

Abstract
Background: A number of recent studies have demonstrated a protective effect of renin-angiotensin system (RAS) antagonism against immune mediated diseases such as myocarditis, chronic allograft rejection, and antiglomerular basement membrane nephritis. To our knowledge, there has been no report on the immunological contribution of the RAS in colonic tissue.
Aims: We evaluated the direct effect of angiotensin II (AII) on the pathogenesis of immune mediated colitis using angiotensinogen deficient homozygous (Atg−/−) mice.
Subjects: 2,4,6-Trinitrobenzene sulphonic acid (TNBS) colitis was induced in Atg−/− and wild-type (Atg+/+) mice.
Methods: Levels of proinflammatory cytokines in the colon were determined by enzyme linked immunosorbent assay. Histological analysis was performed simultaneously.
Results: Although Atg−/− mice developed colitis, the degree was much milder than that in Atg+/+ mice (p<0.05). Colonic cytokine analysis showed that the production of proinflammatory cytokines (interleukin (IL)-1β, interferon γ (IFN-γ)) was impaired in Atg−/− mice. Furthermore, expression of cytokines such as IL-4 and IL-10 in the colon was predominant in Atg−/− compared with Atg+/+ mice after TNBS instillation (p<0.005, p<0.01, respectively). Similarly, subcutaneous infusion of losartan suppressed colitis (p<0.05) and the production of proinflammatory cytokines (IL-1β, IFN-γ). These results indicate that the RAS is directly involved in the pathogenesis of TNBS colitis through regulation of proinflammatory and anti-inflammatory cytokines in the colon.
Conclusions: This study revealed that the RAS is involved in the immune system in the colon. Antagonism of the RAS is a potential prophylactic strategy for the treatment of human inflammatory bowel disease.
Keywords: trinitrobenzene sulphonic acid, colitis, angiotensinogen gene knockout mice, renin-angiotensin system, losartan
Previously, the function of the renin-angiotensin system (RAS) was regarded as control of vascular tonus and maintenance of fluid homeostasis by regulation of water and electrolyte absorption and/or excretion in the kidney and gastrointestinal tract.1,2 However, recent studies have shown that angiotensin II (AII), an octapeptide which is the major biologically active component of the RAS, is also involved in other actions, such as apoptosis, vascular remodelling, and inflammation.3–5
Recently, the potent proinflammatory effects of the RAS have been reported in various types of immune mediated diseases. For example, in mouse antiglomerular basement membrane (GBM) nephritis, AII contributes to expression of glomerular monocyte chemoattractant protein-1 (MCP-1) via the angiotensin II type 1a (AT1a) receptor, resulting in the pathogenesis of anti-GBM nephritis.6 Similarly, in mouse immune complex nephritis induced by intraperitoneal ovalbumin injection, treatment with an angiotensin converting enzyme (ACE) inhibitor, quinapril, reduced MCP-1 expression in the kidney and mononuclear cell infiltration, supporting the hypothesis that the RAS is involved in the pathogenesis of immune complex nephritis.7 Furthermore, in in vitro experiments, AII, acting through nuclear factor κB (NFκB) activation, stimulated the production of inflammatory mediators such as tumour necrosis factor α (TNF-α), transforming growth factor β1, and MCP-1 by mesangial cells.7,8 Stimulation of human monocytes with AII causes activation of NFκB.9 AII can also activate NFκB in cultured vascular smooth muscle cells.10 In some cell types, stimulation of AT1 receptors by AII is coupled to activation of the JAK/STAT kinase systems11–13 which are also involved in the signal transduction of T cell cytokines such as interleukin (IL)-2 and interferon γ (IFN-γ).14 There are also some reports that the calcineurin dependent nuclear factor of activated T cells pathway is more important for regulation of cellular immune responses by AII signals through the AT1 receptor.15–17 These reports are highly suggestive that the RAS is a potent inflammatory mediator.
On the other hand, there is increasing evidence concerning the efficacy of RAS antagonism in immune mediated diseases, such as myocarditis, chronic allograft rejection, and anti-GBM nephritis in humans and other animals.6,7,18–24 These reports also indicate the proinflammatory effect of the RAS.
In the classical pathway of AII synthesis, the precursor of AII is angiotensinogen, which is principally produced in the liver. Angiotensinogen is then subsequently cleaved by renin into angiotensin I (AI). ACE converts AI into AII which binds to its specific membrane receptors, angiotensin II type 1 (AT1) and type 2 (AT2) receptors, expressed by various cells, including inflammatory cells such as T cells and macrophages. In addition to the classical pathway of AII synthesis, tissue renin-angiotensin systems have been identified in many organs, suggesting that various tissues have the ability to synthesise AII independently of circulating RAS. Tissue specific RAS may exist in the kidney, brain, aorta, adrenal gland, heart, stomach, and colon.25–28 Hirasawa and colleagues28 have reported colonic localisation of RAS. AT1 receptors were found in vessel walls, myofibroblasts, macrophages, and surface epithelium, and AT2 receptors in mesenchymal cells and weakly in parts of the surface epithelium. Renin and ACE were distributed in vessel walls, mesenchymal cells, and in parts of the surface epithelium. However, the functions of these local RAS have not been determined.
To our knowledge, there has been no report on the immunological modulating effect of the RAS in the colon. To elucidate the pathophysiological role of AII in immune mediated enteropathy, we induced 2,4,6-trinitrobenzene sulphonic acid (TNBS) colitis,29 an immune mediated acute colitis, in angiotensinogen deficient homozygous (Atg−/−) and wild-type (Atg+/+) mice. Using this model, we evaluated the role of AII in the pathogenesis of immune mediated colitis. We also treated TNBS colitis mice with an AT1 specific angiotensin II receptor blocker (ARB), losartan, to examine the feasibility of RAS inhibitors (ACE inhibitors and ARB) for the treatment of inflammatory bowel disease.
METHODS
Mice
Mice homozygous for targeted disruption of the angiotensinogen gene (Atg−/−) were generated from Jcl:ICR (ICR) mice at the Institute of Applied Biochemistry, University of Tsukuba (Ibaraki, Japan), as described previously.30 They were backcrossed seven times to the ICR strain. These animals were bred and maintained in the animal research facility at Yokohama City University, Graduate School of Medicine, under standard conditions. Male ICR mice were purchased from Japan Clea (Tokyo, Japan). Mice were housed in the animal research facility at Yokohama City University, Graduate School of Medicine, under standard environmental conditions for at least four days. All animals received standard pelleted chow and tap water ad libitum.
Injection of losartan
For the ARB treatment study, eight week old male ICR mice were purchased from Japan Clea. They were given losartan (Lsr; Merck, USA) 30 μg/kg/day subcutaneously from 21 days before instillation of TNBS or phosphate buffered saline (PBS), using a continuous infusion pump (mini-osmotic pump; Alzet, USA), to examine the prophylactic effect of Lsr. Control mice received 0.9% normal saline (NS) subcutaneously.
Similarly, to examine the therapeutic effect of Lsr, eight week old male ICR mice were given Lsr orally at 10 μg/kg/day every 24 hours from two hours after TNBS or PBS administration. Control mice received 0.9% NS orally. For oral administration, a polyethylene catheter was used (flexible intraoral tube; Japan Clea).
Induction of colitis
TNBS colitis was induced in eight week old male ICR and Atg−/− mice and 11 week old male ICR mice, after subcutaneous pretreatment with Lsr or NS for three weeks, as described previously.29 Briefly, mice were anaesthetised by intraperitoneal injection of xyladine (50 mg/kg) and ketamine (50 mg/kg). A polyethylene catheter (flexible intraoral tube; Japan Clea) was inserted into the colon 3.5 cm from the anal verge and a 100 μl enema of TNBS (Wako Chemical, Japan) in 40% ethanol was then infused into the colonic lumen through a 1 ml syringe (Terumo, Japan) connected to the polyethylene catheter. In our preliminary examination, the liquid instilled reached almost the entire way along the colon. Our preliminary experiments also showed that a TNBS dose of 5 mg/mouse in the ICR strain produced a good inflammatory response adequate for laboratory investigations. Subsequently, this dose of TNBS was used for all experiments. Control mice received 100 μl PBS.
Assessment of inflammation
Body weight of the mice was recorded every day for assessment of wasting. Three days after TNBS or PBS enema, mice were sacrificed by cervical dislocation under light anaesthesia induced by inhalation of chloroform. After collecting blood samples, the colon was removed and washed with PBS to remove faecal material. The distal 1.5 cm of the colon was used for histological examination. The rest of the colon was used for determination of local cytokine levels. Briefly, the colon was homogenised in ice cold tissue protein extraction reagent (Pierce, USA) containing phenylmethylsulfonyl fluoride (10 μg/ml), aprotinin (10 μg/ml), leupeptin (10 μg/ml), N-p-tosyl-L-lysine chloromethyl ketone (10 μg/ml), and L-1-tosylamiido-2-phenylethyl chloromethyl ketone (10 μg/ml). The homogenate was then centrifuged at 18 000 g for 20 minutes at 4°C. The supernatant was stored at −80°C until use. Then, TNF-α, IL-1β, IFN-γ, IL-4, and IL-10 levels were assayed using commercially available enzyme linked immunosorbent assay (ELISA) kits (R&D Systems, USA). Also, total protein concentration was assayed using the Bradford method. In addition, corticosterone levels in serum samples were assayed by radioimmunoassay (SRL, Japan).
Histological scoring
The distal 1.5 cm of the colon was used for histological assessment. Haematoxylin-eosin staining was performed in the usual way. Sections were scored blindly based on a semiquantitative scoring system, as described previously,31 where features were graded as follows: extent of destruction of normal mucosal architecture (0, normal; 1, mild; 2, moderate; and 3, extensive damage), presence and degree of cellular infiltration (0, normal; 1, mild; 2, moderate; and 3, transmural infiltration), degree of muscle thickening (0, normal; 1, mild; 2, moderate; and 3, marked thickening), presence or absence of crypt abscesses (0, absent; 1, present), and the presence or absence of goblet cell depletion (0, absent; 1, present). The scores for each feature were summed, with a maximum possible score of 11.
Statistical analyses
Results are expressed as mean (SEM). Group data for Atg+/+ and Atg−/− were compared using the Mann-Whitney U test. Differences were considered significant at p<0.05. For the Lsr treatment study, multiple comparisons were carried out by one way analysis of variance and the Tukey-Kramer method for post hoc analyses. Statistical significance was accepted at p<0.05.
RESULTS
Body weight loss was less in Atg−/− mice
Before TNBS was given, Atg+/+ mice were significantly heavier than Atg−/− mice (36.8 (0.5) v 31.4 (0.6) g; p<0.0001), as reported previously.32 In Atg+/+ mice, intrarectal instillation of TNBS resulted in diarrhoea and wasting while in Atg−/− mice there was no significant body weight loss compared with control mice that received PBS. Moreover, body weight loss after TNBS instillation was significantly milder than that in Atg+/+ mice, from day 1 to day 3 after TNBS treatment (fig 1 ▶). These findings demonstrate that weight loss due to TNBS induced colitis in Atg−/− mice is ameliorated compared with that in Atg+/+ mice.
Figure 1.
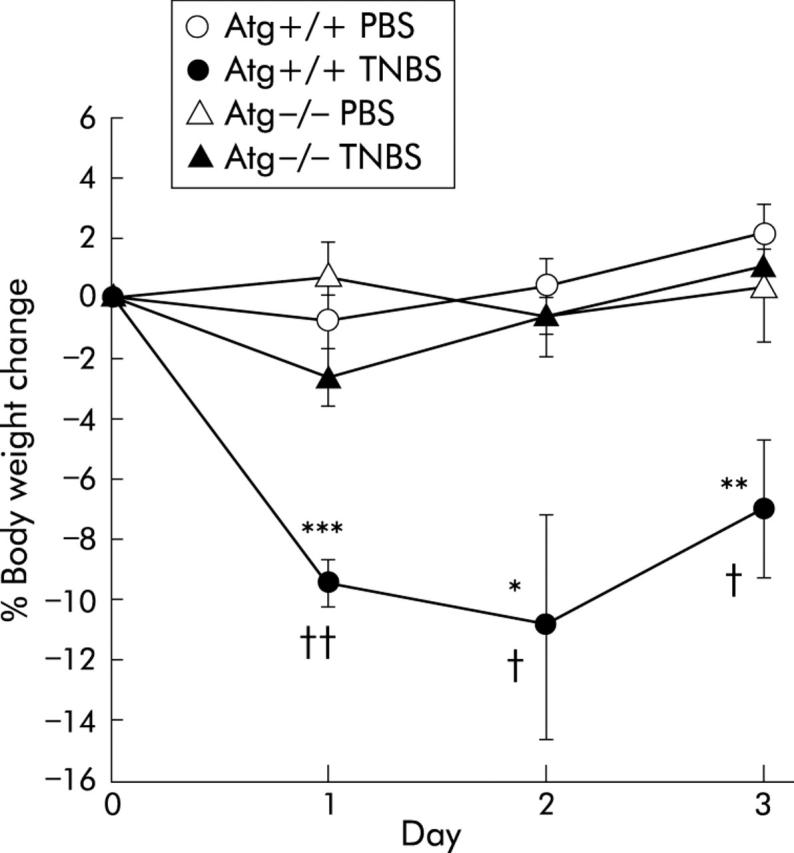
Changes in body weight after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS). Results are shown for PBS instilled wild-type mice (Atg+/+) (n = 9) and angiotensinogen gene knockout mice (Atg−/−) (n = 7) and for TNBS instilled Atg+/+ (n = 13) and Atg−/− (n = 12) mice. Results are expressed as mean (SEM). *p<0.05, **p<0.01, ***p<0.001 versus PBS instilled Atg+/+ mice; †p<0.05, ††p<0.01 versus TNBS instilled Atg−/− mice.
Induction of inflammatory cytokines is prevented in Atg−/− mice
To investigate the role of the RAS in mucosal immunity, local cytokine levels (TNF-α, IL-1β, IFN-γ, IL-4, and IL-10) were quantified by ELISA (fig 2 ▶).
Figure 2.
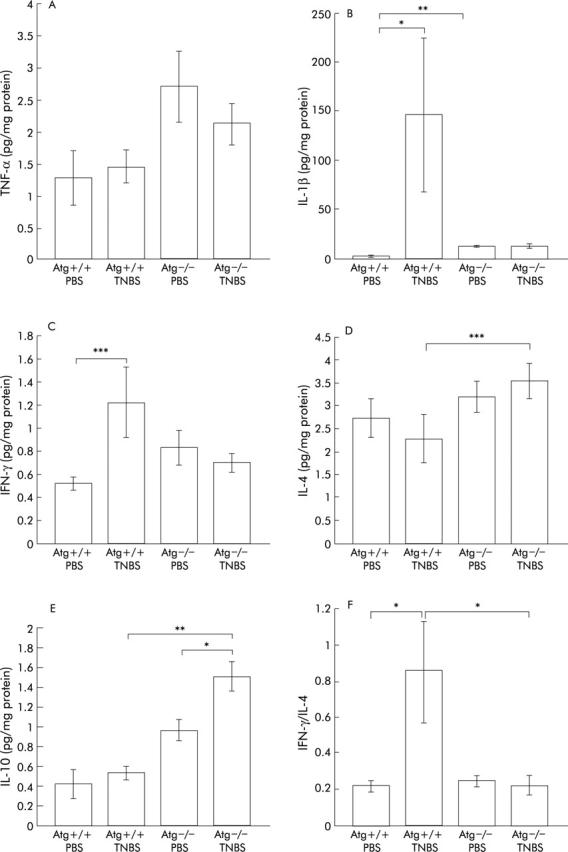
Levels of cytokines in the colon assayed by ELISA. tumour necrosis factor α (TNF-α) (A), interleukin (IL)-1β (B), interferon γ (IFN-γ) (C), IL-4 (D), and IL-10 (E) levels were measured three days after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS). Bars represent PBS instilled wild-type mice (Atg+/+) (n = 8), PBS instilled angiotensinogen gene knockout mice (Atg−/−) (n = 7), TNBS instilled Atg+/+ (n = 13), and TNBS instilled Atg−/− (n = 12), as indicated. To investigate the proinflammatory and anti-inflammatory cytokine balance, the IFN-γ/IL-4 ratio was also calculated (F). The IFN-γ level of each mouse was divided by the IL-4 level of the same mouse and the mean calculated. Results are expressed as mean (SEM). *p<0.05, **p<0.01, ***p<0.005.
In Atg+/+ mice, as expected, levels of proinflammatory cytokines such as IL-1β and IFN-γ were significantly elevated in the TNBS instilled group compared with the PBS group (fig 2B ▶, C). In contrast, there were no differences in colonic TNF-α, IL-4, and IL-10 levels between the TNBS and PBS instilled groups (fig 2A ▶, D, E).
In Atg−/− mice, IL-10 levels were higher in the TNBS instilled group than in the PBS group (fig 2E ▶). Moreover, there were no significant differences in levels of colonic proinflammatory cytokines such as TNF-α, IL-1β, and IFN-γ between the PBS instilled and TNBS instilled groups (fig 2A ▶–C), indicating that little inflammatory change was induced by TNBS instillation in Atg−/− mice.
On the other hand, levels of cytokines such as IL-4 and IL-10 in colonic tissue were higher in Atg−/− mice than in Atg+/+ mice after TNBS instillation (fig 2D ▶, E). There was a tendency for IL-1β and IFN-γ levels to be higher in TNBS instilled Atg+/+ mice than in Atg−/− mice with TNBS colitis (fig 2B ▶, C). In the two PBS instilled groups, the level of IL-1β in Atg−/− mice was higher than that in Atg+/+ mice (fig 2B ▶).
In acute colitis such as TNBS colitis, macrophages are thought to be involved. IFN-γ acts on macrophages as a stimulator and IL-4 acts as a suppressor.33,34 To investigate the condition of the colon, the IFN-γ/IL-4 ratio was also calculated (fig 2F ▶). In Atg−/− mice, there was no significant difference between the PBS instilled and TNBS instilled groups. Furthermore, Atg−/− mice showed a lower ratio than Atg+/+ mice after TNBS instillation. These results indicate that the RAS is directly involved in upregulation of proinflammatory cytokines in the inflamed colon, and the colon of Atg−/− mice was resistant to TNBS colitis.
Histological damage is milder in Atg−/− mice
Atg−/− and Atg+/+ mice were given TNBS or PBS, and three days later the colons of these mice were examined histologically (figs 3 ▶, 4 ▶). In TNBS instilled Atg+/+ mice, ulceration with marked inflammatory infiltrates was observed (fig 3C ▶, D). Fibrosis was also present. These histological changes were markedly reduced in Atg−/− mice (fig 3G ▶, H). In PBS instilled Atg+/+ and Atg−/− mice, no histological alteration was observed (fig 3A ▶, B, E, F). The degree of colitis was quantitatively assessed using the scoring system described in materials and methods (fig 4 ▶). In Atg−/− mice, the histological score after TNBS instillation was significantly lower than that in Atg+/+ mice (fig 4 ▶). These results are consistent with the changes in colonic levels of cytokines described above, indicating that Atg−/− mice are resistant to TNBS colitis.
Figure 3.
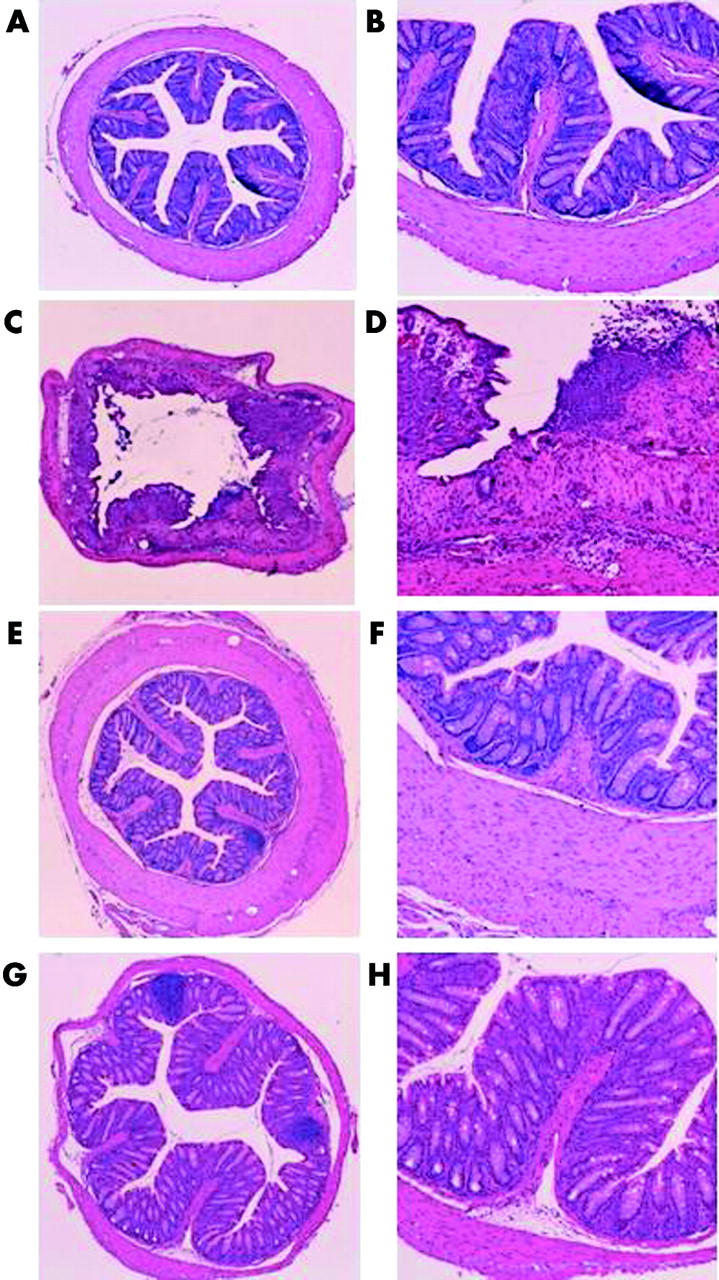
Histological findings in the colon three days after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS). (C, D) TNBS instilled wild-type mice (Atg+/+), (G, H) TNBS instilled angiotensinogen gene knockout mice (Atg−/−), (A, B) PBS instilled Atg+/+ (E, F), and PBS instilled Atg−/− mice. Magnification: A, C, E, G ×25; B, D, F, H ×100.
Figure 4.

Histological damage score of the colon. Severity of inflammation was scored as described in materials and methods. Bars represent phosphate buffered saline (PBS) instilled wild-type mice (Atg+/+) (n = 5), PBS instilled angiotensinogen gene knockout mice (Atg−/−) (n = 4), 2,4,6-trinitrobenzene sulphonic acid (TNBS) instilled Atg+/+ (n = 13), and TNBS instilled Atg−/− (n = 9), as indicated. *p<0.05, **p<0.005.
No significant difference in wasting between NS treated and Lsr treated mice
To investigate the prophylactic effect of Lsr against TNBS colitis, mice were pretreated with Lsr for 21 days before instillation of TNBS. Before induction of TNBS colitis, there was no significant difference in body weight between Lsr treated mice and NS treated mice. After TNBS instillation, both Lsr treated mice and NS treated mice showed diarrhoea and body weight loss. There was no significant difference in body weight loss between Lsr treated and NS treated mice with TNBS colitis (fig 5 ▶).
Figure 5.

Changes in body weight after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS). The groups were: PBS instilled 0.9% normal saline (NS) treated (n = 5) mice, PBS instilled losartan (Lsr) treated (n = 5) mice, TNBS instilled NS treated (n = 7) mice, and TNBS instilled Lsr treated (n = 9) mice. Results are expressed as mean (SEM). *Significant difference versus PBS instilled NS treated mice; †significant difference versus PBS instilled Lsr treated mice.
Additionally, to investigate the therapeutic effect of Lsr, we administered Lsr orally after TNBS or PBS instillation. There was slight but significant suppression of body weight loss in the Lsr treated group one day after TNBS instillation (fig 6A ▶). However, there was no significant difference between TNBS instilled NS treated mice and TNBS instilled Lsr treated mice after two and three days.
Figure 6.
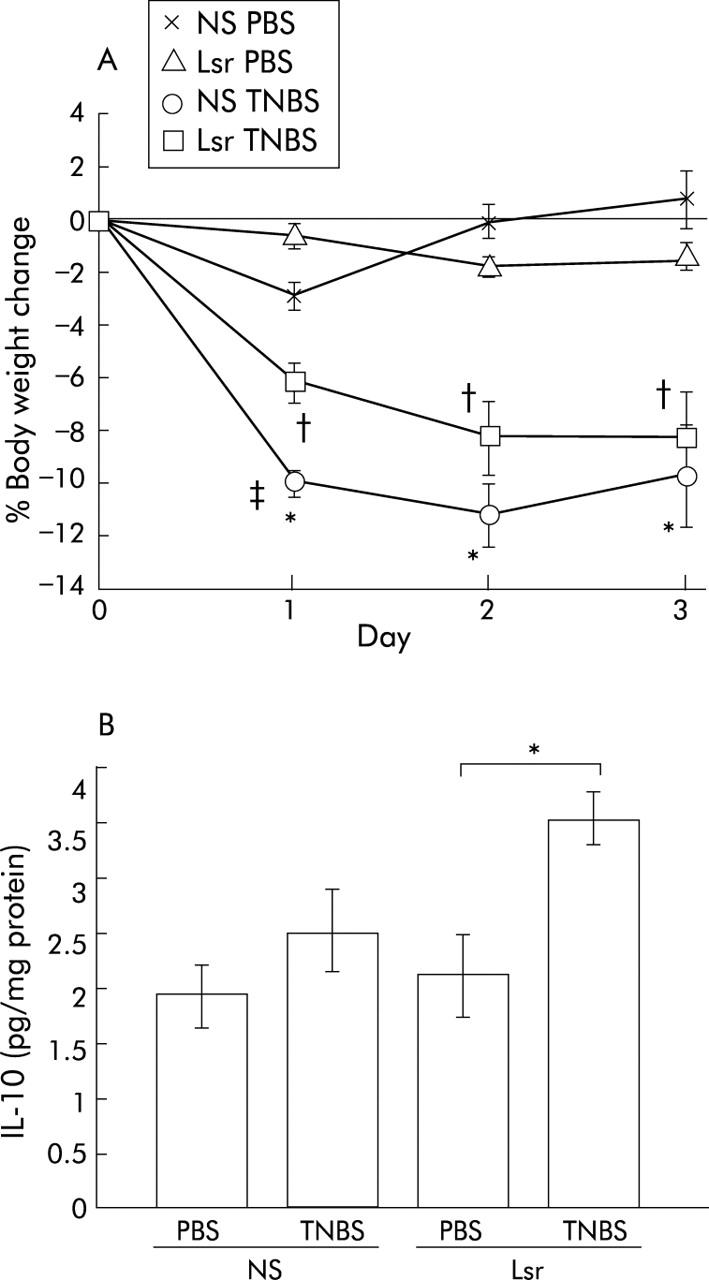
Results of oral administration of losartan (Lsr) after 2,4,6-trinitrobenzene sulphonic acid (TNBS) instillation. Body weight change (A) and interleukin 10 (IL-10) levels in the colon (B) are shown. (A) The groups were: phosphate buffered saline (PBS) instilled 0.9% normal saline (NS) treated (n = 5) mice, PBS instilled Lsr treated (n = 5) mice, TNBS instilled NS treated (n = 8) mice, and TNBS instilled Lsr treated (n = 11) mice. Results are expressed as mean (SEM). *Significant difference versus PBS instilled NS treated mice; †significant difference versus PBS instilled Lsr treated mice;‡ significant difference versus TNBS instilled Lsr treated mice. (B) IL-10 levels in NS treated PBS instilled (n = 5), NS treated TNBS instilled (n = 8), Lsr treated PBS instilled (n = 5), and Lsr treated TNBS instilled (n = 11) mice, as indicated. *p<0.05.
Induction of inflammatory cytokines is prevented by losartan
Colonic cytokine levels quantified by ELISA revealed that in NS treated mice proinflammatory cytokines such as IL-1β and IFN-γ were significantly elevated in the TNBS instilled group compared with the PBS group (fig 7B ▶, C). In contrast, in Lsr treated mice, there were no significant differences in levels of colonic proinflammatory cytokines such as TNF-α, IL-1β, and IFN-γ between the TNBS and PBS instilled groups (fig 7A ▶–C). Moreover, levels of IL-1β and IFN-γ were significantly lower in Lsr treated than in NS treated mice after TNBS instillation (fig 7B ▶, C) while there was no difference between the two PBS instilled groups. The IFN-γ/IL-4 ratio was also calculated (fig 7F ▶). Although there were no significant differences among the four groups, the ratio in NS treated mice with TNBS instillation tended to be higher than in the others. These results indicate that long term administration of Lsr significantly downregulated levels of proinflammatory cytokines in TNBS colitis. However, in the oral treatment study, only IL-10 levels were significantly increased in Lsr treated mice after TNBS instillation (fig 6B ▶) and there were no significant differences in other cytokine levels among the four groups (data not shown).
Figure 7.
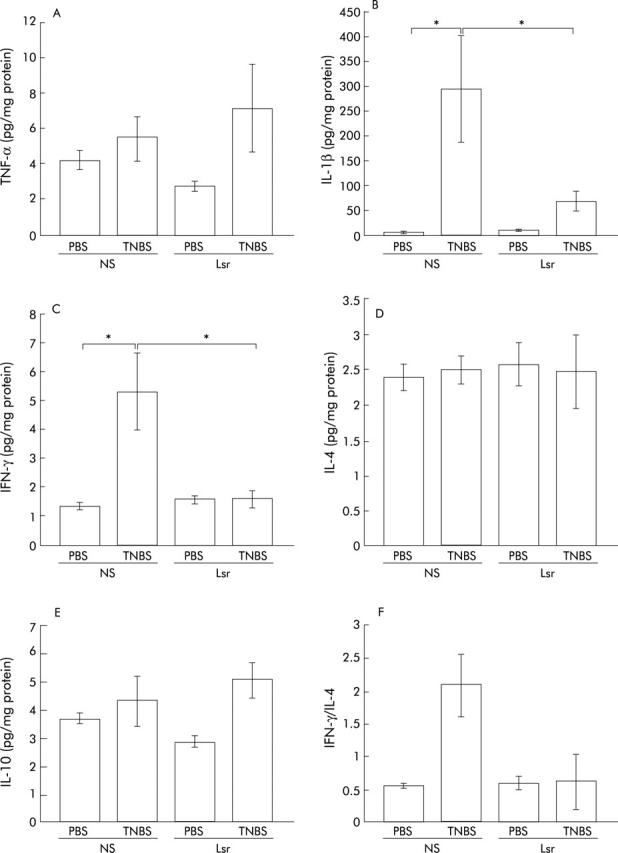
Levels of cytokines in the colon in the subcutaneous administration study. tumour necrosis factor α (TNF-α) (A), interleukin (IL)-1β (B), interferon γ (IFN-γ) (C), IL-4 (D), and IL-10 (E) levels were assayed by ELISA three days after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS). Bars represent 0.9% normal saline (NS) treated PBS instilled (n = 5), NS treated TNBS instilled (n = 7), losartan (Lsr) treated PBS instilled (n = 5), and Lsr treated TNBS instilled (n = 9) mice, as indicated. The IFN-γ/IL-4 ratio was also calculated (F). The IFN-γ level of each mouse was divided by the IL-4 level of the same mouse and the mean calculated. Results are expressed as mean (SEM). *p<0.05.
No difference in corticosterone levels between NS treated and Lsr treated mice
Because there are multiple links between the RAS and the hypothalamic-pituitary-adrenal axes, we assayed corticosterone levels in serum samples in the subcutaneous treatment study to investigate whether or not the immunomodulating effect of the RAS is mediated by the hypothalamic-pituitary axis (table 1 ▶). There was no significant difference between NS treated and Lsr treated mice. This suggests that the immunomodulating effect of the RAS is not mediated by the hypothalamic-pituitary axis.
Table 1.
Corticosterone levels in serum samples
| NS treated | Lsr treated | |
| PBS instilled | 191.14 (16.35) | 315.02 (62.67) |
| TNBS instilled | 299.59 (73.40) | 387.68 (63.82) |
Data are expressed as mean (SEM) (ng/ml). There were no significant differences between the four groups.
PBS, phosphate buffered saline; TNBS, 2,4,6-trinitrobenzene sulphonic acid; NS, 0.9% normal saline; Lsr, losartan.
Histological damage is milder in Lsr treated mice
In NS treated TNBS colitis mice, ulceration with marked infiltration of inflammatory cells was observed (fig 8C ▶, D). While Lsr treated mice also showed similar inflammatory changes, these histological changes were reduced (fig 8G ▶, H). Histological score showed lower activity of TNBS colitis in Lsr treated mice than in NS treated mice (fig 9 ▶). In particular, scores for the features of presence and degree of cellular infiltration, degree of muscle thickening, and presence or absence of goblet cell depletion in Lsr treated mice were significantly lower than those in NS treated mice with TNBS instillation (1.56 (0.29) v 2.57 (0.20), 1.56 (0.18) v 2.71 (0.18), 0 v 0.43 (0.20), respectively). Both Lsr treated and NS treated mice with PBS instillation showed almost normal features (fig 8A ▶, B, E, F).
Figure 8.
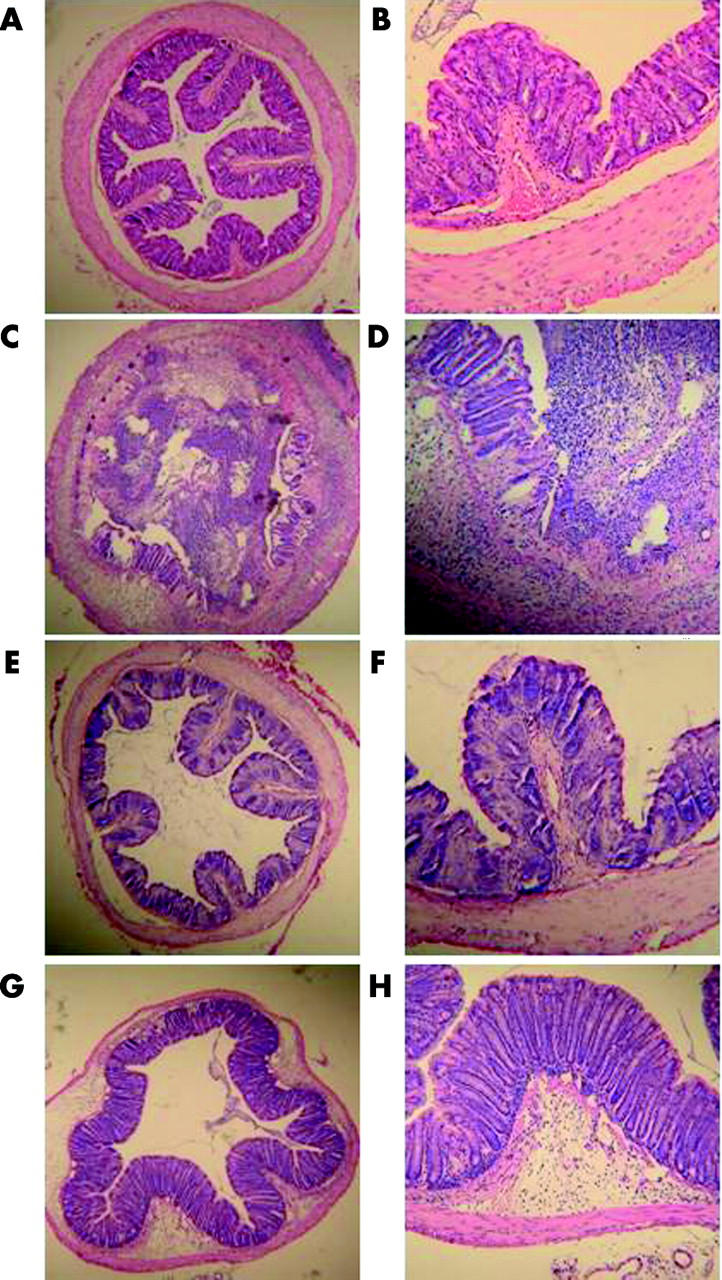
Histological findings in the colon three days after administration of phosphate buffered saline (PBS) or 2,4,6-trinitrobenzene sulphonic acid (TNBS) in the subcutaneous administration study. (A, B) 0.9% Normal saline (NS) treated PBS instilled mice, (C, D) NS treated TNBS instilled mice, (E, F) losartan (Lsr) treated PBS instilled mice, and (G, H) Lsr treated TNBS instilled mice. Magnification: A, C, E, G ×25; B, D, F, H ×100.
Figure 9.

Histological damage score of the colon in the subcutaneous administration study. Severity of inflammation was scored as described in materials and methods. Bars represent 0.9% normal saline (NS) treated phosphate buffered saline (PBS) instilled (n = 5) mice, NS treated 2,4,6-trinitrobenzene sulphonic acid (TNBS) instilled (n = 7) mice, losartan (Lsr) treated PBS instilled (n = 5) mice, and Lsr treated TNBS instilled (n = 9) mice, as indicated. *p<0.05.
DISCUSSION
In this study we assessed the role of the RAS in a murine model of intestinal inflammation using mice genetically deficient in angiotensinogen, which is the precursor of AII. Under control conditions with PBS instillation, IL-1β was increased in baseline knockout mice compared with wild-type baseline mice. Despite elevated baseline levels of this proinflammatory cytokine, these mice had no histological evidence of inflammation. Under inflamed conditions induced by TNBS instillation, Atg−/− mice exhibited reduced activity of intestinal inflammation in contrast with Atg+/+ mice. Although Atg+/+ mice showed severe body weight loss and elevation of mucosal proinflammatory cytokines, Atg−/− mice did not exhibit these changes. Consistently, histological examination showed significantly reduced inflammatory activity in Atg−/− mice. Furthermore, expression of cytokines such as IL-4 and IL-10 was predominant in the colon of Atg−/− mice compared with Atg+/+ mice, and the IFN-γ/IL-4 ratio was lower in Atg−/− mice, indicating that the RAS is involved in the pathogenesis of TNBS colitis via regulation of proinflammatory cytokines. This immunomodulating effect of the RAS may be due to AII which is the major active component of the RAS.
To determine which receptor is involved in the immunomodulating effect, we then treated TNBS colitis mice with an AT1 specific ARB, Lsr. We confirmed that the AT1 receptor is involved in this immunomodulating effect of AII by showing the effectiveness of Lsr subcutaneous infusion against TNBS colitis. As in Atg−/− mice, Lsr treated mice showed lower colonic levels of proinflammatory cytokines (IL-1β and IFN-γ) than NS treated mice, which resulted in reduced histological activity of TNBS colitis.
However, in the oral administration study, which examined the therapeutic effect of AT1 blockade, only IL-10 levels were significantly increased in Lsr treated mice after TNBS instillation, and there were no significant differences among the four groups for other cytokine levels. This suggests that in the TNBS colitis model, which is an acute colitis model, AT1 inhibition does not have sufficient therapeutic efficacy although it has a distinct prophylactic effect. As it takes approximately one week for Lsr to have any effect,35 and 3–6 weeks to have its maximal effect36–38 in vivo, the negative results in the oral administration study do not necessarily indicate that Lsr has no therapeutic effect as the study examined only the short term effects of Lsr. Long term administration may be needed for the treatment of chronic colitis.
Although previous reports showed an increase in TNF-α levels after TNBS instillation, we did not observe this in present study or in our previous study39 using BALB/c mice. The reason is unclear but we may speculate that the timing of sacrifice, degree of colitis, and genetic background of the mice would strongly affect the response of TNF-α to TNBS instillation.
Concerning the immunomodulating effect of the RAS, some reports have shown an effect of AII on inflammatory cells. Nataraj and colleagues17 reported that AII, acting through AT1 receptors on the cell membrane of lymphocytes, induces a cellular immune response through a calcineurin dependent pathway. Fernandez-Castelo and colleagues40 reported Th1 predominance in human lymphocytes stimulated by AII, which is consistent with the report of Nataraj et al that AII induces a cellular immune response. A recent report by Shao and colleagues41 that splenic T cells of AII infused rats show Th1 predominance also supports the immunomodulating effect of the RAS.
There have been few reports on the effect of the RAS on the colon. Jaszewski and colleagues42 reported that AII, the major active component of the RAS, is upregulated in inflamed lesions in the colon of Crohn’s disease patients while Hirasawa and colleagues28 recently reported that all components of the RAS exist in the human colon. Now we have shown that blockade of AT1 receptors results in protection against TNBS colitis. In the colon, many cells such as vascular endothelial cells, colonic epithelial cells, and inflammatory cells express AII receptors. Which cells are the major target cells for Lsr in TNBS colitis is still unclear. We speculate that Lsr acts on each of these cells and the overall effects are expressed as reduced activity of TNBS colitis. To determine whether the AT2 receptor has any effect on TNBS colitis, further study is required. Similarly, additional studies using other chronic colitis models are needed to determine whether this immunomodulating effect of the RAS is limited to TNBS colitis, a model of acute colitis.
In conclusion, we demonstrated that the RAS is involved in TNBS colitis, and the absence of angiotensinogen resulted in amelioration of the effect of TNBS stimulation. Consistently, Lsr treatment suppressed TNBS colitis. Antagonism of the RAS may be a prophylactic strategy for immune mediated intestinal bowel disease.
Abbreviations
Atg−/−, angiotensinogen gene knockout mice
Atg+/+, wild-type mice
TNBS, 2,4,6-trinitrobenzene sulphonic acid
RAS, renin-angiotensin system
AI, angiotensin I
AII, angiotensin II
ACE, angiotensin converting enzyme
AT1 receptor, angiotensin II type 1 receptor
AT2 receptor, angiotensin II type 2 receptor
AT1a receptor, angiotensin II type 1a receptor
ARB, angiotensin II receptor blocker
GBM, glomerular basement membrane
ELISA, enzyme linked immunosorbent assay
MCP-1, monocyte chemoattractant protein 1
NFκB, nuclear factor κB
TNF-α, tumour necrosis factor α
IL, interleukin
IFN-γ, interferon γ
Lsr, losartan
NS, 0.9% normal saline
PBS, phosphate buffered saline
Conflict of interest: None declared.
REFERENCES
- 1.Levens NR. Local control of jejunal absorption by the renin-angiotensin system. J Cardiovasc Pharmacol 1986;8 (suppl 10) :S17–22. [DOI] [PubMed] [Google Scholar]
- 2.Levens NR. Control of intestinal absorption by the renin-angiotensin system. Am J Physiol 1985;249:G3–15. [DOI] [PubMed] [Google Scholar]
- 3.Rossig L, Dimmeler S, Zeiher AM. Apoptosis in the vascular wall and atherosclerosis. Basic Res Cardiol. 2001;96 :11–22. [DOI] [PubMed]
- 4.Wesselman JP, De Mey JG. Angiotensin and cytoskeletal proteins: role in vascular remodeling. Curr Hypertens Rep. 2002;4 :63–70. [DOI] [PubMed]
- 5.Wolf G, Wenzel U, Burns KD, et al. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors. Kidney Int. 2002;61 :1986–95. [DOI] [PubMed]
- 6.Hisada Y, Sugaya T, Yamanouchi M, et al. Angiotensin II plays a pathogenic role in immune-mediated renal injury in mice. J Clin Invest 1999;103:627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz-Ortega M, Bustos C, Hernandez-Presa MA, et al. Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-κB activation and monocyte chemoattractant protein-1 synthesis. J Immunol 1998;161:430–9. [PubMed] [Google Scholar]
- 8.Klahr S, Morrissey J. Angiotensin II and gene expression in kidney. Am J Kidney Dis 1998;31:171–6. [DOI] [PubMed] [Google Scholar]
- 9.Kranzhofer R, Browatzki M, Schmidt J, et al. Angiotensin II activates the proinflammatory transcription factor nuclear factor-κB in human monocytes. Biochem Biophys Res Commun 1999;257:826–8. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Ortega M, Lorenzo O, Ruperez M, et al. Angiotensin II activates nuclear transcription factor κB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ Res 2000;86:1266–72. [DOI] [PubMed] [Google Scholar]
- 11.Marrero MB, Schieffer B, Paxton WG, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995;375:247–50. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein K, Ali M, Sayeski P, et al. New insights into the cellular signaling of seven transmembrane receptors: the role of tyrosine phosphorylation. Lab Invest 1998;78:3–7. [PubMed] [Google Scholar]
- 13.Duff JL, Marrero MB, Paxton WG, et al. Angiotensin II signal transduction and the mitogen-activated protein kinase pathway. Cardiovasc Res 1995;30:511–17. [PubMed] [Google Scholar]
- 14.Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Curr Opin Immunol 1998;10:271–8. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki Y, Gomez-Guerrero C, Shirato I, et al. Susceptibility to T cell-mediated injury in immune complex disease is linked to local activation of renin-angiotensin system: the role of NF-AT pathway. J Immunol 2002;169:4136–46. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki E, Nishimatsu H, Satonaka H, et al. Angiotensin II induces myocyte enhancer factor 2- and calcineurin/nuclear factor of activated T cell-dependent transcriptional activation in vascular myocytes. Circ Res 2002;90:1004–11. [DOI] [PubMed] [Google Scholar]
- 17.Nataraj C, Oliverio MI, Mannon RB, et al. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 1999;104:1693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuzawa M, Satoh J, Sagara M, et al. Angiotensin converting enzyme inhibitors suppress production of tumor necrosis factor-α in vitro and in vivo. Immunopharmacology 1997;36:49–55. [DOI] [PubMed] [Google Scholar]
- 19.Andersson P, Cederholm T, Johansson AS, et al. Captopril-impaired production of tumor necrosis factor-α-induced interleukin-1β in human monocytes is associated with altered intracellular distribution of nuclear factor-κB. J Lab Clin Med 2002;140:103–9. [DOI] [PubMed] [Google Scholar]
- 20.Furukawa Y, Matsumori A, Hirozane T, et al. Angiotensin II receptor antagonist TCV-116 reduces graft coronary artery disease and preserves graft status in a murine model. Circulation 1996;93:333–9. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka A, Matsumori A, Wang W, et al. An angiotensin II receptor antagonist reduces myocardial damage in an animal model of myocarditis. Circulation 1994;90:2051–5. [DOI] [PubMed] [Google Scholar]
- 22.Amuchastegui SC, Azzollini N, Mister M, et al. Chronic allograft nephropathy in the rat is improved by angiotensin II receptor blockade but not by calcium channel antagonism. J Am Soc Nephrol 1998;9:1948–55. [DOI] [PubMed] [Google Scholar]
- 23.Benediktsson H, Chea R, Davidoff A, et al. Antihypertensive drug treatment in chronic renal allograft rejection in the rat. Effect on structure and function. Transplantation 1996;62:1634–42. [DOI] [PubMed] [Google Scholar]
- 24.Mackenzie HS, Ziai F, Nagano H, et al. Candesartan cilexetil reduces chronic renal allograft injury in Fisher-Lewis rats. J Hypertens Suppl 1997;15:S21–5. [DOI] [PubMed] [Google Scholar]
- 25.Campbell DJ, Habener JF. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J Clin Invest 1986;78:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul M, Wagner J, Dzau VJ. Gene expression of the renin-angiotensin system in human tissues. Quantitative analysis by the polymerase chain reaction. J Clin Invest 1993;91:2058–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips MI, Speakman EA, Kimura B. Levels of angiotensin and molecular biology of the tissue renin angiotensin systems. Regul Pept 1993;43:1–20. [DOI] [PubMed] [Google Scholar]
- 28.Hirasawa K, Sato Y, Hosoda Y, et al. Immunohistochemical localization of angiotensin II receptor and local renin-angiotensin system in human colonic mucosa. J Histochem Cytochem 2002;50:275–82. [DOI] [PubMed] [Google Scholar]
- 29.Elson CO, Sartor RB, Tennyson GS, et al. Experimental models of inflammatory bowel disease. Gastroenterology 1995;109:1344–67. [DOI] [PubMed] [Google Scholar]
- 30.Tanimoto K, Sugiyama F, Goto Y, et al. Angiotensinogen-deficient mice with hypotension. J Biol Chem 1994;269:31334–7. [PubMed] [Google Scholar]
- 31.McCafferty DM, Miampamba M, Sihota E, et al. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut 1999;45:864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massiera F, Seydoux J, Geloen A, et al. Angiotensinogen-deficient mice exhibit impairment of diet-induced weight gain with alteration in adipose tissue development and increased locomotor activity. Endocrinology 2001;142:5220–5. [DOI] [PubMed] [Google Scholar]
- 33.Varesio L, Blasi E, Thurman GB, et al. Potent activation of mouse macrophages by recombinant interferon-gamma. Cancer Res 1984;44:4465–9. [PubMed] [Google Scholar]
- 34.Oswald IP, Gazzinelli RT, Sher A, et al. IL-10 synergizes with IL-4 and transforming growth factor-beta inhibit macrophage cytotoxic activity. J Immunol 1992;148:3578–82. [PubMed] [Google Scholar]
- 35.Gradman AH, Arcuri KE, Goldberg AI, et al. A randomized, placebo-controlled, double-blind, parallel study of various doses of losartan potassium compared with enalapril maleate in patients with essential hypertension. Hypertension 1995;25:1345–50. [DOI] [PubMed]
- 36.Ikeda LS, Harm SC, Arcuri KE, et al. Comparative antihypertensive effects of losartan 50 mg and losartan 50 mg titrated to 100 mg in patients with essential hypertension. Blood Press 1997;6:35–43. [DOI] [PubMed] [Google Scholar]
- 37.Byyny RL. Antihypertensive efficacy of the angiotensin II AT1-receptor antagonist losartan: results of a randomized, double-blind, placebo-controlled, parallel-group trial using 24-hour blood pressure monitoring. Ambulatory Blood Pressure Monitoring Study Group. Blood Press Suppl 1996;2:71–7. [PubMed] [Google Scholar]
- 38.Dahlof B, Keller SE, Makris L, et al. Efficacy and tolerability of losartan potassium and atenolol in patients with mild to moderate essential hypertension. Am J Hypertens 1995;8:578–83. [DOI] [PubMed] [Google Scholar]
- 39.Morohashi T, Iwabushi K, Watano K, et al. Allograft inflammatory factor-1 regulates trinitrobenzene sulphonic acid-induced colitis. Immunology 2003;110:112–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Castelo S, Arzt ES, Pesce A, et al. Angiotensin II regulates interferon-γ production. J Interferon Res 1987;7:261–8. [DOI] [PubMed] [Google Scholar]
- 41.Shao J, Nangaku M, Miyata T, et al. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 2003;42:31–8. [DOI] [PubMed] [Google Scholar]
- 42.Jaszewski R, Tolia V, Ehrinpreis MN, et al. Increased colonic mucosal angiotensin I and II concentrations in Crohn’s colitis. Gastroenterology 1990;98:1543–8. [DOI] [PubMed] [Google Scholar]