

Abstract
Background and Aims: The roles of the virD4 and the cagG genes in the cag pathogenicity island of Helicobacter pylori for gastroduodenal pathogenesis are unclear and their roles in vivo have not been examined.
Methods: Seven week old male Mongolian gerbils were inoculated with the wild type H pylori TN2GF4, its isogenic virD4, or cagG mutants. Animals were sacrificed at 4, 12, and 24 weeks after inoculation. Gastric inflammation and H pylori density were evaluated by histology, inflammatory response (as measured by interleukin (IL)-1β mRNA levels), proliferative activity (as assessed by 5′-bromo-2′deoxyuridine labelling indices), and host systemic reaction (as measured by anti-H pylori IgG antibody).
Results: Degree of gastric inflammation, proliferative activity, and mucosal IL-1β mRNA levels remained low throughout the first 12 weeks in gerbils infected with the virD4 mutants. Degree of gastric inflammation and proliferative activity increased at 24 weeks with the virD4 mutants reaching levels comparative with those seen at four weeks with the wild-type strains. Mucosal IL-1β mRNA levels were also increased at 24 weeks with the virD4 mutants and levels at 24 weeks were similar between the wild-type and virD4 mutants. In contrast, gerbils infected with the cagG mutants had reduced ability to colonise gerbils, and no or little gastric inflammation or proliferative activity was observed.
Conclusions: Loss of the virD4 gene temporally retarded but did not abrogate gastric inflammation. Loss of the cagG gene abolished gastric inflammation partially via reduced ability to colonise gerbils. Unknown factors related to the type IV secretion system other than CagA may influence gastric inflammation.
Keywords: Helicobacter pylori, cag pathogenicity island, cagG, virD4, Mongolian gerbils
The presence of the cag pathogenicity island (PAI) in Helicobacter pylori is associated with increased mucosal inflammation and an increased risk of the development of gastric cancer or peptic ulcer disease.1–4 The cag PAI is a 40 kbp cluster of approximately 27 genes that encodes a type IV secretary apparatus (a molecular syringe) which injects the CagA protein and possibly other unknown proteins into eukaryotic cells.5,6,7,8,9,10,11 Defining the roles of the various genes in the cag PAI in the pathogenesis of H pylori related diseases is an area of active research interest. In vitro experiments using gastric cancer cells cocultured with H pylori indicate that several genes in the island are involved in induction of a proinflammatory cytokine; interleukin (IL)-8 (for example, cagE but not cagA).3,12 IL-8 is a potent neutrophil chemotactic and activating peptide produced by gastric epithelial cells and is thought to play a major role in the pathogenesis of H pylori associated diseases. Recent in vivo studies using Mongolian gerbils (Meriones unguiculatus) showed that cagE knockout mutants were associated with reduced gastric inflammation13–15 and did not induce gastric ulcers or gastric cancer.14 In contrast, cagA knockout mutants caused gastric inflammation similar to the parental strain.16 The in vivo function of other genes in the cag PAI has not been examined. This study therefore involves two genes in the cag PAI (virD4 and cagG), both of which have been suggested to play unique roles based on in vitro studies,12,17–19 but their roles in vivo have not been examined.
virD4 (hp0524; hp number from GenBank: AE000511) is one of seven genes in the cag PAI that are virulent (vir) gene homologues.20virD4 is a key component of the type IV secretion system. In the plant pathogen Agrobacterium tumefaciens, VirD4 is thought to mediate introduction of the nucleoprotein complex into the transporter by an energy dependent mechanism.11,21 In H pylori, VirD4 is thought to act as an adapter protein for the transfer of CagA protein and possibly other unknown proteins into the transfer channel formed by other Vir proteins in the cag PAI.17 This is based on previous reports showing that knockout of the virD4 gene resulted in loss of CagA translocation/phosphorylation as well as loss of H pylori induced host cytoskeletal rearrangement.17 Although the role of VirD4 in relation to IL-8 secretion from host cells remains unclear,12,17,18 the consensus is that loss of VirD4 does not parallel the reduction in IL-8 in contrast with other Vir factors in the cag PAI.
The second gene we examined was the cagG gene which is not a vir homologue gene but has weak homology to the flagellar motor switch protein gene or toxin coregulated pilus biosynthesis protein gene.3,20 The cagG gene has recently been reported to be involved in adherence to gastric epithelial cells.19 As the roles of these two gene have not been investigated in vivo, we used the Mongolian gerbil model to examine their functions in vivo in relation to gastric mucosal inflammation.
MATERIALS AND METHODS
Bacterial strains
We used a clinical isolate of H pylori strain TN2GF4 (kind gift from Masafumi Nakao, Takeda Chemical Industries Ltd, Osaka, Japan) and its isogenic knockout mutants for cagG and virD4. Strain TN2GF4 was isolated from Japanese gastric ulcer patients and is reported to induce gastric ulcer and gastric cancer in gerbils over 62 weeks.13
Isogenic mutant strains were constructed from a single colony from stock frozen H pylori. A portion of the genes encoding the cagG and virD4 genes was amplified by polymerase chain reaction (PCR) and the amplified fragment was inserted into the EcoRV restriction enzyme site of pBluescriptSK+ (Stratagene, La Jolla, California, USA). A chloramphenicol resistance gene cassette (a gift from DE Taylor, University of Alberta, Edmonton, Canada) was inserted into BsmI and HindIII sites of the insert DNA for the cagG and virD4 genes, respectively. All plasmids (1–2 μg) were used for inactivation of chromosomal genes by natural transformation, as previously described.22 Inactivation of the genes was confirmed by PCR amplification followed by Southern blot hybridisation.
IL-8 levels from gastric cancer cells cocultured with H pylori
In vitro IL-8 measurement was performed as previously described.23 Briefly, the human gastric cell line MKN 45 (Japanese Cancer Research Bank, Tsukuba, Japan) (1×105/ml) was plated onto 24 well plates and cultured for two days. H pylori was added to the cultured cells (bacterium to cell ratio of 100:1) and incubated for 24 hours. IL-8 in the supernatant was measured by an enzyme linked immunosorbent assay (R&D Systems, Minneapolis, Minnesota, USA) in triplicate.
Animal, housing, and H pylori challenge
Specific pathogen free seven week old male Mongolian gerbils (MGS/Sea; Seac Yoshitomi, Fukuoka, Japan) were used in this study. They were housed in an air conditioned biohazard room designed for infectious animals, with a 12 hour light/12 hour dark cycle. They were provided with a rodent diet and water ad libitum. All experimental protocols were approved by the Animal Experiment Committee of Shinshu University School of Medicine, Matsumoto, Japan.
H pylori were grown in Brucella broth supplemented with 10% (vol/vol) horse serum for 40 hours at 37°C under microaerobic conditions and saturated humidity, with shaking at 150 rpm. After fasting for 24 hours, each animal was orogastrically inoculated with 1.0 ml of an inoculum preparation of H pylori (108 colony forming units (CFU)/ml) or sterile Brucella broth (as an uninfected control) using gastric intubation needles. No specific pretreatments (for example, acid inhibition or antibiotics) were used before orogastric H pylori inoculation. Four hours after administration, animals were again allowed free access to water and food.
Time course and euthanasia
Mongolian gerbils were assigned to one of three groups: inoculated with the wild-type H pylori strains, with its cagG mutants, or with its virD4 mutants. Infected gerbils were killed and underwent necropsy at 4, 12, and 24 weeks after H pylori inoculation. Eight to nine gerbils were used for each time point. Uninfected control gerbils were killed at 11, 19, and 31 weeks of age (to serve as controls for the infected animals 4, 12, and 24 weeks after H pylori inoculation) (n = 6 each). Thirty minutes before being killed, gerbils were given 200 mg/kg of 5′-bromo-2′deoxyuridine (BrdU) intraperitoneally.
At necropsy, stomachs were opened along the greater curvature, beginning at the gastro-oesophageal junction and ending at the proximal portion of the duodenum, and observed macroscopically. Stomachs were then divided longitudinally into two parts and one half was fixed in 20% phosphate buffered formalin fixative for histological examination. The other part was further divided into the pyloric gland mucosa (antrum) and the fundic gland mucosa (corpus). The gastric mucosa was separated as much as possible from the underlying muscle using sharp dissection. Each specimen was placed on dry ice and stored at −80°C for cytokines mRNA analysis.
H pylori cultures
A 1 mm2 piece of gastric mucosa from the pyloric part of the stomach was used for culture of H pylori. These fragments were minced with Brucella broth and several diluted aliquots were spread on commercially available H pylori selective agar plates (Eiken Chemical Co., Tokyo, Japan). Cultures were incubated for seven days and the number of H pylori colonies per plate was counted.
Histological examination
Half of the stomach was stapled onto paper and fixed in 20% phosphate buffered formalin for 24 hours at 4°C. The fixed gastric tissue was processed for histopathological examination, and paraffin embedded sections were sliced and stained with haematoxylin-eosin or May-Grunwald-Giemsa. The degree of inflammation was graded according to the updated Sydney system.24
Analysis of IL-1β mRNA expression by real time quantitative PCR
Total RNA was extracted from the gastric mucosa using an RNA extraction kit (Isogen; Nippon Gene, Tokyo, Japan). After DNase treatment, 5 μg of total RNA were subjected to reverse transcription (RT) using 200 U of Moloney murine leukaemia virus reverse transcriptase (Life Technologies, Inc., Gaithersburg, Maryland, USA). Partial gerbil specific IL-1β cDNA sequences were recently cloned in our group (GenBank accession number AB164705) and we normalised IL-1β mRNA levels to the gerbil specific glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA identified previously.13 Specific primers and TaqMan probes are listed in table 1 ▶. Real time PCR was performed using an ABI Prism 7700 Sequence-Detection System (Perkin-Elmer Applied Biosystems) at 50°C for two minutes, 95°C for 10 minutes, followed by 50 cycles of 95°C for 15 seconds and 60°C for 60 seconds. IL-1β mRNA levels were expressed as the ratio of IL-1β mRNA to GAPDH mRNA (100 000 × IL-1β mRNA (unit/μl)/GAPDH mRNA (unit/μl)). Each assay was performed in triplicate.
Table 1.
Primers and probes used in this study
| GAPDH |
| Forward: 5′- CATGGCCTTCCGAGTTCCT-3′ |
| Reverse: 5′-TTCTGCAGTCGGCATGTCA-3′ |
| Probe: 5′-VIC-CCCCCAACGTGTCTGTCGTGGA-TAMURA-3′ |
| IL-1β |
| Forward: 5′-GGTGACACAAGCAGCAACAAA-3′ |
| Reverse: 5′-CATCACACAGGACAGGTACAGATTCT -3′ |
| Probe: 5′-FAM-TACCGGTGGCCTTGGGCCTCA-TAMURA-3′ |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin; FAM, 6-carboxyfluorescein; TAMURA, 6-carboxy-N, N, N′, N′-tetramethylrhodamine.
Serology
Before the animals were killed, blood samples were obtained from the orbital plexus using haematocrit tubes. Sera were used to measure the titre of anti-H pylori IgG antibody, as previously described.25,26 Antibody titre was expressed as an arbitrary index (AI) with values greater than 1.37 (⩽15 weeks of age) or 1.90 (>15 weeks) being scored as positive for H pylori based on our system.26
Statistical analyses
Results are presented as medians when the data were not distributed normally, and mean (SEM) when they were. Statistical analyses included the Student’s t test or the Mann-Whitney rank sum test, depending on whether the data were normally distributed. Prevalence of infection was analysed using Fisher’s extract test. A p value of <0.05 was considered significant.
RESULTS
In vitro IL-8 production from MKN45 cells cocultured with H pylori
The wild-type strain (TN2GF4) containing a complete set of the cag PAI genes induced greater secretion of IL-8 from MKN45 cells (mean (SEM) 3162 (147) pg/ml) than the virD4 mutants (2318 (43) pg/ml) (p<0.01) or the cagG mutants (325 (9) pg/ml) (p<0.001). In agreement with reports by Selbach and colleagues,17 the virD4 knockout mutants induced intermediate levels of IL-8 whereas the cagG mutants induced less than one tenth of IL-8 produced by the wild-type strain (control IL-8 levels without H pylori infection were 113 (5) pg/ml).
Establishment of H pylori infection in Mongolian gerbils
Ninety two gerbils were used. Bacteriological, histological, and serological examination showed no detectable H pylori in control gerbils. Infection status in inoculated gerbils was assessed using bacteriological and histological examination (table 2 ▶). Gerbils were classified as H pylori positive if culture and/or histology yielded positive results. With the exception of five gerbils infected with the cagG mutants for four weeks, all gerbils were successfully infected (table 2 ▶). The five gerbils with failed infection were excluded from further analyses.
Table 2.
Prevalence of H pylori colonisation in gerbils evaluated with different methods
| Wild-type | cagG mutants | virD4 mutants | ||||||||
| 4W (n = 8) | 12W (n = 8) | 24W (n = 8) | 4W (n = 8) | 12W (n = 8) | 24W (n = 9) | 4W (n = 8) | 12W (n = 8) | 24W (n = 9) | ||
| Culture (pyloric) | 88% | 100% | 88% | 0% | 75% | 11% | 25% | 88% | 100% | |
| Serology | 75% | 100% | 100% | 13% | 0% | 22% | 100% | 100% | 100% | |
| Histology (pyloric) | 100% | 100% | 100% | 38% | 88% | 100% | 100% | 75% | 100% | |
| Histology (fundic) | 100% | 100% | 100% | 38% | 88% | 89% | 88% | 88% | 100% | |
| Colonisation (total) | 100% | 100% | 100% | 38% | 100% | 100% | 100% | 100% | 100% | |
W, weeks post infection.
Gerbils were classified as H pylori positive if culture and/or histology yielded positive results.
H pylori IgG antibody titres were significantly increased in gerbils inoculated with the wild-type strains at 12 and 24 weeks compared with those at four weeks (27.9 (4.3) at 12 weeks and 111.9 (21.7) at 24 weeks compared with 2.2 (0.3) at four weeks) (p<0.001 for each) (fig 1 ▶). Although seroconversion occurred in all gerbils inoculated with the virD4 mutants, antibody titres were significantly lower than those of gerbils infected with the wild-type strains (8.6 (1.2) at 12 weeks and 17.0 (3.1) at 24 weeks for the virD4 mutants; p<0.001 for each). Antibody titres of gerbils infected with the cagG mutants were very low (maximum 3.5), and even seroconversion occurred.
Figure 1.

Titre of serum anti-Helicobacter pylori IgG antibodies of Mongolian gerbils inoculated orally with H pylori or without H pylori infection (control). Mean (SEM) values are presented. **p<0.01 compared with control; †††p<0.001 compared with the cagG knockout mutants; ‡‡p<0.01, ‡‡‡p<0.001 compared with the virD4 knockout mutants. AI, arbitrary index.
Histopathological findings
Histopathological changes at 4, 12, and 24 weeks after inoculation of Mongolian gerbils with H pylori and in controls are shown in fig 2 ▶. Inflammatory cell infiltration in the lamina propria was negligible in controls. At four weeks after inoculation, gerbils infected with the wild-type strains showed chronic active gastritis in the antrum, with marked mucosal infiltration by neutrophilic polymorphonuclear cells (PMN) (infiltration score 1.0 (0.3)) and by mononuclear cells (MNC) (1.9 (0.3)) (figs 3 ▶, 4 ▶). At 12 weeks, with the wild-type strains, dense PMN infiltration was seen throughout the mucosa with a dense MNC infiltration in the lamina propria and submucosa in the antrum, with the normal mucosal architecture being almost completely replaced with hyperplastic epithelium (PMN 2.5 (0.2) and 0.7 (0.2); MNC 2.8 (0.1) and 1.0 (0.2) for the antrum and corpus, respectively). At 24 weeks with the wild-type strains, numerous irregularly branched dilated mucous glands were seen in the lower portion of the proper muscle layer and the PMN and MNC infiltration scores reached their maximal levels (PMN 3.0 and 1.0 (0.2); MNC 3.0 and 1.1 (0.1) for the antrum and corpus, respectively).
Figure 2.
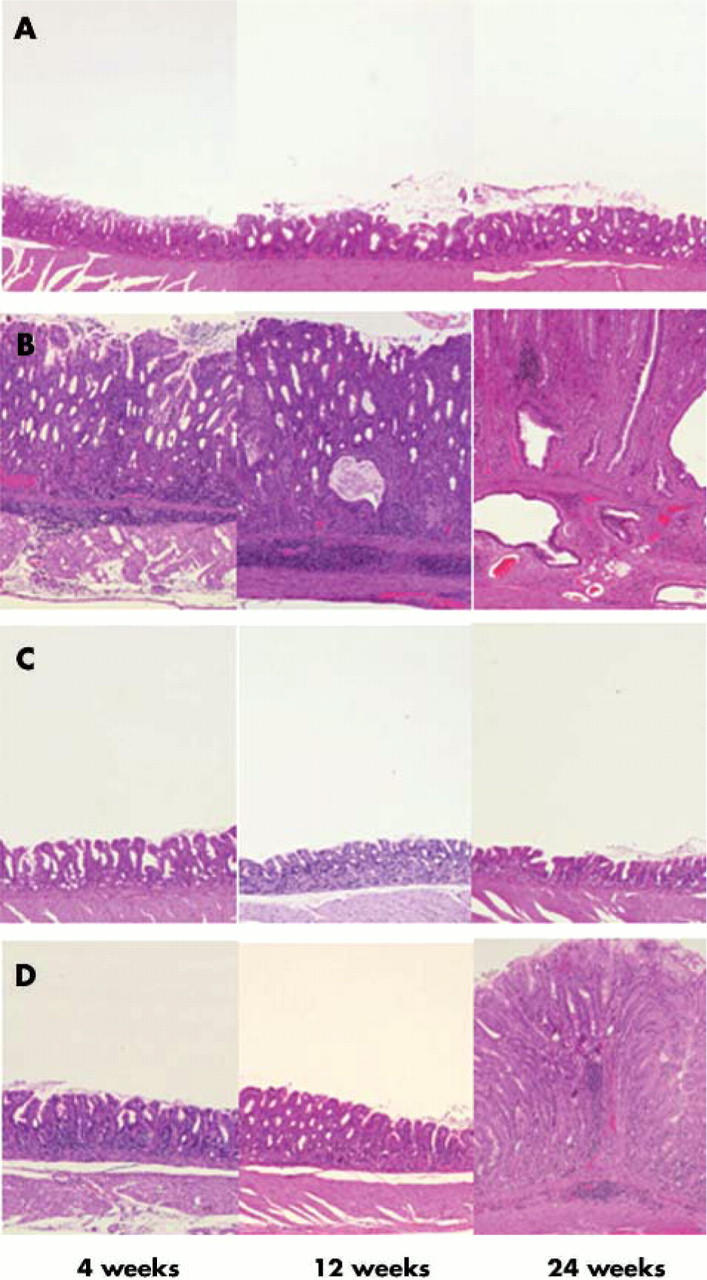
Histology of the gastric pyloric mucosa of (A) control, (B) wild-type Helicobacter pylori strain TN2GF4, (C) its isogenic cagG knockout mutant, or (D) virD4 knockout mutant. Haematoxylin and eosin stain, original magnification ×200. (A) In controls, inflammatory cell infiltration in the lamina propria was negligible throughout the experimental periods. (B) In gerbils infected with the wild-type strain, pyloric mucosa showed marked infiltration by neutrophilic polymorphonuclear cells and mononuclear cells at four weeks after inoculation and the inflammatory response increased with the duration of infection. The pyloric mucosa became thickened from four weeks after inoculation, and irregularly branched and dilated mucous glands appeared at 24 weeks after inoculation. (C) In gastric mucosa infected with the cagG knockout mutants, inflammatory cell infiltration in the lamina propria was negligible throughout the experimental periods. (D) Pyloric mucosa of gerbils infected with the virD4 knockout mutants showed mild inflammatory inflammation at four and 12 weeks after inoculation. At 24 weeks after inoculation, pyloric mucosa showed increased degrees of inflammatory cell infiltration and became thickened.
Figure 3.
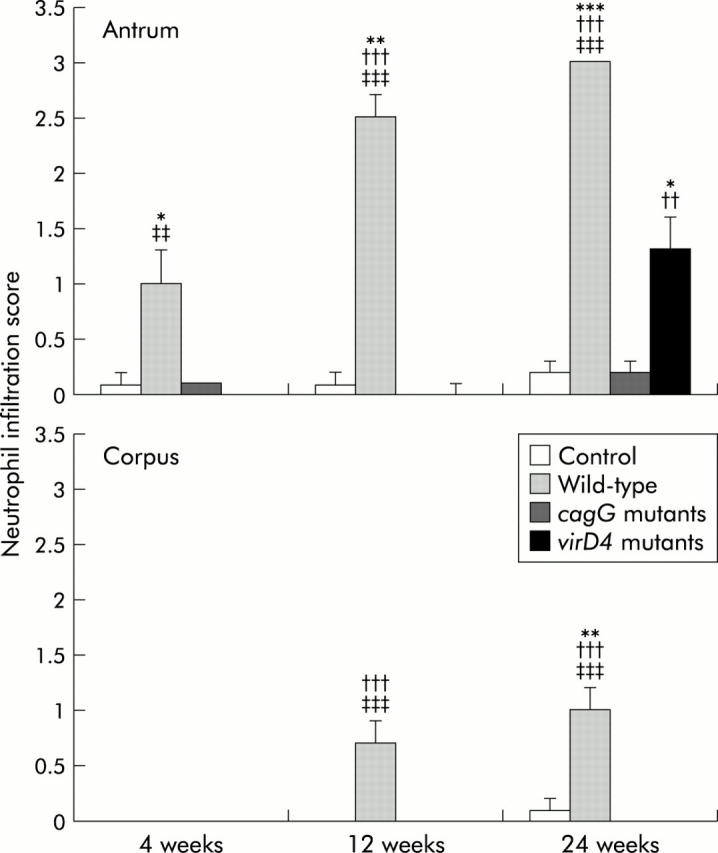
Neutrophil infiltration scores at 4, 12, and 24 weeks after inoculating with Helicobacter pylori or without H pylori (control). Mean (SEM) values are presented. *p<0.05, **p<0.01, ***p<0.001 compared with control; ††p<0.01, †††p<0.001 compared with the cagG knockout mutants; ‡‡p<0.01, ‡‡‡p<0.001 compared with the virD4 knockout mutants.
Figure 4.
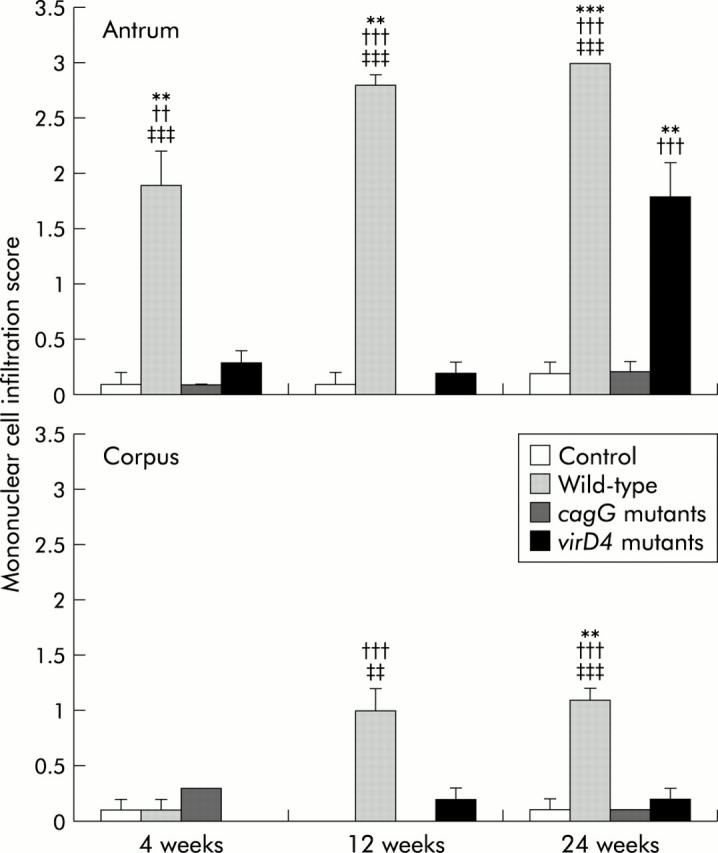
Mononuclear cell infiltration scores at 4, 12, and 24 weeks after inoculating with Helicobacter pylori or without H pylori (control). Mean (SEM) values are presented. **p<0.01, ***p<0.001 compared with control; ††p<0.01, †††p<0.001 compared with the cagG knockout mutants; ‡‡p<0.01, ‡‡‡p<0.001 compared with the virD4 knockout mutants.
In contrast, gerbils infected with the cagG mutants showed almost no inflammation at any time after inoculation (figs 3 ▶, 4 ▶). MNC and PMC infiltration scores in gerbils infected with the cagG mutants were significantly lower than those with the wild-type strain throughout the observation periods.
Gerbils infected with the virD4 mutants showed mild cellular inflammation four and 12 weeks after inoculation (MNC and PMC infiltration scores less than 0.5). MNC and PMC infiltration scores in gerbils infected with the virD4 mutants were significantly lower than those with the wild-type strains throughout the observation periods. Interestingly, however, at 24 weeks after inoculation, gerbils infected with the virD4 mutants showed chronic active gastritis with marked mucosal infiltration in the antrum (MNC 1.8 (0.3) and PMN 1.3 (0.3)) whereas mucosal infiltration in the corpus remained very mild (figs 3 ▶, 4 ▶). The amount of cellular infiltration in the antrum increased in gerbils infected with the virD4 mutants at 24 weeks compared with 12 weeks, and the pyloric mucosa appeared expanded similar to that observed with the wild-type strains. The grade of mucosal inflammation observed in gerbils infected with the virD4 mutants at 24 weeks was similar to those with the wild-type strains at four weeks.
H pylori density score, as evaluated by histology in the antrum of gerbils infected with the virD4 mutants, was significantly greater than that in animals infected with the wild-type strains at 24 weeks (fig 5 ▶) (p<0.01). Importantly, gerbils infected with the cagG mutants showed no inflammation at any time after inoculation. H pylori density score was mostly equivalent to the wild-type strains or virD4 mutants (fig 5 ▶).
Figure 5.
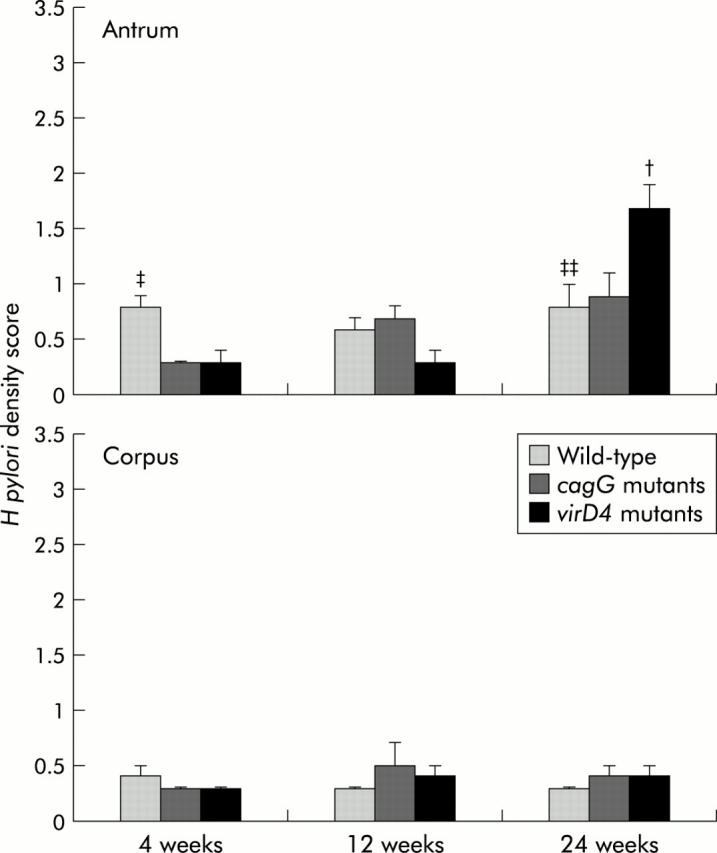
Scores for Helicobacter pylori density evaluated by histology at 4, 12, and 24 weeks after inoculation with H pylori. Mean (SEM) values are presented. †p<0.05 compared with the cagG knockout mutants, ‡p<0.05, ‡‡p<0.01 compared with the virD4 knockout mutants.
BrdU labelling indices
Detectable BrdU labelling indices were not observed in control gerbils without H pylori infection. BrdU labelling indices in the antrum were independent of the duration of H pylori infection both in gerbils infected with the wild-type strains (mean 26.3 (3.1) to 34.6 (3.3)) and the cagG mutants (mean 3.2 (0.5) to 11.3 (7.3)) (fig 6 ▶). In contrast, BrdU labelling indices were significantly increased in the virD4 mutants at 24 weeks after inoculation (29.3 (3.9) at 24 weeks compared with 6.1 (0.8) at four weeks and 3.8 (0.5) at 12 weeks) (p<0.001 for each). Overall, BrdU labelling indices were higher in gerbils infected with the wild-type strains compared with those with the cagG mutants or the virD4 mutants at four or 12 weeks. Indices were also significantly higher in gerbils infected with the wild-type strains compared with those with the cagG mutants at 24 weeks (p<0.001) whereas the indices were similar among gerbils infected with the wild-type strains and the virD4 mutants.
Figure 6.

5′-Bromo-2′deoxyuridine (BrdU) labelling indices in the pyloric mucosa. Mean (SEM) values are presented. **p<0.01 compared with control; †††p<0.001 compared with the cagG knockout mutants; ‡‡p<0.01, ‡‡‡p<0.001 compared with the virD4 knockout mutants.
Mucosal IL-1β mRNA levels
In the control group, mucosal IL-1β mRNA levels were very low throughout the observation periods (10 000× mean (SEM); IL-1β/GAPDH 1.8 (0.4) to 6.6 (0.8)) (fig 7 ▶). At four weeks after inoculation, mucosal IL-1β mRNA levels were significantly greater in gerbils infected with the wild-type strains compared with the virD4 or cagG mutants. Mucosal IL-1β mRNA levels at 12 weeks with the wild-type strains were also significantly higher than those with the virD4 or cagG mutants. At 24 weeks after inoculation, IL-1β levels decreased in gerbils infected with the wild-type strains.
Figure 7.
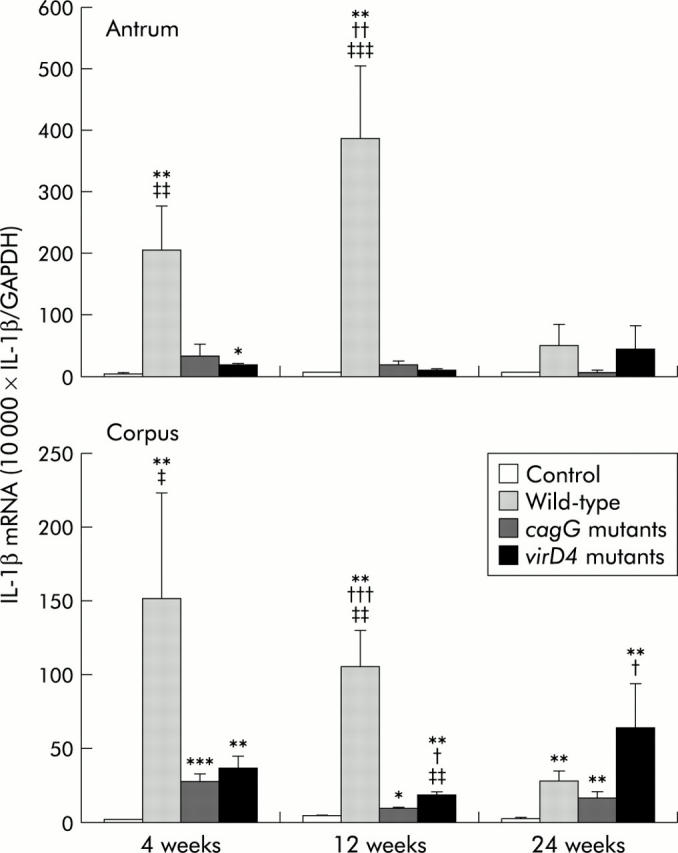
Mucosal interleukin (IL)-1β mRNA levels in gerbils at 4, 12, and 24 weeks after inoculating with Helicobacter pylori or without H pylori (control) in the pyloric mucosa (antrum) and fundic mucosa (corpus). Mean (SEM) values are presented. *p<0.05, **p<0.01, ***p<0.001 compared with control; †p<0.05, ††p<0.01, †††p<0.001 compared with the cagG knockout mutants; ‡‡p<0.01, ‡‡‡p<0.001 compared with the virD4 knockout mutants.
Mucosal IL-1β levels were very low in gerbils infected with the virD4 mutants throughout the first 12 weeks; however, these levels tended to increase at 24 weeks (45.6 (36.0) for the antrum and 63.0 (30.6) for the corpus). In contrast, IL-1β levels were very low in gerbils infected with the cagG mutants throughout the observation periods (fig 7 ▶).
DISCUSSION
We used the Mongolian gerbil model to examine the effect of two previously unstudied genes in the cag PAI (virD4 and cagG) on gastric inflammation in vivo. Wild-type H pylori caused typical severe gastritis in gerbils whereas the virD4 mutants caused very low levels of gastric inflammation, mucosal proliferative activity, and mucosal IL-1β levels throughout the first 12 weeks. H pylori density was similar with the different inocula, confirming that the differences were not due to bacterial load. At 24 weeks, the degree of gastric inflammation and proliferative activity in gerbils infected with the virD4 mutants increased, reaching levels comparative with those seen at four weeks with the wild-type strains. H pylori density in the antrum at 24 weeks in gerbils infected with the virD4 mutants was significantly higher than that with wild-type H pylori (p = 0.03). Lack of acute inflammation might help growth of the virD4 mutants; however, it remains unclear whether it is sufficient to explain the results.
Mucosal IL-1β levels at 24 weeks were similar for virD4 mutants and wild-type infections. IL-1β levels with the wild-type strains were maximal at four weeks in the corpus and at 12 weeks in the antrum. In gerbils and in mice, IL-1β mRNA levels do not mirror chronic mucosal inflammation.27,28 In contrast, in humans, IL-1β levels are consistently elevated in H pylori infected gastric mucosa.23 IL-1β mRNA levels in the corpus were very low in the chronic phase of the infection, suggesting that induction of acute inflammation rather than inhibition of gastric acid secretion29,30 is the main role of IL-1β in gerbils. Probably the most important proinflammatory cytokine in the gastric mucosa is IL-8. Gerbils do not encode an IL-8 gene, as cross species RT-PCR techniques failed to identify an IL-8 gene (unpublished observation). We selected IL-1β based on the fact that in humans, mucosal IL-8 levels were closely correlated with mucosal IL-1β levels.23,31 Future studies will examine cytokine expression using IL-8 families such as KC which behave like IL-8 in mice.
With the wild-type strains, the degree of gastric inflammation reached maximal levels at 12–26 weeks and proliferative activity at four weeks.25,32,33 Loss of the virD4 gene temporally retarded but did not abrogate H pylori induced gastric inflammation, and proliferative activity with the virD4 mutants was similar to that with the wild-type strains at 24 weeks. We did not examine animals beyond 24 weeks and can only speculate regarding later time points.
The virB4 (cagE) knockout mutants produce mild gastritis and not gastric ulcers.13–15 VirB4 is a major component of the type IV secretion system such that loss of the cagE gene results in loss of CagA translocation/phosphorylation as well as loss of host cytoskeletal rearrangement and IL-8 induction.3,12,17 Although the virD4 mutants also lose the ability to translocate CagA into host cells, cagA mutants can produce inflammation16 consistent with in vitro studies showing that CagA is not responsible for IL-8 induction. Lack of inflammation with the virD4 mutants in the first 12 weeks suggests the absence of as yet unidentified factors that translocate into epithelial cells using the type IV secretory pathway or interact with the type IV secretion system. Several factors other than the cag PAI, in particular OipA as one of the outer membrane proteins, are related to induction of mucosal IL-8 and gastric inflammation.22,34,35 In addition, cag PAI status is closely related to OipA status (for example, if the strains possess the cag PAI, strains almost always possess functional OipA).34 However, possible interactions between OipA and cag PAI were not examined in these experiments.
The cagG mutants did not produce an inflammatory response or increase proliferative activity, most likely related to their poor ability to colonise gerbil gastric mucosa. The cagG gene is not a vir homologue gene and has a weak homology to the flagellar motor switch protein gene or toxin coregulated pilus biosynthesis protein gene.3,20 The current consensus is that loss of the cagG gene also results in loss of CagA translocation/phosphorylation.3,12 Recent reports suggest that isolates lacking cagG genes have decreased adherence to epithelial cell lines.19 An in vivo study has shown no relationship between cagG and clinical outcome36; the population studied (Chinese) were predominantly infected with cag PAI positive strains such that the effect of the cagG gene could not be examined. Most reports, including our present study, suggest that loss of the cagG gene results in almost complete elimination of H pylori induced IL-8 induction.3,37 However, a recent report suggested that precise deletion of the cagG gene resulted in no reduction in IL-8 induction.12 Gerbils infected with cagG mutants showed no inflammation although the H pylori density score was generally equivalent to wild-type strains or virD4 mutants (fig 5 ▶). From these data it is not possible to define whether or not the lack of inflammation with cagG mutants is related to reduced colonisation, loss of the type IV secretion system, or both. Complementation experiments will be needed to resolve this issue.
Our current hypothetical model is presented in fig 8 ▶. VirD4 is thought to act as an adapter protein for the transfer of CagA protein and possibly other unknown proteins (D4 dependent) into the transfer channel formed by other Vir proteins in the cag PAI. We also hypothesise the presence of unknown proteins independent of VirD4. As virD4 mutants are unable to translocate CagA as well as any D4 dependent factors, loss of CagA effects and loss of D4 dependent factors occurs. However, it is possible that D4 independent factors may be translocated into cells or interact with the type IV secretion system, inducing chronic inflammation. cagG mutants have decreased adherence to epithelial cells and reduced ability to colonise gerbils. In addition, they are unable to translocate CagA and any D4 dependent or independent factors due to loss of functional transporter system such that inflammation would not be expected.
Figure 8.

Hypothetical model for induction of host responses by Helicobacter pylori.
Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research C-15590482 (to HO) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, by a grant from the Hokuto Foundation for Bioscience (to HO), and by National Institutes of Health grants R01 DK62813 (to YY) Drs Graham and Yamaoka are supported in part by the Office of Research and Development Medical Research Service Department of Veterans Affairs. We thank M Nakao, Takeda Chemical Industries, Ltd, Osaka, Japan, for providing the H pylori isolate TN2GF4 and DE Taylor, University of Alberta, Edmonton, Canada, for providing a chloramphenicol resistance gene cassette.
Abbreviations
AI, arbitrary index
BrdU, 5′-bromo-2′deoxyuridine
CFU, colony forming units
GAPDH, glyceraldehyde-3-phosphate dehydrogenase
IL, interleukin
MNC, mononuclear cells
PAI, pathogenicity island
PCR, polymerase chain reaction
PMN, polymorphonuclear cells, RT, reverse transcription
vir, virulent
Conflict of interest: None declared.
REFERENCES
- 1.Graham DY. Helicobacter pylori infection in the pathogenesis of duodenal ulcer and gastric cancer: a model. Gastroenterology 1997;113:1983–91. [DOI] [PubMed] [Google Scholar]
- 2.Graham DY, Yamaoka Y. H. pylori and cagA: relationships with gastric cancer, duodenal ulcer, and reflux esophagitis and its complications, Helicobacter 1998;3:145–51. [DOI] [PubMed] [Google Scholar]
- 3.Censini S, Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 1996;93:14648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaser MJ, Perez-Perez GI, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995;55:2111–15. [PubMed] [Google Scholar]
- 5.Segal ED, Cha J, Lo J, et al. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A 1999;96:14559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A 2000;97:1263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asahi M, Azuma T, Ito S, et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med 2000;191:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Backert S, Ziska E, Brinkmann V, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2000;2:155–64. [DOI] [PubMed] [Google Scholar]
- 9.Odenbreit S, Puls J, Sedlmaier B, et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000;287:1497–500. [DOI] [PubMed] [Google Scholar]
- 10.Selbach M, Moese S, Hauck CR, et al. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem 2002;277:6775–8. [DOI] [PubMed] [Google Scholar]
- 11.Covacci A, Telford JL, Del Giudice G, et al. Helicobacter pylori virulence and genetic geography. Science 1999;284:1328–33. [DOI] [PubMed] [Google Scholar]
- 12.Fischer W, Puls J, Buhrdorf R, et al. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 2001;42:1337–48. [DOI] [PubMed] [Google Scholar]
- 13.Sakai T, Fukui H, Franceschi F, et al. Cyclooxygenase expression during Helicobacter pylori infection in Mongolian gerbils. Dig Dis Sci 2003;48:2139–46. [DOI] [PubMed] [Google Scholar]
- 14.Ogura K, Maeda S, Nakao M, et al. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med 2000;192:1601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Israel DA, Salama N, Arnold CN, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest 2001;107:611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirth HP, Beins MH, Yang M, et al. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect Immun 1998;66:4856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selbach M, Moese S, Meyer TF, et al. Functional analysis of the Helicobacter pylori cag pathogenicity island reveals both VirD4-CagA-dependent and VirD4-CagA-independent mechanisms. Infect Immun 2002;70:665–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crabtree JE, Kersulyte D, Li SD, et al. Modulation of Helicobacter pylori induced interleukin-8 synthesis in gastric epithelial cells mediated by cag PAI encoded VirD4 homologue. J Clin Pathol 1999;52:653–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima T, Sugiyama T, Kobayashi T, et al. Decreased adherence of cagG-deleted Helicobacter pylori to gastric epithelial cells in Japanese clinical isolates. Helicobacter 2002;7:22–9. [DOI] [PubMed] [Google Scholar]
- 20.Tomb JF, White O, Kerlavage AR, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997;388:539–47. [DOI] [PubMed] [Google Scholar]
- 21.Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol 2000;8:354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci U S A 2000;97:7533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaoka Y, Kita M, Kodama T, et al. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 1997;41:442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon MF, Genta RM, Yardley JH, et al. Classification and grading of gastritis: the updated Sydney system. Am J Surg Pathol 1996;20:1161–81. [DOI] [PubMed] [Google Scholar]
- 25.Ikeno T, Ota H, Sugiyama A, et al. Helicobacter pylori-induced chronic active gastritis, intestinal metaplasia, and gastric ulcer in Mongolian gerbils. Am J Pathol 1999;154:951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumagai T, Yan J, Graham DY, et al. Serum immunoglobulin G immune response to Helicobacter pylori antigens in Mongolian gerbils. J Clin Microbiol 2001;39:1283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takashima M, Furuta T, Hanai H, et al. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut 2001;48:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 2003;124:1879–90. [DOI] [PubMed] [Google Scholar]
- 29.Uehara A, Okumura T, Sekiya C, et al. Interleukin-1 inhibits the secretion of gastric acid in rats: possible involvement of prostaglandin. Biochem Biophys Res Commun 1989;162:1578–84. [DOI] [PubMed] [Google Scholar]
- 30.Wallace JL, Cucala M, Mugridge K, et al. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol 1991;261:G559–64. [DOI] [PubMed] [Google Scholar]
- 31.Yamaoka Y, Kodama T, Kita M, et al. Relation between clinical presentation, Helicobacter pylori density, interleukin-1β and -8 production and cagA status. Gut 1999;45:804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, Tada M, Nagai H, et al. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998;115:642–8. [DOI] [PubMed] [Google Scholar]
- 33.Sawada Y, Kuroda Y, Sashio H, et al. Pathological changes in glandular stomach of Helicobacter pylori-infected Mongolian gerbil model. J Gastroenterol 1998;33 (suppl 10) :22–5. [PubMed] [Google Scholar]
- 34.Yamaoka Y, Kikuchi S, El-Zimaity HMT, et al. Importance of Helicobacter pylori OipA in clinical presentation, gastric inflammation, and mucosal interleukin-8 production. Gastroenterology 2002;123:414–24. [DOI] [PubMed] [Google Scholar]
- 35.Yamaoka Y, Kudo T, Lu H, et al. Role of interferon stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology 2004;126:1030–43. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Li ZS, Tu ZX, et al. Distribution of cagG gene in Helicobacter pylori isolates from Chinese patients with different gastroduodenal diseases and its clinical and pathological significance. World J Gastroenterol 2003;9:2258–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu PI, Hwang IR, Cittelly D, et al. Clinical presentation in relation to diversity within the Helicobacter pylori cag pathogenicity island. Am J Gastroenterol 2002;97:2231–8. [DOI] [PubMed] [Google Scholar]