factor as a function of [Mg2+] gives an equilibrium dissociation constant KD = 240 μM, consistent with the value determined from the gel electrophoresis mobility experiment by using unlabeled RNA. Because koff > 1/(integration time) = 1/(5 ms), a lower limit can be placed on kon at 8 × 105 M−1 s−1.
The width of the RNA conformation distribution is narrow for both extremes of [Mg2+] but is broad and asymmetric for intermediate [Mg2+] (Fig. 3). The contribution of shot noise, caused by finite photon counts from each molecule (≈200), is not significant and should decrease when P deviates from 0.5 in contrast to what was observed here. The individual dye molecules have different fluorescent intensities (measured independently on donor-only and acceptor-only RNA) and can account for the width of both very low and very high [Mg2+]. In principle, inhomogeneities in the distribution of distance and orientation of the two dyes and spectral properties can be maximally amplified near 50% energy transfer efficiency (corresponding to intermediate [Mg2+] here), broadening the width of the proximity factor distribution. However, if the two-state model is correct, RNA never spends time in the half-folded state (≈50% energy transfer), and dye inhomogeneities cannot be responsible for the observed width at intermediate [Mg2+].
We do not know the reason for the broad distribution at intermediate [Mg2+]. One possibility is that some RNA molecules are more foldable than others because of differences in microenvironments or intrinsic molecular properties. For a given [Mg2+], some may spend a longer time in the folded conformation than others and vice versa, resulting in the broadened distribution. We can model this molecular inhomogeneity by assuming that the free energy change ΔG on Mg2+ binding, related to KD by 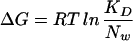 (Nw = molar concentration of water = 55.6 M) is not described by a fixed number but has a distribution of values among molecules. Best agreement with the width and asymmetry of the experimental distribution for all [Mg2+] was achieved for ΔG = −7.2 ± 0.8 kcal/mol or −12.1 ± 1.3 kT per molecule, where k is the Boltzmann constant and T is the absolute temperature in Kelvin (solid line in Fig. 3). In comparison, ignoring the molecular inhomogeneity of ΔG in our model cannot reproduce the data (dashed line in Fig. 3). Another possibility is that the two-state model is not adequate. For example, Mg2+ may induce folding because of a specific ion-binding site in the junction but also because of the screening of the repulsive electrostatic forces between the negatively charged phosphates on the RNA backbone. A deviation from the two-state model, such as presence of many different conformations at intermediate [Mg2+], could broaden the distribution. Future investigation with improved time resolution may clarify the sources of the distribution by directly measuring the Mg2+ binding-dissociation kinetics on a single-molecule level.
(Nw = molar concentration of water = 55.6 M) is not described by a fixed number but has a distribution of values among molecules. Best agreement with the width and asymmetry of the experimental distribution for all [Mg2+] was achieved for ΔG = −7.2 ± 0.8 kcal/mol or −12.1 ± 1.3 kT per molecule, where k is the Boltzmann constant and T is the absolute temperature in Kelvin (solid line in Fig. 3). In comparison, ignoring the molecular inhomogeneity of ΔG in our model cannot reproduce the data (dashed line in Fig. 3). Another possibility is that the two-state model is not adequate. For example, Mg2+ may induce folding because of a specific ion-binding site in the junction but also because of the screening of the repulsive electrostatic forces between the negatively charged phosphates on the RNA backbone. A deviation from the two-state model, such as presence of many different conformations at intermediate [Mg2+], could broaden the distribution. Future investigation with improved time resolution may clarify the sources of the distribution by directly measuring the Mg2+ binding-dissociation kinetics on a single-molecule level.
The conformational dynamics of a single RNA junction can be observed by taking time traces of the donor and acceptor fluorescence signal from individual RNA molecules. Time traces for individual molecules representing the protein-bound RNA, the free RNA, and the RNA at the intermediate [Mg2+], are shown in Fig. 4. In most cases, the relative intensity between the donor and the acceptor signal remains constant within shot noise (Fig. 4 a, c, and d). Some time traces show correlated fluctuations between the two signals (Fig. 4b). Fluctuations such as those shown in (Fig. 4b) are similar to intensity fluctuations seen with donor-only RNA. Correlation with the acceptor channel may arise from donor signal fluorescence in the acceptor channel (“leakage”; see ref. 23) and from energy transfer from the donor molecule. None of the protein-bound RNA showed anticorrelated signal fluctuations, consistent with the assumption of a static configuration formed by protein binding. Likewise, free RNA did not show anticorrelated signal fluctuations so that the spontaneous folding of free RNA junction, if any, must be very brief or infrequent. Anticorrelated signal fluctuations for intermediate [Mg2+] were seen before donor photobleaching for ≈5% of the molecules studied (Fig. 4 e and f). Because the Mg2+ dissociation rate is much faster, this change is not likely caused by the association or dissociation of an individual Mg2+. Because the broadened distribution of P with intermediate [Mg2+] could be attributed to the inhomogeneities in RNA foldability and/or conformations, one may speculate that these RNA properties may also change with time and are responsible for the observed anticorrelated fluctuations.
Figure 4.

Time traces of donor (solid lines) and acceptor (dotted lines) intensities from single RNA molecules. (a and b) Protein-bound RNA; (c and d) free RNA. (e) 1 mM Mg2+; (f) 300 μM Mg2+. Because fluorescein (donor) is much less photostable than Cy3 (acceptor), it photobleaches first most of the time, resulting in a sudden signal drop (marked by arrows). Acceptor signal caused by the direct excitation remains until its own photobleaching (not shown). Data points are taken every 20 ms and three-point averaging is used to reduce noise.
Having established that different conformational states can be seen by using fluorescent indicators, the real-time response of single molecules can now be followed in changing environments. As a demonstration, we exchanged Mg2+ free buffer and 1 mM Mg2+ buffer every 200 ms and watched for switching between open and mostly folded conformations (Fig. 5a). Fluorescence time traces show donor quenching with Mg2+ buffer that is recovered after Mg2+ free buffer is introduced (Fig. 5b). We note that for the slow-flow rate used here, the transition between the open and folded forms is not abrupt. Because the RNA junction responds to Mg2+ concentration change instantaneously within our time resolution, the gradual change seen is likely caused by the finite mixing time of two buffers. A higher flow rate resulted in a faster mixing time (<20 ms; data not shown). Fig. 5c shows the proximity factor averaged over seven periods and clearly shows that the individual RNA molecule can measure varying Mg2+ concentration with 20-ms time resolution.
Figure 5.

Real-time observation of single RNA molecule conformational changes on buffer exchange. (a) Schematics of the experimental apparatus showing how buffer is directly introduced on the area under observation to reduce the mixing time. (b) Time traces (integration time, 5 ms) of donor (solid line) and acceptor signal (dotted line) on buffer exchange. [Mg2+] was alternated between 0 and 1 mM every 200 ms (starting from 0). Significant donor signal reduction is seen every time Mg2+ buffer is present. Vertical grids denote buffer exchange periods (400 ms). Three-point averaging was applied to reduce noise. Donor photobleaching is marked by an arrow. (c) The average proximity factor over seven periods. The nominal arrival times for the buffer without Mg2++ and with 1 mM Mg2++ are at time = 0 and 200 ms. The transition is not abrupt because of the finite mixing time between the two buffers.
In summary, we have demonstrated that single-molecule FRET can be used to unambiguously identify conformational changes of RNA at a single-molecule level. In the case of S15 protein binding to the three-way junction, we observed the exchange of molecules between the folded and the open conformations as a result of the slow protein dissociation. However, for Mg2+ binding where the equilibrium time scale is much faster, a single broad distribution of RNA conformations was observed at intermediate [Mg2+], which could be described by assuming that some RNA are more foldable than others or could be a signature of deviation from a two-state model. Furthermore, by changing the Mg2+ concentration in the buffer, we demonstrated that the response of an individual molecule to a time-dependent external perturbation can be measured in real time. Direct extension of the scheme outlined here could allow one to study the effects of protein binding or enzymatic activity on nucleic acids at the single-molecule level.
Acknowledgments
Financial support for this work was provided by the Air Force Office of Scientific Research and the National Science Foundation (S.C.) and the National Institutes of Health (GM-53757, J.R.W.). X.Z. is a Marvin Chodorow Fellow of the Applied Physics Department of Stanford University. H.D.K. is a Stanford Graduate Fellow. J.W.O. is a Fellow of the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation. We thank Thilo Lacoste for helpful tips in the flow-cell design and Hazen Babcock for experimental contributions and careful proofreading.
ABBREVIATION
- FRET
fluorescence resonance energy transfer
References
- 1.Perkins T T, Smith D E, Chu S. Science. 1997;276:2016–2021. doi: 10.1126/science.276.5321.2016. [DOI] [PubMed] [Google Scholar]
- 2.Smith D E, Chu S. Science. 1998;281:1335–1340. doi: 10.1126/science.281.5381.1335. [DOI] [PubMed] [Google Scholar]
- 3.Selvin P R. Methods Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- 4.Stryer L, Haugland R P. Proc Natl Acad Sci USA. 1967;58:719–730. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ha T, Enderle T, Ogletree D F, Chemla D S, Selvin P R, Weiss S. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schutz G J, Trabesinger W, Schmidt T. Biophys J. 1998;74:2223–2226. doi: 10.1016/S0006-3495(98)77931-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ha T, Ting A Y, Liang J, Caldwell W B, Deniz A A, Chemla D S, Schultz P G, Weiss S. Proc Natl Acad Sci USA. 1999;96:893–898. doi: 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deniz A A, Dahan M, Grunwell J R, Ha T, Faulhaber A E, Chemla D S, Weiss S, Schultz P G. Proc Natl Acad Sci USA. 1999;96:3670–3675. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nie S M, Zare R N. Annu Rev Biophys Biomol Struct. 1997;26:567–596. doi: 10.1146/annurev.biophys.26.1.567. [DOI] [PubMed] [Google Scholar]
- 10.Xie X S, Trautman J K. Annu Rev Phys Chem. 1998;49:441–480. doi: 10.1146/annurev.physchem.49.1.441. [DOI] [PubMed] [Google Scholar]
- 11.Moerner W E, Orrit M. Science. 1999;283:1670–1676. doi: 10.1126/science.283.5408.1670. [DOI] [PubMed] [Google Scholar]
- 12.Weiss S. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 13.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. Nature (London) 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 14.Sase I, Miyata H, Ishiwata S, Kinosita K. Proc Natl Acad Sci USA. 1997;94:5646–5650. doi: 10.1073/pnas.94.11.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vale R D, Funatsu T, Pierce D W, Romberg L, Harada Y, Yanagida T. Nature (London) 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu H P, Xun L Y, Xie X S. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 17.Held W A, Ballou B, Mizushim S, Nomura M. J Biol Chem. 1974;249:3103–3111. [PubMed] [Google Scholar]
- 18.Batey R T, Williamson J R. J Mol Biol. 1996;261:536–549. doi: 10.1006/jmbi.1996.0481. [DOI] [PubMed] [Google Scholar]
- 19.Batey R T, Williamson J R. J Mol Biol. 1996;261:550–567. doi: 10.1006/jmbi.1996.0482. [DOI] [PubMed] [Google Scholar]
- 20.Orr J W, Hagerman P J, Williamson J R. J Mol Biol. 1998;275:453–464. doi: 10.1006/jmbi.1997.1489. [DOI] [PubMed] [Google Scholar]
- 21.Batey R T, Williamson J R. RNA. 1998;4:984–997. doi: 10.1017/s1355838298980426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith L M, Fung S, Hunkapiller M W, Hunkapiller T J, Hood L E. Nucleic Acids Res. 1985;13:2399–2412. doi: 10.1093/nar/13.7.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickson R M, Cubitt A B, Tsien R Y, Moerner W E. Nature (London) 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 24.Vandenbout D A, Yip W T, Hu D H, Fu D K, Swager T M, Barbara P F. Science. 1997;277:1074–1077. [Google Scholar]
- 25.Lu H P, Xie X S. Nature (London) 1997;385:143–146. [Google Scholar]
- 26.Ha T, Enderle T, Chemla D S, Selvin P R, Weiss S. Phys Rev Lett. 1996;77:3979–3982. doi: 10.1103/PhysRevLett.77.3979. [DOI] [PubMed] [Google Scholar]