

Abstract
Background: Several animal models for human ulcerative colitis (UC) associated neoplasia have been reported. However, most neoplasias developed in these models have morphological and genetic characteristics different from UC associated neoplasia.
Aims: To establish a new colitis associated neoplasia model in p53 deficient mice by treatment with dextran sulphate sodium (DSS).
Methods: DSS colitis was induced in homozygous p53 deficient mice (p53−/−-DSS), heterozygous p53 deficient mice (p53+/−-DSS) and wild-type mice (p53+/+-DSS) by treatment with 4% DSS. Numbers of developed neoplasias were compared among the experimental groups, and macroscopic and microscopic features of the neoplasias were analysed. Furthermore, K-ras mutation and beta-catenin expression were assessed.
Results: p53−/−-DSS mice showed 100% incidence of neoplasias whereas the incidences in p53+/−-DSS and p53+/+-DSS mice were 46.2% and 13.3%, respectively. No neoplasias were observed in the control groups. The mean numbers of total neoplasias per mouse were 5.0 (p53−/−-DSS), 0.62 (p53+/−-DSS), and 0.2 (p53+/+-DSS). The number of neoplasias per mouse in the p53−/−-DSS group was significantly higher than that in the other DSS groups. The incidences of superficial type neoplasias were 91.7% in p53−/−-DSS mice, 75.0% in p53+/−-DSS mice, and 33.3% in p53+/+-DSS mice. The K-ras mutation was not detected in any of the neoplasias tested. Translocation of beta-catenin from the cell membrane to the cytoplasm or nucleus was observed in 19 of 23 (82.6%) neoplasias.
Conclusions: The p53−/−-DSS mice is an excellent animal model of UC associated neoplasia because the morphological features and molecular genetics are similar to those of UC associated neoplasia. Therefore, this model will contribute to the analysis of tumorigenesis related to human UC associated neoplasia and the development of chemopreventive agents.
Keywords: colitis associated neoplasia, p53 deficient mice, dextran sulphate sodium, ulcerative colitis
Colorectal neoplasia is one of the known complications of ulcerative colitis (UC).1,2 The incidence risk of colorectal neoplasia increases with duration of the disease and is greater in patients with extensive colitis.3,4 UC associated colorectal neoplasia has several characteristics different from sporadic colorectal neoplasia. Clinicopathologically, the main macroscopic characteristic of UC associated neoplasia at the early stage (dysplasia or early cancer) is a flat configuration, and multiple synchronous neoplasias occur much more frequently in UC associated neoplasia than in sporadic colorectal neoplasia.5–7 Genetically, alterations of the adenomatous polyposis coli (APC) gene and the K-ras gene are less frequent than in sporadic colorectal neoplasia.8–15 Beta-catenin is a key component of the cadherin mediated cell-cell adhesion system and an important molecule in the Wnt-APC signal transduction system.16 Mutation in beta-catenin is detected infrequently and translocation of beta-catenin is shown frequently in both colorectal neoplasia and UC associated neoplasia. One of the major tumour suppressor genes, p53, plays a critical role in the development of many types of neoplasia, including colorectal neoplasia. Although alteration of the p53 gene is frequently observed in both UC associated neoplasia and sporadic neoplasia, this alteration is an early event in UC associated neoplasia whereas it is occurs late in sporadic neoplasia.13,14,17–20 Therefore, it is generally agreed that the tumorigenesis pathway in UC associated neoplasia is different from that in sporadic neoplasia; the former is called the chronic colitis dysplasia sequence and the latter the adenoma-carcinoma sequence.
There are many experimental animal models of human UC. In recent reports, colitis was induced mainly in mice and rats by colitis inducing agents such as dextran sulphate sodium (DSS) and trinitrobenzene sulphonic acid (TNB).21–26 A few of these reports have noted colonic tumorigenetic effects of colitis inducing agents, particularly DSS, and suggested they would be useful tumorigenesis models for human UC associated neoplasia.24–26 However, the incidence of colonic tumorigenesis in the models was not high24,25 and the developed neoplasias were predominantly protruded-type tumours.26 Furthermore, the occurrence of p53 gene alteration, which is found at high frequency in human UC associated neoplasia, was reported to be rare.26 When carcinogens such as azoxymethane (AOM) and 1,2-dimethylhydrazine (DMH) were added to colitis inducing agents, the incidence of developed neoplasias increased.27–29 However, in colonic neoplasia induced by these carcinogens, several reports have revealed that the K-ras gene mutation, which is considered infrequent in human UC associated neoplasia, was found frequently.30–32 Thus neoplasias developed in the previous experimental tumorigenesis models have properties different from those of human UC associated neoplasia with respect to both morphology and genetic alterations.
It has been reported that mice deficient in the p53 gene are developmentally normal but susceptible to spontaneous tumours.33–35 Homozygous p53 deficient mice develop neoplasia at high frequency and most die by six months of age. Heterozygous p53 deficient mice also have a significantly high incidence of neoplasia and poor survival when compared with wild-type mice. Most neoplasias that develop in homozygous and heterozygous deficient mice are lymphomas and sarcomas. The incidence of colonic neoplasia developing spontaneously in p53 deficient mice is low: the rate in the heterozygous deficient mice is 2%35 and no case has been reported in the homozygous deficient mice.33–35
In the present study, we developed a new colitis associated neoplasia model using p53 deficient mice. We then compared the morphology and genetic alterations in the neoplasia developed in p53 deficient mice with those in human UC associated neoplasia.
MATERIALS AND METHODS
Animals
Eight week old specific pathogen free homozygous p53 deficient mice (p53−/−, n = 12), heterozygous p53 deficient mice (p53+/−, n = 13), and wild-type C57BL/6 x CBA mice (p53+/+ n = 15; Charles River, Kanagawa, Japan) were used for the study. p53−/− mice with a C57BL/6 and CBA background were produced by Tsukada and colleagues36 and kindly provided by Dr Norio Ishida (Clock Cell Biology Group, Institute for Biological Resources and Functions, National Institute of Advanced Industrial Science and Technology, Tsukuba, Japan). p53+/− mice were obtained by crossing p53−/− mice and wild-type C57BL/6 mice.
Induction of colitis
The design for inducing colitis is shown in fig 1 ▶. All mice were eight weeks old at the beginning of the experiment. Colitis was induced by feeding 4% DSS (ICN Biomedicals Inc., Aoraro, Ohio, USA), molecular weight 36 000–50 000, dissolved in the drinking water which was given ad libitum. One cycle was defined as seven days of DSS followed by 14 days of distilled water. DSS colitis was induced for two cycles in p53+/+ mice (p53+/+-DSS), p53+/− mice (p53+/−-DSS), and p53−/− mice (p53−/−-DSS). After two cycles, mice in each DSS group were given distilled water for the next 84 days. As controls for each DSS group, mice given distilled water alone were divided into three groups (p53−/−-water, p53+/−-water, and p53+/+-water). At 126 days, mice in all groups were killed by cervical dislocation. Experiments were carried out under the control of Animal Care and Use Committee, Dokkyo University School of Medicine, in accordance with Guidelines for the Care and Use of Laboratory Animals, Dokkyo University School of Medicine.
Figure 1.

Overview of the experimental design. All mice were eight weeks old at the beginning of the experiment. Colitis was induced by feeding 4% dextran sulphate sodium (DSS) ad libitum. One cycle was defined as seven days of DSS followed by 14 days of distilled water. DSS colitis was induced for two cycles in p53+/+ mice (p53+/+-DSS), p53+/− mice (p53+/−-DSS), and p53−/−- mice (p53−/−-DSS). After two cycles, mice in each DSS group were given distilled water for the next 84 days. As controls for each DSS group, mice given distilled water alone were divided into three groups (p53−-/−-water, p53+/−-water, and p53+/+-water). At 126 days, mice in all groups were killed.
Pathological evaluation
The colons of the mice were removed and cut open along the longitudinal median axis. These colons were rinsed with saline and fixed in a neutral aqueous phosphate buffered 4% solution of formaldehyde for eight hours. The entire part of the colon was then stained with alcian blue (pH 2.5), and the surface microstructure was observed using stereomicroscopy (Olympus, Tokyo, Japan) and examined for the presence of neoplasias. All gross lesions that were suspected neoplasias were sectioned under stereomicroscopy; remaining tissues were sectioned at intervals of 2 mm, embedded in paraffin, and stained with haematoxylin and eosin.
Macroscopically, neoplasias were classified into two types: superficial and protruded. The superficial type was defined as a lesion with a height not greater than twice the thickness of the adjacent non-neoplastic epithelium. The protruded type was defined as a lesion that clearly projected above the surface of the adjacent non-neoplastic epithelium.
Histologically, each sample was examined by three experienced pathologists in a blinded fashion, and neoplasias were classified according to the Vienna classification of gastrointestinal epithelial neoplasia.37 This new classification is practical and has been recommended to resolve large discrepancies between Western and Japanese pathologists in the diagnosis of gastrointestinal epithelial neoplasias and aid in better understanding of research data in the fields of gastroenterology. This classification has five categories: category 1, negative for neoplasia; category 2, indefinite for neoplasia; category 3, non-invasive low grade neoplasia; category 4, non-invasive high grade neoplasia; and category 5, invasive neoplasia. The numbers of neoplasias, and the macroscopic and microscopic features of the neoplasias were recorded.
Preparation of tissue sections and DNA extraction
To obtain neoplastic cell samples for DNA extraction, we conducted laser capture microdissection. Serial sections (5 μm) of formalin fixed paraffin embedded tissue were mounted onto a clear polyethylene membrane which was attached to an aluminium frame slide (NikonInstech, Kanagawa, Japan). Microdissection was performed using a UV laser microdissection system (NikonInstech) and dissected tissues were collected on each plastic cap. Caps were placed in a microcentrifuge tube and DNA was extracted using a DNA isolator PS kit (Wako Pure Chemical, Osaka, Japan) according to the supplied protocol.
Analysis of K-ras codon 12 mutations
Extracted DNA was amplified using the polymerase chain reaction (PCR) which was carried out using the following amplification profile: five minutes at 94°C once; one minute at 94°C, one minute at 60°C, two minutes at 72°C for 40 cycles; then four minutes at 72°C. The reaction mixture (50 μl) contained 0.1 μg genomic DNA, 5 μl 10× PCR Gold Buffer, 4 μl 25 mM MgCl2, 5 μl dNTP mixture, 1.25 units AmpliTaq Gold (Applied Biosystems, Tokyo, Japan), 10 pmol of forward mismatch primer (5′-AACTTGTGGTTGGAcCTG-3′), and 10 pmol of reverse primer (5′-AGCGTTACCTCTATCGTAGG-3′). The forward mismatch primer produced Mva I site in the amplified fragment.38
Mutations of K-ras codon 12 in the amplified DNA were screened using the PCR-restriction fragment length polymorphism (PCR-RFLP) method. In brief, PCR products were digested with Mva I (Takara, Kyoto, Japan) to distinguish the mutant allele from the wild-type allele, and electrophoresed on 3% agarose gels, followed by staining with ethidium bromide. PCR products encoding the wild-type and mutant were detected as 86 bp and 106 bp fragments, respectively.
Direct DNA sequencing of amplified DNA was performed to confirm the results of PCR-RFLP. PCR products were purified using the QIAquick PCR purification kit (Qiagen KK, Tokyo, Japan). Purified PCR products were sequenced on an ABI Prism 3700 DNA Analyzer (Applied Biosystems) using the ABI Prism Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems). The same primers were used for both amplification and sequencing. The resulting sequencing data were analysed using the Gene Scan Analysis software Analyzer (Applied Biosystems) in accordance with the manufacturer’s protocol. All sequences were verified in both the forward and reverse directions.
Immunohistochemistry
Immunohistochemical analysis was carried out with primary antibodies against beta-catenin (diluted 1:1000; Transduction Laboratories, California, USA) in formalin fixed paraffin embedded tissue sections using a labelled streptavidin biotin kit (Dako Japan, Kyoto Japan), as described previously.39 Faint membrane staining for beta-catenin was interpreted as a normal staining pattern which was observed in all of the normal colon epithelial specimens. Intense cytoplasmic or nuclear staining for beta-catenin was interpreted as an abnormal staining pattern.
Statistical analysis
The numbers of neoplasias in each group were expressed as mean (SEM), and differences between groups were analysed using the non-parametric Mann-Whitney U test; p<0.05 was considered significant. The χ2 test was used to compare types of neoplasias in each group; p<0.05 was considered significant.
RESULTS
Number and incidence of neoplasias
Numbers of neoplasias and incidences of mice with neoplasia in each group are summarised in table 1 ▶. Sixty neoplasias were found in the p53−/−-DSS mice group; 44 were low grade neoplasias, 11 were high grade neoplasias, and five were invasive neoplasias. Eight neoplasias were found in the p53+/−-DSS mice group; five were low grade neoplasias, one was a high grade neoplasia, and two were invasive neoplasias. Three neoplasias were found in the p53+/+-DSS mice group; one was a low grade neoplasia and two were invasive neoplasias. No neoplasias were observed in the p53−/−-water, p53+/−-water, or p53+/+-water groups. The incidences of mice with neoplasias were 100% in p53−/−-DSS mice, 46.2% in p53+/−-DSS mice, and 13.3% in p53+/+-DSS mice
Table 1.
Incidence of neoplasia and numbers of neoplasias in each group of mice
| Group | n | Incidence* | No of neoplasias | |||
| Total† | Category 3 | Category 4 | Category 5 | |||
| p53−/−-DSS | 12 | 12/12 | 60 | 44 | 11 | 5 |
| p53+/−-DSS | 13 | 6/13 | 8 | 5 | 1 | 2 |
| p53+/+-DSS | 15 | 2/15 | 3 | 1 | 0 | 2 |
| p53−/−-water | 10 | 0/10 | 0 | 0 | 0 | 0 |
| p53+/−-water | 10 | 0/10 | 0 | 0 | 0 | 0 |
| p53+/+-water | 10 | 0/10 | 0 | 0 | 0 | 0 |
*Number of mice with neoplasia/total number of mice.
†Total neoplasias: categories 3, 4, and 5.
DSS, dextran sulphate sodium.
Mean numbers of neoplasias per mouse are presented in fig 2 ▶. Mean (SEM) numbers of total neoplasias (categories 3–5) per mouse were 5.0 (0.82) in the p53−/−-DSS group, 0 (0) in the p53−/−-water group, 0.62 (0.17) in the p53+/−-DSS group, 0 (0) in the p53+/−-water group, 0.2 (0.14) in the p53+/+-DSS group, and 0 (0) in the p53+/+-water group. Mean (SEM) numbers of low grade neoplasias (category 3) per mouse were 3.7 (0.63) in the p53−/−-DSS group, 0.38 (0.15) in the p53+/−-DSS group, and 0.07 (0.06) in the p53+/+-DSS group. Mean (SEM) numbers of high grade neoplasias (category 4) per mouse were 0.92 (0.41), 0.08 (0.07), and 0 (0), respectively. Mean (SEM) numbers of invasive neoplasias (category 5) per mouse were 0.42 (0.20), 0.15 (0.10), and 0.13 (0.08), respectively. The number of total neoplasias per mouse in the p53−/−-DSS group was significantly higher than that in the p53+/−-DSS, p53+/+-DSS, and p53−/−-water groups (p<0.001). The number of total neoplasias per mouse in the p53+/−-DSS group was significantly higher than that in the p53+/−-water group (p = 0.017).
Figure 2.

Mean numbers of total neoplasias per mouse and mean numbers in each category. The number of total neoplasias per mouse in the p53−/−-dextran sulphate sodium (DSS) group was significantly higher than that in the p53+/−-DSS, p53+/+-DSS, and p53−/−-water groups. The number of total neoplasias per mouse in the p53+/−-DSS group was significantly higher than that in the p53+/−-water group. *p = 0.017, ***p<0.001.
Pathology
Table 2 ▶ summarises the macroscopic classification of neoplasias in each group. In the p53−/−-DSS group, 55 of 60 total neoplasias (91.7%) were of the superficial type and five (8.3%) were the protruded type. In the p53+/−-DSS group, six of eight total neoplasias (75.0%) were superficial and two (25.0%) were protruded. In the p53+/+-DSS group, one of three total neoplasias (33.3%) was superficial and two (66.7%) were protruded. In the p53−/−-DSS group, 47 of 55 superficial type lesions were flat lesions with the same height as the adjacent non-neoplastic epithelium (fig 3 ▶). In the p53+/−-DSS group, three of six superficial type lesions were flat. In the p53+/+-DSS group, flat neoplasias were not observed. There was a significant difference in the incidences of superficial versus protruded type lesions between the p53−/−-DSS and p53+/+-DSS groups (p = 0.028). There was no significant difference between the p53−/−-DSS and p53+/−-DSS groups, or between the p53+/−-DSS and p53+/+-DSS groups.
Table 2.
Macroscopic type of developed neoplasia in each dextran sulphate sodium (DSS) group
| Total neoplasias | Macroscopic type | ||
| Superficial | Protruded | ||
| p53−-/−-DSS | 60 | 55 | 5 |
| p53+/−-DSS | 8 | 6 | 2 |
| P53+/+-DSS* | 3 | 1 | 2 |
*p = 0.028 compared with p53−/−-DSS.
Figure 3.
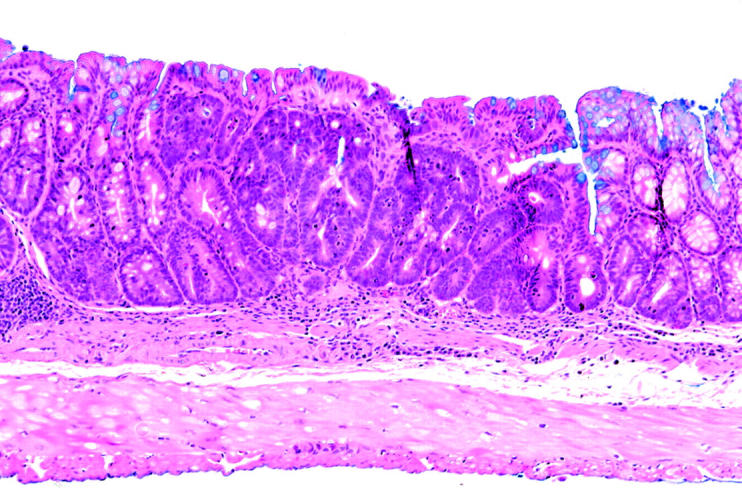
Neoplasia in the p53−/−-dextran sulphate sodium (DSS) group, classified as category 4 (high grade neoplasia). This neoplasia is a flat type with the same height as the adjacent non-neoplastic epithelium.
Histologically, nine invasive neoplasias (category 5) were observed. Seven were well differentiated adenocarcinomas (fig 4 ▶) and two were poorly differentiated adenocarcinomas (fig 5 ▶). Two of them invaded the submucosal layer (fig 6 ▶).
Figure 4.
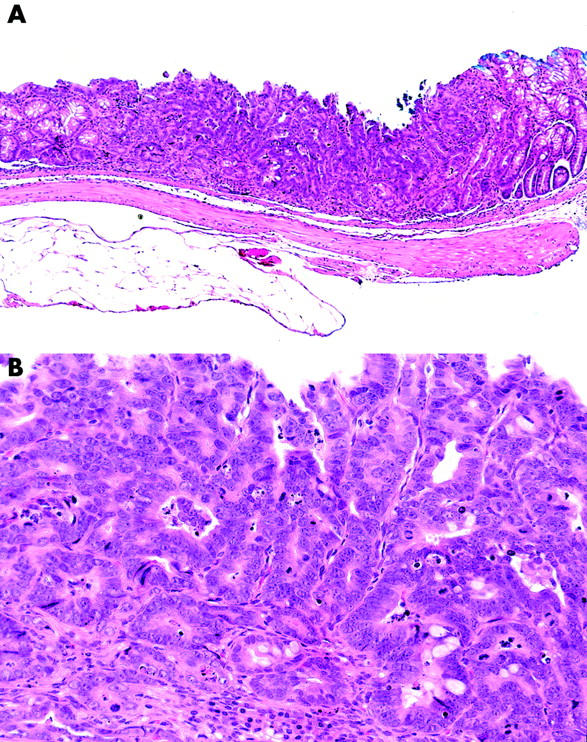
Neoplasia in the p53−/−-dextran sulphate sodium (DSS) group, classified as category 5 (invasive neoplasia). This neoplasia is of the superficial type macroscopically (A) and a well differentiated adenocarcinoma histologically (B).
Figure 5.
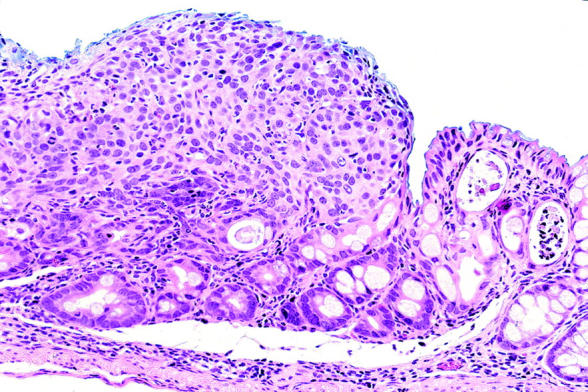
Neoplasia in the p53−/−-dextran sulphate sodium (DSS) group, classified as category 5 (invasive neoplasia). This neoplasia is a poorly differentiated adenocarcinoma histologically.
Figure 6.
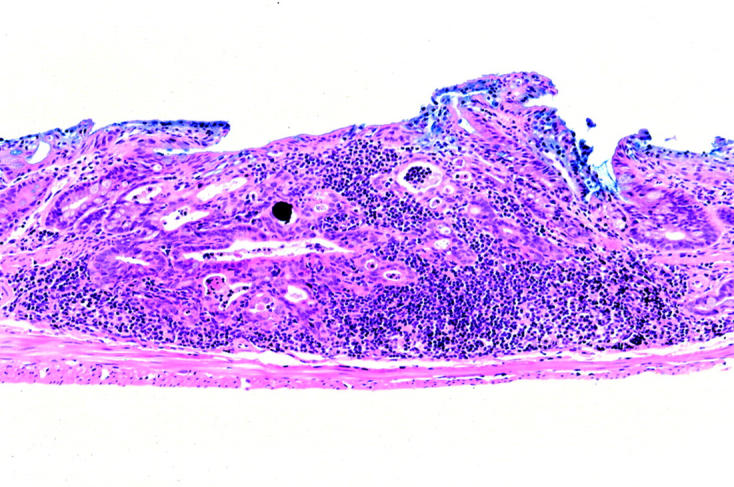
Neoplasia in the p53−/−-dextran sulphate sodium (DSS) group, classified as category 5 (invasive neoplasia). This neoplasia invades through the muscularis mucosae into the submucosa.
Incidence of K-ras codon 12 mutation
PCR-RFLP analysis was performed on 32 neoplasias; eight were invasive neoplasias, 10 were high grade neoplasias, and 14 were low grade neoplasias. No mutation of K-ras codon 12 was detected in any neoplasia. Direct DNA sequencing was performed on 10 lesions to confirm the results of PCR-RFLP. No mutation was detected in the K-ras gene.
Immunohistochemistry for beta-catenin
Analysis of immunohistochemistry for beta-catenin was performed on 23 neoplasias: seven were invasive neoplasias, seven were high grade neoplasias, and nine were low grade neoplasias. In non-neoplastic colonic epithelial cells, beta-catenin was localised at the cell membrane (fig 7A ▶). In contrast, translocation of beta-catenin from the cell membrane to the cytoplasm or nucleus was observed in 19 of 23 (82.6%) neoplasias (fig 7B ▶). There was no difference in beta-catenin localisation for any group, any category, or any macroscopic type.
Figure 7.
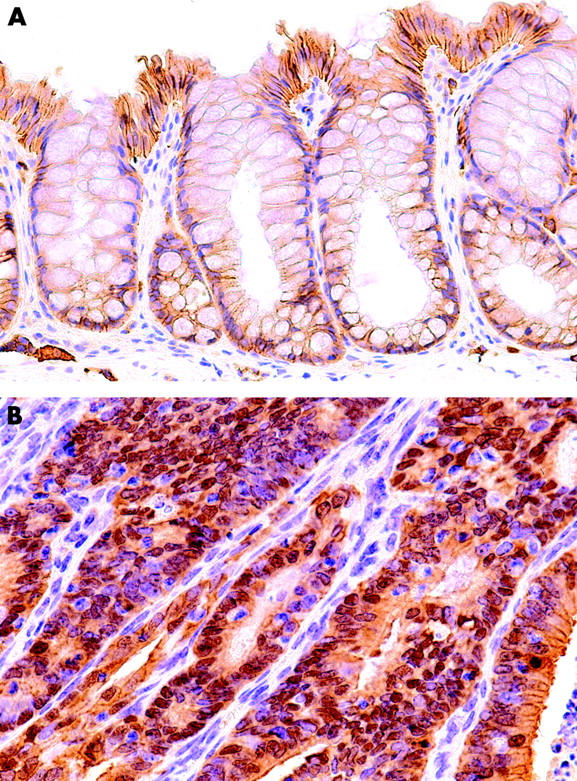
Immunohistochemical staining for beta-catenin in normal colon epithelium and neoplasia. In normal colon epithelium (A), beta-catenin was localised at the cell membrane. In most neoplasias (B), translocation of beta-catenin from the cell membrane to the cytoplasm or nucleus was observed.
DISCUSSION
We have succeeded in developing colonic neoplasias at a high rate by inducing colitis with DSS in p53 deficient mice. The number of neoplasias was significantly higher in p53−/−-DSS mice than in p53+/−-DSS or p53+/+-DSS mice. Most of the neoplasias that developed in p53−/− deficient mice were of the flat type and accompanied multiple synchronous lesions. In these neoplasias, K-ras mutation was not detected and translocation of beta-catenin was frequently observed. Therefore, this animal model is suitable to study human UC associated neoplasia because the morphological features and molecular genetics are similar to those of human UC associated neoplasia.
In animal models of experimental colitis, the incidence of colonic neoplasia increased when a colitis inducing agent and a carcinogen were used in combination. Okayasu and colleagues29 reported the development of 10.5 lesions of neoplasia per mouse by combined use of DSS and AOM. According to Karlin and colleagues,27 neoplasias developed in all mice treated with DSS and DMH, and 5.5 neoplasias were found per mouse. As shown above, DSS induced colitis was reported to act as a promoter of tumorigenesis in carcinogen induced colonic neoplasia. On the other hand, in an experimental colonic tumorigenesis animal model using only a colitis inducing agent, Cooper and colleagues23 reported that the incidence of neoplasia in mice treated with DSS was 37.5% (dysplasia 5/16; cancer 4/16) in 204 days of observation after four cycles of 5% DSS administration. In the study of Okayasu and colleagues,22,29 the incidence was 0–13.3%. Thus in experimental inflammatory tumorigenesis induced by a colitis inducing agent alone, the incidence of neoplasia was not high enough to conduct experiments for developing chemopreventive agents or analysing the molecular events in colitis associated neoplasia.
The macroscopic type of neoplasia that developed in the present study was as follows. The number of superficial type neoplasias in p53−/−-DSS mice was significantly greater than that in p53+/+-DSS mice (91.7% (55/60) v 33.3% (1/3); p = 0.028). Cooper and colleagues26 reported that among developed neoplasias in a DSS colitis induced tumorigenesis model, 67% (10/15) were of the polypoid type and 33% (5/15) were of the flat type. In our study, 67% (2/3) of neoplasias that developed in p53+/+-DSS mice were polypoid, indicating good agreement with their report. In human UC associated neoplasia, flat type neoplasias are often observed at an early stage (dysplasia and/or early cancer).5–7 Therefore, neoplasias developed by DSS colitis in p53 deficient mice are similar to human UC associated neoplasias with respect to morphology.
In human UC associated neoplasia, several immunohistochemical and molecular studies have demonstrated that p53 is frequently altered in both carcinoma and dysplasia. For example, Brentnall and colleagues20 detected p53 mutations and/or loss of heterozygosity in 83% of cancers and 76% of dysplasias. In our previous report, nuclear abnormal expression of p53 protein was found in 61.1% of cancers and 58.3% of dysplasias by immunohistochemical staining, and p53 mutations within exons 5–8 were found in 100% of cancers and 92.3% of dysplasias by PCR-single stranded conformation polymorphism (SSCP).14 In addition, p53 alterations have been reported in non-neoplastic colonic epithelium adjacent to neoplasias.40,41 Therefore, p53 gene alteration is an early event in UC associated tumorigenesis, in contrast with sporadic colorectal tumours in which the p53 gene alteration is a late event.
There have been several reports on the incidence of p53 gene alterations in colonic tumorigenesis animal models.26,28,42–44 p53 gene mutations were rare in carcinogen induced colonic neoplasias in animal models.42–44 There are two reports in which p53 gene mutations in a colitis induced tumorigenesis model using a colitis inducing agent were examined.26,28 Suzui and colleagues28 analysed p53 gene mutations in colonic neoplasia developed in a rat colitis induced tumorigenesis model using 1-hydroxyanthraquinone, the colitis inducing agent, and methylazoxymethanol in combination, and detected no p53 alterations by the PCR-SSCP method. Cooper and colleagues26 analysed p53 gene alterations in colonic neoplasias developed in a mouse colitis induced tumorigenesis model induced only by DSS. They found that only 7.4% of neoplasias showed abnormal nuclear accumulation of p53 protein in an immunohistochemical study. From these findings, colonic neoplasias in previous animal models would be different from human UC associated neoplasias with respect to p53 alterations.
In our preliminary study, we used immunohistochemistry to evaluate p53 expression in neoplasias developed in p53+/−-DSS and p53+/+-DSS mice. However, we could not detect expression or abnormal nuclear accumulation of p53 protein in any neoplasias (data not shown). At present, we are analysing genetic alterations of the remaining allele of the p53 gene in neoplasias developed in p53+/− mice, and both alleles of the p53 gene in neoplasias developed in p53+/+ mice. The negative results of p53 expression based on immunohistochemistry may indicate that these p53 genes are inactivated by genetic deletion or truncated mutation. As a result, complete loss of function of the p53 gene would eliminate an important defence mechanism against oncogenic events induced by inflammation, such as production of reactive oxygen intermediates and genotoxic cytokines, and enhancement of cell proliferation.45,46 Then, loss of function of the p53 gene would play a causative role in the development of colitis associated neoplasias.
In most sporadic colorectal neoplasias, mutational activation of the K-ras oncogene is considered to play a role in the progression of size and grade of atypia in the adenoma-carcinoma sequence, and the K-ras mutation is found in approximately 50% of polypoid adenomas larger than 1 cm in diameter.15 In the past 10 years, two morphologically distinct subtypes in sporadic colorectal neoplasias have been found, the polypoid type and the superficial type, and the incidence of K-ras mutations is lower in the superficial type than in the polypoid type.47,48 In human UC associated neoplasia, which is mostly the superficial type, K-ras mutations are reported to be rare.9–11,13,14 Thus K-ras mutations may play an important role in polypoid growth of colorectal neoplasias. In animal models, Jacoby and colleagues30 found K-ras mutations in 66% of colonic neoplasias developed in rats treated with DMH and DSS. Endo and colleagues32 reported a rate of 57% in a similar experiment. Thus these neoplasias, which were developed by treatment with carcinogens and DSS as the inflammation inducer, showed different genetic alterations from those of human UC associated colorectal neoplasia. In our present model, mutation of the K-ras gene was not found in colonic neoplasias associated with DSS induced colitis in any group. Therefore, the colonic neoplasias developed in our model are similar to human UC associated colorectal neoplasias with respect to K-ras gene mutations.
It is reported that 4–15% of sporadic colorectal neoplasias in humans harbour beta-catenin mutations,49,50 and that translocation of beta-catenin is shown in 65–100% of sporadic colorectal neoplasias by immunohistochemical studies.51–54 In human UC associated neoplasia, Aust et al reported that none of their neoplasias showed a beta-catenin mutation55 but they showed translocation of beta-catenin in most neoplasias (79%) by immunohistochemistry.56 We previously reported that translocation of beta-catenin from the cell membrane to the cytoplasm or nucleus was observed in all invasive neoplasias developed in a rat tumorigenesis model using TNB and DMH.39 In the present study, we found translocation of beta-catenin in 82.6% of neoplasias. Therefore, the colonic neoplasias developed in our model are also similar to human UC associated colorectal neoplasias with respect to the abnormality of the Wnt-APC-beta-catenin system.
We are currently studying other molecular alterations that occur in the neoplasias developed in this model by microarray analysis. Moreover, we are analysing the type of p53 gene alteration (complete loss of function by genetic deletion or truncated mutation, or gain of function by the activated point mutation) in superficial type neoplasias and polypoid type neoplasias in p53+/−-DSS and p53+/+-DSS mice. These attempts will contribute to the elucidation of tumorigenesis related to human UC associated neoplasia and the development of chemopreventive agents.
Acknowledgments
This study was supported by a grant in aid for Scientific Research (C: 14570157) from the Japan Society for the Promotion of Science (JSPS). The authors thank Dr M Shinoda (Professor of Laboratory Animal Research Center, Dokkyo University School of Medicine) for supporting our experiment. The authors also thank Ms C Sato-Matsuyama, A Shimizu, T Ohtsuki, M Matsuura, and S Miyahara (Department of Surgical and Molecular Pathology, Dokkyo University School of Medicine) for technical assistance.
Abbreviations
UC, ulcerative colitis
DSS, dextran sulphate sodium
APC, adenomatous polyposis coli
TNB, trinitrobenzene sulphonic acid
AOM, azoxymethane
DMH, 1,2-dimethylhydrazine
PCR, polymerase chain reaction
RFLP, restriction fragment length polymorphism
SSPC, single stranded conformation polymorphism
REFERENCES
- 1.Brostrom O, Lofberg R, Nordenvall B, et al. The risk of colorectal cancer in ulcerative colitis. An epidemiologic study. Scand J Gastroenterol 1987;22:1193–9. [DOI] [PubMed] [Google Scholar]
- 2.Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med 1990;323:1228–33. [DOI] [PubMed] [Google Scholar]
- 3.Lennard-Jones JE, Melville DM, Morson BC, et al. Precancer and cancer in extensive ulcerative colitis: findings among 401 patients over 22 years. Gut 1990;31:800–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillen CD, Walmsley RS, Prior P, et al. Ulcerative colitis and Crohn’s disease: a comparison of the colorectal cancer risk in extensive colitis. Gut 1994;35:1590–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butt JH, Konishi F, Morson BC, et al. Macroscopic lesions in dysplasia and carcinoma complicating ulcerative colitis. Dig Dis Sci 1983;28:18–26. [DOI] [PubMed] [Google Scholar]
- 6.Greenstein AJ, Slater G, Heimann TM, et al. A comparison of multiple synchronous colorectal cancer in ulcerative colitis, familial polyposis coli, and de novo cancer. Ann Surg 1986;203:123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Herbay A, Herfarth C, Otto HF. Cancer and dysplasia in ulcerative colitis: a histologic study of 301 surgical specimen. Z Gastroenterol 1994;32:382–8. [PubMed] [Google Scholar]
- 8.Tarmin L, Yin J, Harpaz N, et al. Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer Res 1995;55:2035–8. [PubMed] [Google Scholar]
- 9.Umetani N, Sasaki S, Watanabe T, et al. Genetic alterations in ulcerative colitis-associated neoplasia focusing on APC, K-ras gene and microsatellite instability. Jpn J Cancer Res 1999;90:1081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burmer GC, Levine DS, Kulander BG, et al. c-Ki-ras mutations in chronic ulcerative colitis and sporadic colon carcinoma. Gastroenterology 1990;99:416–20. [DOI] [PubMed] [Google Scholar]
- 11.Bell SM, Kelly SA, Hoyle JA, et al. c-Ki-ras gene mutations in dysplasia and carcinomas complicating ulcerative colitis. Br J Cancer 1991;64:174–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaubert P, Benhattar J, Saraga E, et al. K-ras mutations and p53 alterations in neoplastic and nonneoplastic lesions associated with longstanding ulcerative colitis. Am J Pathol 1994;144:767–75. [PMC free article] [PubMed] [Google Scholar]
- 13.Holzmann K, Klump B, Borchard F, et al. Comparative analysis of histology, DNA content, p53 and Ki-ras mutations in colectomy specimens with long-standing ulcerative colitis. Int J Cancer 1998;76:1–6. [DOI] [PubMed] [Google Scholar]
- 14.Fujii S, Fujimori T, Chiba T. Usefulness of analysis of p53 alteration and observation of surface microstructure for diagnosis of ulcerative colitis-associated colorectal neoplasia. J Exp Clin Cancer Res 2003;22:107–15. [PubMed] [Google Scholar]
- 15.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319:525–32. [DOI] [PubMed] [Google Scholar]
- 16.Miller JR, Moon RT. Signal transduction through b-catenin and specification of cell fate during embryogenesis. Genes Dev 1996;10:2527–39. [DOI] [PubMed] [Google Scholar]
- 17.Baker SJ, Preisinger AC, Jessup JM, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 1990;50:7717–22. [PubMed] [Google Scholar]
- 18.Yin J, Harpaz N, Tong Y, et al. p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993;104:1633–9. [DOI] [PubMed] [Google Scholar]
- 19.Harpaz N, Peck AL, Yin J, et al. p53 protein expression in ulcerative colitis-associated colorectal dysplasia and carcinoma. Hum Pathol 1994;25:1069–74. [DOI] [PubMed] [Google Scholar]
- 20.Brentnall TA, Crispin DA, Rabinovitch PS, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994;107:369–78. [DOI] [PubMed] [Google Scholar]
- 21.Morris GP, Beck PL, Herridge MS, et al. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 1989;96:795–803. [PubMed] [Google Scholar]
- 22.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990;98:694–702. [DOI] [PubMed] [Google Scholar]
- 23.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 1993;69:238–49. [PubMed] [Google Scholar]
- 24.Kullmann F, Messmann H, Alt M, et al. Clinical and histopathological features of dextran sulfate sodium induced acute and chronic colitis associated with dysplasia in rats. Int J Colorectal Dis 2001;16:238–46. [DOI] [PubMed] [Google Scholar]
- 25.Okayasu I, Yamada M, Mikami T, et al. Dysplasia and carcinoma development in a repeated dextran sulfate sodium-induced colitis model. J Gastroenterol Hepatol 2002;17:1078–83. [DOI] [PubMed] [Google Scholar]
- 26.Cooper HS, Murthy S, Kido K, et al. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation, Carcinogenesis 2000;21:757–68. [DOI] [PubMed] [Google Scholar]
- 27.Karlin DA, O’Donnell RT, Jensen WE. Effect of dioctyl sodium sulfosuccinate feeding on rat colorectal 1,2-dimethylhydrazine carcinogenesis. J Natl Cancer Inst 1980;64:791–3. [PubMed] [Google Scholar]
- 28.Suzui M, Yoshimi N, Ushijima T, et al. No involvement of Ki-ras or p53 gene mutations in colitis-associated rat colon tumors induced by 1-hydroxyanthraquinone and methylazoxymethanol acetate. Mol Carcinog 1995;12:193–7. [DOI] [PubMed] [Google Scholar]
- 29.Okayasu I, Ohkusa T, Kajiura K, et al. Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 1996;39:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacoby RF, Llor X, Teng BB, et al. Mutations in the K-ras oncogene induced by 1,2-dimethylhydrazine in preneoplastic and neoplastic rat colonic mucosa. J Clin Invest 1991;87:624–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llor X, Jacoby RF, Teng BB, et al. K-ras mutations in 1,2-dimethylhydrazine-induced colonic tumors: effects of supplemental dietary calcium and vitamin D deficiency. Cancer Res 1991;51:4305–9. [PubMed] [Google Scholar]
- 32.Endo T, Ookawa K, Tanaka M, et al. Differences in carcinogenesis by the length of carcinogen exposure period in rat colon. Dig Dis Sci 2001;46:109–17. [DOI] [PubMed] [Google Scholar]
- 33.Harvey M, McArthur MJ, Montgomery CA Jr, et al. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet 1993;5:225–9. [DOI] [PubMed] [Google Scholar]
- 34.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994;4:1–7. [DOI] [PubMed] [Google Scholar]
- 35.Purdie CA, Harrison DJ, Peter A, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 1994;9:603–9. [PubMed] [Google Scholar]
- 36.Tsukada T, Tomooka Y, Takai S, et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 1993;8:3313–22. [PubMed] [Google Scholar]
- 37.Schlemper RJ, Riddell RH, Kato Y, et al. The Vienna classification of gastrointestinal epithelial neoplasia. Gut 2000;47:251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaidi NH, Pretlow TP, O’Riordan MA, et al. Transgenic expression of human MGMT protects against azoxymethane-induced aberrant crypt foci and G to A mutations in the K-ras oncogene of mouse colon. Carcinogenesis 1995;16:451–6. [DOI] [PubMed] [Google Scholar]
- 39.Furihata T, Kawamata H, Kubota K, et al. Evaluation of the malignant potential of aberrant crypt foci by immunohistochemical staining for beta-catenin in inflammation-induced rat colon carcinogenesis. Int J Mol Med 2002;9:353–8. [PubMed] [Google Scholar]
- 40.Lashner BA, Shapiro BD, Husain A, et al. Evaluation of the usefulness of testing for p53 mutations in colorectal cancer surveillance for ulcerative colitis. Am J Gastroenterol 1999;94:456–62. [DOI] [PubMed] [Google Scholar]
- 41.Hussain SP, Amstad P, Raja K, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 2000;60:3333–7. [PubMed] [Google Scholar]
- 42.Okamoto M, Ohtsu H, Miyaki M, et al. No allelic loss at the p53 locus in 1,2-dimethylhydrazine-induced mouse colon tumours: PCR-SSCP analysis with sequence-tagged microsatellite site primers. Carcinogenesis 1993;14:1483–6. [DOI] [PubMed] [Google Scholar]
- 43.Shivapurkar N, Belinsky SA, Wolf DC, et al. Absence of p53 gene mutations in rat colon carcinomas induced by azoxymethane. Cancer Lett 1995;96:63–70. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto M, Ohtsu H, Kominami R, et al. Mutational and LOH analyses of p53 alleles in colon tumors induced by 1,2-dimethylhydrazine in F1 hybrid mice. Carcinogenesis 1995;16:2659–66. [DOI] [PubMed] [Google Scholar]
- 45.Kawai K, Kawamata H, Kemeyama S, et al. Persistence of carcinogen-altered cell population in rat urothelium which can be promoted to tumors by chronic inflammatory stimulus. Cancer Res 1994;54:2630–2. [PubMed] [Google Scholar]
- 46.Okamoto M, Kawamata H, Kawai K, et al. Enhancement of transformation in vitro of a nontumorigenic rat urothelial cell line by interleukin 6. Cancer Res 1995;55:4581–5. [PubMed] [Google Scholar]
- 47.Fujimori T, Satonaka K, Yamamura-Idei Y, et al. Non-involvement of ras mutations in flat colorectal adenomas and carcinomas. Int J Cancer 1994;57:51–5. [DOI] [PubMed] [Google Scholar]
- 48.Yamagata S, Muto T, Uchida Y, et al. Polypoid growth and K-ras codon 12 mutation in colorectal cancer. Cancer 1995;75:953–7. [DOI] [PubMed] [Google Scholar]
- 49.Sparks AB, Morin PJ, Vogelstein B, et al. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130–4. [PubMed] [Google Scholar]
- 50.Samowitz WS, Powers MD, Spirio LN, et al. Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 1999;59:1442–4. [PubMed] [Google Scholar]
- 51.Takayama T, Shiozaki H, Shibamoto S, et al. Beta-catenin expression in human cancers. Am J Pathol 1996;148:39–46. [PMC free article] [PubMed] [Google Scholar]
- 52.Hao X, Tomlinson I, Ilyas M, et al. Reciprocity between membranous and nuclear expression of beta-catenin in colorectal tumours. Virchows Arch 1997;431:167–72. [DOI] [PubMed] [Google Scholar]
- 53.Valizadeh A, Karayiannakis AJ, el-Hariry I, et al. Expression of E-cadherin-associated molecules (alpha-, beta-, and gamma-catenins and p120) in colorectal polyps. Am J Pathol 1997;150:1977–84. [PMC free article] [PubMed] [Google Scholar]
- 54.El-Bahrawy MA, Poulsom R, Jeffery R, et al. The expression of E-cadherin and catenins in sporadic colorectal carcinoma. Hum Pathol 2001;32:1216–24. [DOI] [PubMed] [Google Scholar]
- 55.Aust DE, Terdiman JP, Willenbucher RF, et al. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: a mutational analysis. Cancer 2002;94:1421–7. [DOI] [PubMed] [Google Scholar]
- 56.Aust DE, Terdiman JP, Willenbucher RF, et al. Altered distribution of beta-catenin, and its binding proteins E-cadherin and APC, in ulcerative colitis-related colorectal cancers. Mod Pathol 2001;14:29–39. [DOI] [PubMed] [Google Scholar]