

Abstract
Background: Although previous studies have shown that the hepatic sympathetic nerve controls various physiological functions in the liver, the role of this nerve in liver injury has yet to be clarified.
Aims: The purpose of this study was to elucidate the role of this nerve, based on our newly developed technique for selectively removing the activities of the hepatic sympathetic nerve.
Subjects and methods: Male C57BL/6 mice were operated on for hepatic sympathetic denervation. Thereafter, mice were intravenously administered 0.25 or 0.35 μg/g weight of the Fas agonist antibody, Jo-2, after which mortality by fulminant hepatitis was evaluated. Apoptosis in the liver was also examined by both terminal deoxynucleotidyl transferase mediated dUTP nick end labelling and caspase-3 assay.
Results: Mortality in sympathectomised mice was significantly higher than that in sham operated mice following administration of Jo-2. This result was also supported by apoptosis data in which sympathectomised livers exhibited a significant elevation in the number of apoptotic hepatocytes and caspase-3 activity after Jo-2 treatment compared with sham operated livers. Moreover, pretreatment with norepinephrine dose dependently inhibited the hepatic sympathectomy induced increase in mortality after Jo-2 injection. Antiapoptotic protein levels of FLICE inhibitory protein, Bcl-xL, and Bcl-2 in the liver were significantly lower in sympathectomised mice at one and two hours following Jo-2 treatment than in sham operated animals. In addition, interleukin 6 supplementation dose dependently suppressed the hepatic sympathectomy induced increase in mortality after Jo-2 treatment.
Conclusions: These results suggest that norepinephrine released from the hepatic sympathetic nerve plays a critical role in protecting the liver from Fas mediated fulminant hepatitis, possibly via mechanisms including antiapoptotic proteins and interleukin 6.
Keywords: apoptosis, fas, hepatitis, norepinephrine, sympathetic nervous system
Approximately 150 years ago, interaction between the autonomic nervous system and liver function was first observed by Claude Bernard who demonstrated that “wounding” on the floor of the fourth ventricle in the brain stimulated the release of glucose from the liver.1 Since then, a number of researchers have provided overwhelming evidence that the hepatic sympathetic nerve participates in a variety of liver functions, such as hepatic circulation, metabolism (glycogenolysis, glycogen synthesis, urea output, ammonia uptake, and ketogenesis), and bile formation.1,2 In addition to these functions, associated mainly with normal physiological states, recent reports have demonstrated that the hepatic nervous system is also involved in pathological states of the liver. Indeed, it was reported that intrahepatic sympathetic nerve fibres change their distribution, accompanying structural alterations in hepatic lobules, in human livers with chronic active hepatitis or cirrhosis.3 Furthermore, recent animal studies have revealed that central corticotrophin releasing factor triggered activation of the hepatic sympathetic nerve aggravated carbon tetrachloride induced acute liver injury.4,5 Hence these findings suggest a close relationship between the hepatic sympathetic nerve and various liver disorders; however, the exact mechanism regulating this remains to be elucidated.
In animal studies, surgical or chemical denervation of the hepatic sympathetic nerves is generally used as the standard method for investigating the function of these nerves. Several researchers have developed procedures such as orthotopic liver transplantation,6,7 intraportal or intraperitoneal infusion of 6-hydroxydopamine (6-OHDA),7,8,9,10 which can delete catecholamines in peripheral tissues, microsurgery,11,12 and local chemical sympathectomy, which is performed by applying phenol solution to the hepatic nervous plexus.2,4,5 These procedures are effective in large animals but are difficult when applied to small animals, such as mice. For example, both surgical transplantation and chemical sympathectomy with 6-OHDA often elicit liver abscesses and pan peritonitis and, moreover, microsurgery and local chemical sympathectomy with phenol solution cannot satisfactorily ablate the activities of the hepatic sympathetic nerve. Therefore, we have modified the standard methods and established a new method applicable to mice which combines microsurgery with application of phenol solution onto the hepatic nervous plexus.
Fas on the cell surface interacts with Fas ligand and consequently induces cell apoptosis. Fas induced apoptosis has been implicated in hepatic failure from specific drugs and toxins, viral infection, autoimmune disorders, ischaemia, allograft rejection, and sepsis.13–17 In addition, when mice are injected with anti-Fas antibody, such as Jo-2, binding of this antibody to the Fas receptor also directly causes hepatic injury by stimulating the apoptotic cell death programme.16 Recently, we have demonstrated that the central nervous system affects the severity of Fas induced liver injury in mice via endogenous glucocorticoids18 whereas few data are available on peripheral sympathetic nervous affection on Fas induced liver injury. Therefore, we believe that this Jo-2 animal model is useful for investigating whether or not the hepatic sympathetic nerve is involved in the pathological process of Fas induced liver injury.
The purpose of this study was to clarify the effect of the hepatic sympathetic nerve on Fas induced fulminant hepatitis in mice and thereby reveal the precise mechanism by which this effect can be mediated, based on our newly devised hepatic sympathectomy technique.
MATERIALS AND METHODS
Mice
Male C57BL/6 (B6) mice (8–10 weeks of age; Charles River Japan, Shizuoka, Japan) were kept on a 12 hour light/12 hour dark cycle with food and water freely available. Mice were allowed to acclimatise themselves to the colony for seven days before the experiments began. The temperature of the colony room was maintained at 22–23°C. This experiment was reviewed by the ethics committee on animal experiments of the Graduate School of Medical Sciences, Kyushu University, and was carried out under the control of the guidelines for animal experiments of the Graduate School of Medical Sciences, Kyushu University, and the Law (No 105) and Notification (No 6) of the government.
Experimental procedures
Postganglionic sympathetic nerves to the liver are derived from splanchnic nerves originating in the coeliac and superior mesenteric ganglia. These nerves form two communicating plexuses—an anterior plexus running along the common hepatic artery and a posterior plexus branching around the portal vein and bile duct—and via two major plexuses the liver takes delivery of sympathetic nerves.2 Based on this anatomical knowledge, we devised a method for selectively ablating the activities of the hepatic sympathetic nerves, as described below.
All mice were anaesthetised by intraperitoneal injection of sodium pentobarbital (50 μg/g weight; Dainippon Pharmaceutical Co, Osaka, Japan) and thereafter were administered penicillin G intramuscularly (9000 U/mice; Banyu Pharmaceutical Co, Tokyo, Japan). The abdomen was cut along the midline by scissors and kept wide open by retractors. After the intestines were displaced to the lower outside of the abdomen, the hepatoduodenal ligament superior to the duodenum was cut transversely so that the proper hepatic artery, bile duct, and portal vein could all be visualised at the point where they entered the liver. In order to achieve hepatic denervation, all of the connective tissues surrounding these vessels were carefully isolated using an operating binocular microscope at 20× magnification, and resected with small forceps. At the end of the operation, 10 μl of phenol solution (85%; Katayama Chemical, Osaka, Japan) were applied to a swab and painted over the resected area. Thereafter, the intestines were returned and the abdominal cavity was closed with ophthalmological sutures in two layers. The procedure was achieved with minimal blood loss and took approximately 15–20 minutes.
In the experiments examining the effect of hepatic sympathectomy on liver injury, mice were intravenously injected with either hamster antimouse Fas antigen monoantibody (clone Jo-2; PharMingen, San Diego, California, USA) following dilution in a 500 μl total volume solution with sterile normal saline or a corresponding vehicle two weeks after the operation. In some experiments, referring to previous reports,19,20 mice were pretreated with norepinephrine (NE) intraperitoneally (0.5 or 5 μg/g weight; Sankyo, Tokyo, Japan) and were pretreated with recombinant human interleukin 6 (IL-6) subcutaneously (10 or 20 ng/g weight; PeproTech EC, London, UK) in a 100 μl total volume solution with sterile normal saline or a corresponding vehicle 30 minutes before Jo-2 injection. Mice were monitored on the hour for 24 hours and time of death was recorded.
High performance liquid chromatography for norepinephrine
Plasma and tissue NE levels were measured by a method described previously.8 Each animal was killed by cervical dislocation according to our institutional guidelines for animal experiments, blood was obtained by cardiac puncture, and the animal was perfused with 20 ml of phosphate buffered saline (PBS, 0.1 M, pH 7.2) via the left ventricle of the heart. Liver, spleen, and kidney samples were removed from one end of each organ, and samples weighing 80–120 mg were homogenised in 0.1 M HClO4 (100 μl/10 mg tissue; Sigma-Aldrich, Tokyo, Japan) at 4°C. The homogenate was centrifuged at 15 000 rpm for 15 minutes and aliquots of the resulting supernatant were treated with alumina. NE was eluted with 0.2 N acetic acid (Sigma-Aldrich) and assayed using a high performance liquid chromatography system for catecholamine analysis.
Immunohistochemistry for tyrosine hydroxylase
Immunohistochemical examination was performed according to a previously reported method.21 Sham operated or sympathectomised mice were perfused via the left ventricle, with 25 ml of PBS (0.01 M), and then fixed with cold 50 ml phosphate buffered saline, including 4% paraformaldehyde (Sigma-Aldrich), 0.5% glutaraldehyde (Sigma-Aldrich), and 0.2% picric acid (Sigma-Aldrich). The livers were sectioned at 30 μm and then rinsed with 0.1 M PBS overnight at 4°C. Sections were immediately processed for immunohistochemistry by a peroxidase-antiperoxidase technique using rabbit antityrosine hydroxylase (TH) polyclonal antibody (1:1600; Chemicon, Temecula, California, USA) for detecting aminergic innervation.
Histological examination by haematoxylin-eosin and terminal deoxynucleotidyl transferase mediated dUTP nick end labelling staining
Formalin fixed sections of liver tissue (5 μm) collected from mice two hours after treatment with 0.35 mg/g weight Jo-2 were analysed by haematoxylin-eosin (H&E) and terminal deoxynucleotidyl transferase mediated dUTP nick end labelling (TUNEL) staining. In the TUNEL assay, 5 μm liver sections were deparaffinised in xylene and hydrated in graded ethanol. Apoptotic cells were identified using a Cell Death Detection kit (Roche Molecular Biochemicals, Rotekreuz, Switzerland).20 Seven random ×20?fields from three slides/groups were examined, and TUNEL positive brown nuclei (apoptotic) within hepatocytes were counted.
Measurement of liver caspase-3 activity and IL-6 levels
Liver caspase-3 activities and IL-6 levels were analysed by commercially available kits (caspase-3: CaspACE assay system, Promega, Madison, Wisconsin, USA; IL-6: Quantikine mouse IL-6, R&D Systems, Minneapolis, Minnesota, USA). Briefly, 100 μg of mouse liver were homogenised in 200 μl of a cell lysis buffer with a protease inhibitor cocktail (Sigma-Aldrich). The homogenates were centrifuged at 14 000 g at 4°C for 15 minutes. IL-6 levels in the supernatant were measured by the sandwich ELISA technique, as specified by the manufacturer. In evaluating caspase-3 activities, the supernatant was incubated with Ac-DEVD-pNA as a substrate for caspase-3 at 37°C for four hours. Released pNA was determined by changes in absorbance at 405 nm using a microplatemeter (NJ-2300; System Instrument, Tokyo, Japan). To verify the specificity of the reaction, enzyme activity in each sample was measured in the presence or absence of the caspase-3 inhibitor Ac-DEVD-CHO. Results are expressed as pmol of substrate cleaved per mg of liver protein.
Immunoblot analysis for Fas antigen, FLICE inhibitory protein, Bcl-xL, and Bcl-2
Protein levels of Fas, FLICE inhibitory protein (FLIP), Bcl-xL, and Bcl-2 in liver particulate were determined using an immunoblot analysis, as described previously,20 with some modifications. Liver samples were homogenised in RIPA buffer (1% Triton X-100, 10 mM Tris-HCl, pH 7.4, 0.1% sodium dodecyl sulphate, 1 mM EDTA, 150 mM NaCl) with a protein inhibitor cocktail (Sigma-Aldrich). Insoluble material was removed by centrifugation at 12 000 g for 15 minutes at 4°C. Protein concentration of the supernatants was determined by a Bio-Rad protein assay (Bio-Rad, Hercules, California, USA). Cell lysates (100 μg) were mixed with Laemmli’s sample buffer, boiled, and then run on 12.5% sodium dodecyl sulphate-polyacrylamide gels. Full range molecular weight markers (Nacalai Tesque) were used to determine apparent molecular weights. Separated proteins were transferred to nitrocellulose membrane (Amersham, Wikströms, Sweden) which were then blocked with 5% skim milk (Becton-Dickinson, Sparks, Maryland, USA) in PBS containing 0.05% Tween (PBS-T) for two hours. After washing in PBS-T, membranes were incubated overnight at 4°C with Jo-2, mouse anti-FLIP monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, California, USA), mouse anti-Bcl-xL monoclonal antibody (Zymed Laboratories, South San Francisco, California, USA), mouse anti-Bcl-2 monoclonal antibody (Exalpha Biologicals, Boston, Massachusetts, USA), or antimouse β-actin antibody (mouse IgG1; Sigma) in a dilution of 1:1000 in PBS-T. Then, membranes were washed and incubated for one hour at room temperature with secondary goat antihamster (Jackson Immunoresearch, West Grove, Pennsylvania, USA) or rat antimouse IgG1 (PharMingen) horseradish peroxidase antibodies in a dilution of 1:1000 in PBS-T. Next, membranes were washed in PBS-T, incubated in enhanced chemiluminescence plus (ECL-plus) detection reagents (Pierce, Rockford, Illinois, USA) for five minutes at room temperature, and then exposed to an ECL mini camera (Amersham). β-Actin protein expression was used to normalise protein loading for each blot. The immunoblot was scanned densitometrically to quantify protein levels using public domain NIH image software.
Statistical analysis
All data are expressed as means (SEM). Data were analysed by one factor analysis of variance (ANOVA) followed by the Scheffé test. To analyse data on mortality, we used Kaplan-Meier survival analysis (log rank). A value of p<0.05 was considered to be statistically significant.
RESULTS
Selective denervation of the hepatic sympathetic nerve
We first examined the validity and specificity of our procedures based on hepatic NE concentrations. As shown in fig 1 ▶, liver NE content (7.4 (1.7) ng/g wet liver) in mice denervated by mixed sympathectomy was 12% (64.3 (6.4) ng/g wet liver) of that in sham operated mice, 26% (28.4 (5.2) ng/g wet liver) of that in mice treated by microsurgery alone, and 28% (26.2 (11.7) ng/g wet liver) of that in mice who had phenol solution painted onto their hepatic nervous plexus one week after operation. Decreased NE levels in mixed sympathectomised mice were observed for at least two weeks after denervation, and thereafter returned to levels (31.7 (4.5) ng/g wet liver) observed in microsurgical denervated mice four weeks after undergoing the procedure (fig 1 ▶). In addition, treatment with phenol solution on the hepatic surface one week after the operation failed to affect liver NE levels, thus indicating that phenol per se had no denervation effect if applied to anywhere other than sympathetic nerve branches.
Figure 1.

Kinetics of postoperative hepatic norepinephrine (NE) levels in sham operated mice (Sham), in mice who had phenol solution applied to their hepatic surface (Phenol), in mice who had phenol solution painted onto their hepatic nervous plexus (Phenol painted), in microsurgical denervated mice (Sympx), and in mixed denervated mice (Mixed sympx). Liver NE content was measured at one, two, or four weeks after the operative procedures. All values are expressed as mean (SEM); n = 5–11. *p<0.05, **p<0.01, ***p<0.001 compared with the corresponding value.
To further confirm that our techniques ablated the hepatic sympathetic nerve, we performed immunohistochemical analysis using anti-TH antibody. TH is a key enzyme responsible for catecholamine synthesis of the sympathetic nerve.22 Immunohistochemical analysis showed that TH positive nerve fibres in the liver of sham operated animals were richer in the portal vein than in either the bile duct or hepatic artery in the region of the portal triad whereas no such TH positive fibres were detected in the liver of the mixed sympathectomised group (fig 2 ▶). Examination by light microscopy of H&E staining showed no histological differences between the sham operated and mixed sympathectomised groups (data not shown).
Figure 2.

Immunohistochemical analysis of liver tyrosine hydroxylase (TH) positive nerve fibres showing the region of the portal triad in the liver two weeks after the operative procedures. TH positive nerve fibres (arrows) are seen around the portal vein (PV), bile duct (BD), and hepatic artery (HA) in sham operated mice (A) but not in mixed sympathectomised animals (B). Magnification ×400. The results are representative of three sets of experiments performed on two animals each.
Spleen and kidney NE levels in mixed denervated mice were almost identical to those in sham operated and microsurgical denervated mice one week after the procedures (fig 3 ▶), thus indicating that our techniques can specifically affect the branches of the sympathetic nerve in the liver.
Figure 3.

Effects of sympathectomy on spleen and kidney norepinephrine (NE) levels after operation in the sham operated (Sham), microsurgical denervated (Sympx), and mixed denervated (Mixed sympx) animals, respectively. Spleen and kidney NE content was measured two weeks after the operative procedures. All values are expressed as mean (SEM); n = 4–5.
Increased mortality after Fas monoclonal antibody treatment in hepatic sympathectomised mice
In order to clarify the role of the hepatic sympathetic nerve in liver injury, we intravenously injected sham operated and mixed sympathectomised mice with Jo-2, and thus evaluated the extent of liver apoptosis in both groups of mice.
As shown in fig 4 ▶, liver NE levels in sham operated mice significantly increased one hour after 0.15 μg/g weight Jo-2 treatment, but not those in the sympathectomised mice, thus indicating that the hepatic sympathetic nerve is activated in the liver following Jo-2 injection.
Figure 4.

Kinetics of liver norepinephrine (NE) content after Jo-2 treatment in sham operated (Sham) and mixed denervated (Sympx) mice. NE content was measured at the indicated time points after injection of Jo-2 (0.15 μg/g weight). All values are expressed as mean (SEM); n = 6–8. ***p<0.001 compared with the corresponding value in the sham operated group; ††p<0.01 compared with the basal value in the sham operated group.
We next examined the effect of the hepatic sympathetic nerve on the survival course after Jo-2 injection. After administering a high dose (0.35 μg/g) of Jo-2, all sympathectomised mice became lethargic and died rapidly, with an average time to death of 3.4 hours; 6/11 sham operated mice subsequently died with an average time to death of 8.2 hours (p<0.001) (fig 5A ▶). Similarly, after a moderate dose (0.25 μg/g) of Jo-2, mortality of sympathectomised mice (8/10) was also significantly higher than that of sham operated mice (1/10; p<0.005) (fig 5B ▶). These results were also supported by H&E and TUNEL staining histological analysis in which sympathectomised livers revealed massive haemorrhaging, congestion, parenchymal collapse (fig 6A ▶), and a significant elevation in the number of apoptotic hepatocytes (TUNEL stained cells, fig 6B ▶), while few such findings were found in sham operated livers. Likewise, as shown in fig 7 ▶, liver caspase-3 activity, which actually cleaves intracellular substrates resulting in apoptosis, was also significantly higher in sympathectomised mice than that in sham operated mice, at both eight hours (0.15 μg/g of Jo2) and two hours (0.35 μg/g of Jo-2).
Figure 5.
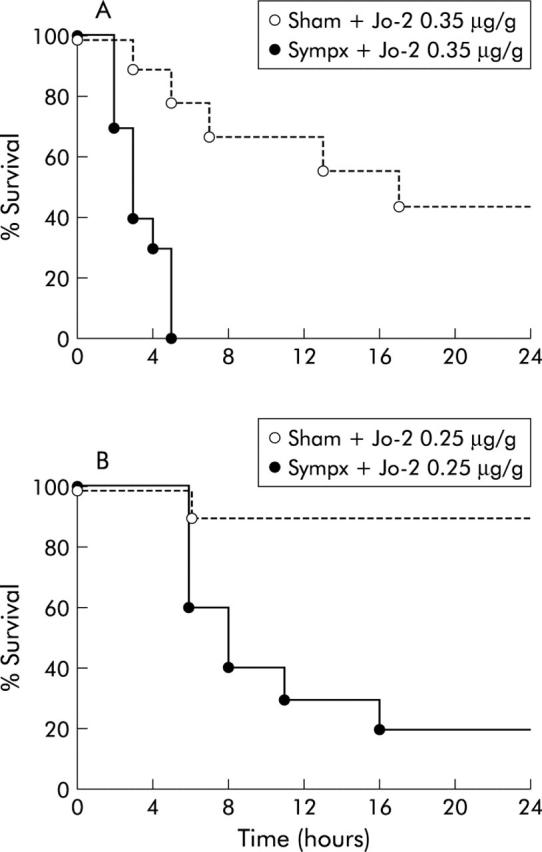
Survival curves showing reduced survival in sympathectomised mice (Sympx) following high or moderate doses of Jo-2 compared with sham operated (Sham) animals. (A) Survival course of mice (n = 11) following a high dose (0.35 μg/g weight) of Jo-2. (B) Survival course of mice (n = 10) following a moderate dose (0.25 μg/g weight) of Jo-2.
Figure 6.
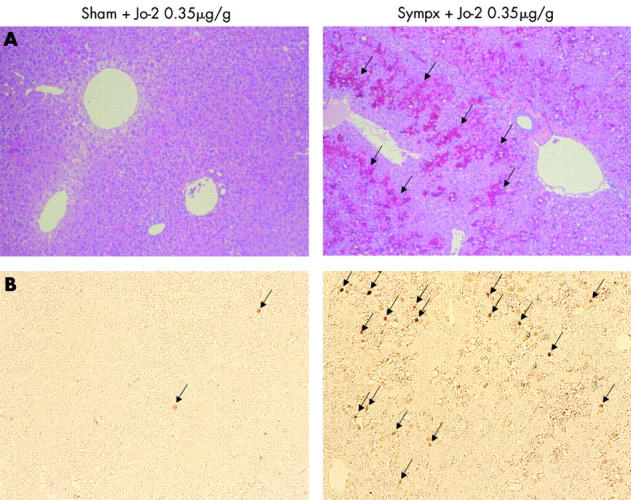
Haematoxylin-eosin (H&E) staining and terminal deoxynucleotidyl transferase mediated UTP nick end labelling (TUNEL) assay of the liver after a high dose of Jo-2. (A) H&E staining of sham operated (Sham) or sympathectomised (Sympx) livers two hours after Jo-2 (0.35 μg/g weight) injection. Massive haemorrhage, congestion, and parenchymal collapse (arrows) are seen in the livers of sympathectomised mice. Magnification ×200. (B) TUNEL staining in sham operated or sympathectomised livers two hours after Jo-2 (0.35 μg/g weight) injection. The number of TUNEL positive brown nuclei (arrows) within hepatocytes in sympathectomised mice is much higher than that of sham operated animals. Magnification ×200. Results are representative of three sets of experiments performed on two animals each.
Figure 7.

Induction of liver caspase-3 activity by Jo-2 treatment in sham operated (Sham) and mixed denervated (Sympx) animals. Caspase-3 activity was measured in the liver two and eight hours after injection of a low dose (0.15 μg/g weight), and two hours after injection of a high dose (0.35 μg/g weight) of Jo-2. All values are expressed as mean (SEM); n = 5–8/group). *p<0.05, ***p<0.001 compared with the corresponding value.
Taken together, these results indicate that activation of the hepatic sympathetic nerve protects against Jo-2 induced hepatic apoptosis.
NE supplementation completely suppressed sympathectomy induced enhancement of mortality in Jo-2 treated mice
The sympathetic nervous system is well documented to contain several subpopulations that can release many other neurological mediators, apart from NE.23 Therefore, in order to substantiate the contribution of NE per se to the protective effect of the hepatic sympathetic nerve in Fas induced fulminant hepatitis, we supplemented sham operated and sympathectomised mice with NE (0.5 or 5 μg/g weight) 30 minutes before Jo-2 treatment.
As shown in fig 8 ▶, injection with a low (0.5 μg/g) or high (5 μg/g) dose of NE dose dependently attenuated the enhancement of mortality after Jo-2 treatment (0.35 μg/g) in sympathectomised mice, with complete attenuation when treated with a high dose of NE. In contrast, no such attenuation was found at any NE dose in sham operated mice.
Figure 8.
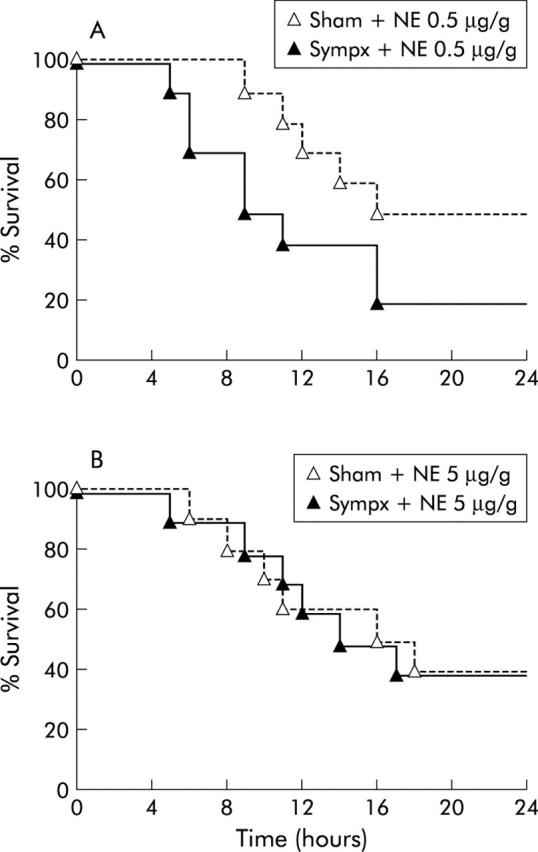
Norepinephrine (NE) supplementation completely suppressed the elevation in mortality in hepatic sympathectomised mice after Jo-2 treatment. (A) Survival course of sympathectomised (Sympx) and sham operated (Sham) mice (n = 10) pretreated with a low dose (0.5 μg/g weight) of NE 30 minutes before Jo-2 injection (0.35 μg/g weight). (B) Survival course of mice (n = 10) pretreated with a high dose (5 μg/g weight) of NE 30 minutes before Jo-2 injection (0.35 μg/g weight).
Sympathectomised livers exhibited decreased expression of antiapoptotic proteins compared with sham operated livers after Jo-2 treatment
Many studies have reported that increased expression of the antiapoptotic proteins FLIP, Bcl-xL, and Bcl-2 was critically involved in protection against Fas mediated hepatocyte apoptosis.24,25 Therefore, we investigated expression levels of FLIP, Bcl-xL, and Bcl-2 in sham operated and sympathectomised mice after Jo-2 injection.
Expression levels of FLIP, Bcl-xL, and Bcl-2 in sympathectomised livers were significantly lower one and two hours following Jo-2 treatment (0.35 μg/g) than corresponding levels in sham operated animals (fig 9 ▶), although no significant difference in expression of Fas antigen was found between sham operated and sympathectomised livers.
Figure 9.

Decreased expression of the antiapoptotic proteins FLICE inhibitory protein (FLIP), Bcl-xL, and Bcl-2 in sympathectomised (Sympx) and sham operated (Sham) livers following Jo-2 treatment. (A) Fas antigen, FLIP, Bcl-xL, Bcl-2, and β-actin expression in the liver one hour after injection of Jo-2 (0.35 μg/g weight). Protein sizes of Fas antigen, FLIP, Bcl-xL, Bcl-2, and β-actin were 48, 52, 30, 28, and 44 kDa, respectively, as indicated. (B) Densitometically analysed immunoblotting data for FLIP, Bcl-xL, and Bcl-2 at the indicated time points after Jo-2 administration. All values are the mean of three mice/group. *p<0.05, **p< 0.01, ***p<0.001 compared with the corresponding value.
A previous study showing that IL-6 protected against Fas mediated hepatocyte apoptosis by maintaining expression of the antiapoptotic proteins FLIP, Bcl-xL, and Bcl-2 in mice,20 and our present finding that IL-6 protein levels in sympathectomised liver significantly decreased after Jo-2 treatment (0.35 μg/g) compared with those in sham operated liver (IL-6: before Jo-2 treatment: sham 2.52 (0.39) pg/mg protein, sympathectomy 2.54 (0.15) pg/mg protein; one hour after Jo-2 treatment: sham 2.35 (0.13) pg/mg protein, sympathectomy 1.65 (0.13) pg/mg protein; p<0.001), led us to hypothesise that IL-6 might play a role in the observed protective effect of the hepatic sympathetic nerve on Fas induced hepatitis. In accordance with this hypothesis, IL-6 pretreatment (10 or 20 ng/g weight) dose dependently inhibited the elevation in mortality after Jo-2 treatment in sympathectomised mice whereas no such inhibition was found at any IL-6 dose in sham operated mice (fig 10 ▶).
Figure 10.
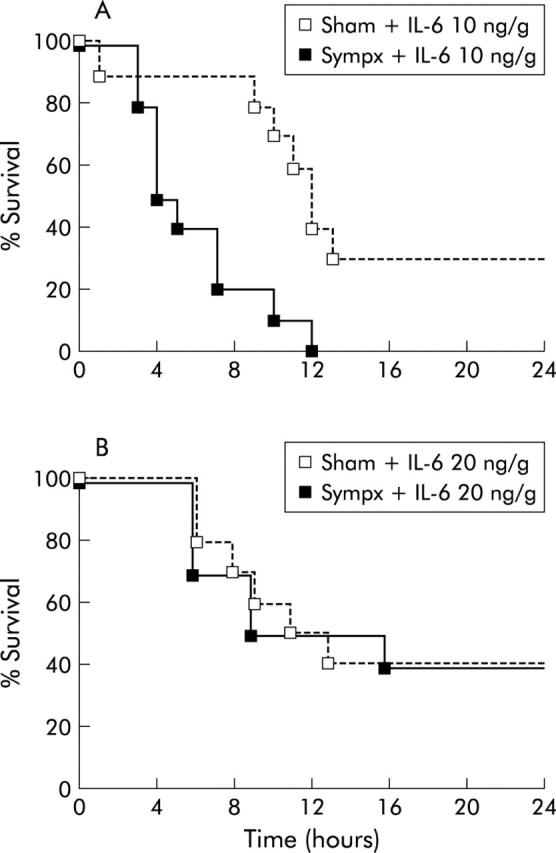
Interleukin 6 (IL-6) supplementation dose dependently inhibited the elevation in mortality in hepatic sympathectomised mice after Jo-2 treatment. (A) Survival course of mice (n = 10) pretreated with a low dose (10 ng/g weight) of recombinant IL-6 30 minutes before Jo-2 injection (0.35 μg/g weight) in sham operated (Sham) and hepatic sympathectomised (Sympx) mice. (B) Survival course of mice (n = 10) pretreated with a high dose (20 ng/g weight) of recombinant IL-6 30 minutes before Jo-2 injection (0.35 μg/g weight).
DISCUSSION
We have developed a quick method of performing sympathectomy in mice. We found that hepatic sympathetic denervation exaggerated Fas mediated apoptosis in the liver and accelerated the mortality of Jo-2 injected mice, while NE supplementation completely inhibited this Fas induced acceleration. We also found that antiapoptotic protein levels of FLIP, Bcl-xL, and Bcl-2 were significantly reduced in the liver of sympathectomised mice after injection of Jo-2 and that pretreatment with IL-6 dose dependently inhibited the sympathectomy induced elevation in mortality in Jo-2 treated mice. Hence our results indicate that NE released from the hepatic sympathetic nerve plays a critical role in preventing Fas mediated hepatocyte apoptosis, probably through mechanisms involving antiapoptotic proteins and IL-6.
In addition to the classic neurotransmitter NE, the sympathetic nervous system contains several subpopulations of target selective and neurochemically coded neurones that express a variety of neuropeptides and, in some cases, adenosine triphosphate, nitric oxide, or lipid mediators of inflammation. Moreover, corticotrophin releasing hormone, neuropeptide Y, and somatostatin are colocalised in noradrenergic vasoconstrictive neurones.23 Therefore, further investigation was needed to clarify whether NE per se mediated the protection against Fas induced fulminant hepatitis. In this study, NE supplementation was shown to completely and dose dependently reverse the sympathectomy induced elevation in mortality in Jo-2 treated mice, thus proving that NE has a crucial role in the protective effect of the sympathetic nervous system in Fas triggered liver injury.
Recent studies4,5 have shown that central corticotrophin releasing hormone triggered activation of the hepatic sympathetic nerve exacerbates carbon tetrachloride induced acute liver injury. In contrast with this injurious effect of the sympathetic nerve, chemical sympathectomy by systemic administration of 6-OHDA was reported to cause deterioration in liver injury induced by either concanavalin A or D-galactosamine plus staphylococcal enterotoxin B, and that this deterioration was prevented by peripheral sympathetic nerve activation by systemic treatment with a β2 adrenergic agonist. In line with the findings of this report, André and colleagues26 also demonstrated that treatment with the β2 adrenergic agonist clenbuterol prevented Fas induced liver apoptosis and death in mice. However, as these experiments were performed by systemic adrenergic stimulation with a pharmacological agonist that could not selectively affect the liver, a definitive answer about how and to what extent the hepatic sympathetic nerve is involved in hepatic susceptibility to Fas induced apoptosis remains to be answered. In our study, selective hepatic sympathectomy significantly increased both Fas induced hepatic apoptosis and mortality in mice, which thus conclusively demonstrates that the hepatic sympathetic nerve is responsible for preventing Fas induced fulminant hepatitis.
It is well established that NE binds principally to α and β1 adrenoceptors.27 Moreover, the distribution of β1 adrenoceptors could not be shown in the murine liver by a radioligand binding assay.28,29 Therefore, together with the present finding that systemic treatment with an α adrenoceptor antagonist (phentolamine 20 μg/g weight intraperitoneally) did not cause any elevation in Fas induced mortality in normal mice (data not shown), it seems likely that local α adrenoceptors in the liver specifically mediate the protective effect of NE released from the hepatic sympathetic nerve on Fas induced fulminant hepatitis. This possibility is further supported by our observation that a Jo-2 induced elevation in NE levels of normal mice was exhibited locally in the liver but not in the general circulation (NE levels in plasma: before Jo-2 treatment 14.3 (1.8) ng/ml; one hour after Jo-2 treatment 14.7 (0.9) ng/ml, n = 5).
Although IL-6 was originally identified as an immunomodulator that regulates the promotion of B lymphocyte differentiation to antibody forming cells and stimulation of T lymphocyte proliferation and differentiation,30 recent reports have also demonstrated that IL-6 exerts the following antiapoptotic effect: pretreatment with IL-6 protects mice from fulminant hepatitis following various types of lethal challenges, such as warm ischaemia/reperfusion,31Staphylococcus aureus endotoxin B following d-galactosamine sensitisation,32 and a high dose of lipopolysaccharide.33 A recent study performed by Kovalovich and colleagues20 has proposed an animal model explaining the antiapoptotic effect of IL-6. Based on their findings that expression levels of FLIP, Bcl-xL, and Bcl-2 were observed in the liver of wild-type mice for 12 hours after Jo-2 treatment while decreasing rapidly in the liver of IL-6 deficient mice, they speculated that IL-6 may maintain adequate levels of FLIP, Bcl-xL, and Bcl-2 proteins in liver cells to render them more resistant to Fas mediated apoptosis, possibly via post-transcriptional mechanisms. Therefore, the present finding that expression levels of FLIP, Bcl-xL, and Bcl-2 following Jo-2 treatment were more profoundly reduced in sympathectomised compared with sham operated livers, suggests that IL-6 may participate in the protective effect of the hepatic sympathetic nervous system on Fas induced fulminant hepatitis. Indeed, IL-6 supplementation was shown to completely suppress the elevation in Fas induced mortality in sympathectomised mice while no such suppression was found in sham operated animals. This indicates that IL-6, at least in part, is involved in the hepatic sympathetic nervous prevention of Fas induced fulminant hepatitis, probably by maintaining expression of FLIP, Bcl-xL, and Bcl-2.
In this study, we found that hepatic sympathectomy significantly reduced IL-6 levels in the liver after Jo-2 treatment, which suggests that NE may maintain liver IL-6 levels to protect against Fas induced liver injury. At present, the precise mechanism involved in such maintenance of IL-6 remains unclear; however, one possible explanation may be that NE maintains IL-6 levels via a Ca2+ regulated protease inhibitor because NE signals have been reported to be mediated through α adrenoceptors as a Ca2+ signal.34 Another explanation may be associated with the possibility that NE decreases clearance of IL-6 from the liver. Clearly, further investigations are needed to clarify the precise mechanisms behind modulation of NE in the production, maintenance, and resolution of IL-6 in the liver.
We cannot exclude the possibility that hepatocyte growth factor, vascular endothelial growth factor, granulocyte colony stimulating factor, and other factors may also be involved in the present findings. In fact, a recent important report by Oben and colleagues35 showed that granulocyte colony stimulating factor and hepatocyte growth factor expression levels in the liver increase in mice fed antioxidant depleted diets to induce liver injury, and that systematic adrenergic blockade with either prazosin, an α1 adrenoceptor antagonist, or 6-OHDA, reduces liver damage in this model. This opposing effect of sympathetic nerve activation leads to the following important question: what effect does the sympathetic nerve system have on liver injury, defensive or harmful? We cannot answer this question at present but one possible explanation is that acute and transient activation of the sympathetic nerve may exert a beneficial effect on liver injury, which is not the case when chronic or prolonged activation of this nerve occurs. A new project in our laboratory is now in progress to further clarify the influence of chronic sympathetic nerve activation in carbon tetrachloride induced liver cirrhosis in mice.
In conclusion, the hepatic sympathetic nerve was found to be involved in the prevention of Fas mediated fulminant hepatitis and death. This new finding provides us with important insights into the pathophysiological significance of the hepatic sympathetic nerve in maintaining normal liver function.
Acknowledgments
This study was supported by grants in aid for General Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (No 16390200, No 17390210, and No 17659205).
Abbreviations
6-OHDA, 6-hydroxydopamine
NE, norepinephrine
IL-6, interleukin 6
PBS, phosphate buffered saline
TH, tyrosine hydroxylase
H&E, haematoxylin-eosin
TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labelling
FLIP, FLICE inhibitory protein
Conflict of interest: None declared.
REFERENCES
- 1.Bhathal PS, Grossman H. Nerve supply and nervous control of liver function. In: Bircher J, Benhamou JP, McIntyre N, et al, eds. In: Oxford textbook of clinical hepatology, 2nd edn. London: Oxford Medical Publications, 1999:165–7.
- 2.Lautt WW. Afferent and efferent neural roles in liver function. Prog Neurobiol 1983;21:323–48. [DOI] [PubMed] [Google Scholar]
- 3.Miyazawa Y, Fukuda Y, Imoto M, et al. Immunohistochemical studies on the distribution of nerve fibers in chronic liver diseases. Am J Gastroenterol 1988;83:1108–14. [PubMed] [Google Scholar]
- 4.Nakade Y, Yoneda M, Nakamura K, et al. Involvement of endogenous CRF in carbon tetrachloride-induced acute liver injury in rats. Am J Physiol 2002;282:R1782–8. [DOI] [PubMed] [Google Scholar]
- 5.Yokohama S, Yoneda M, Nakamura K, et al. Effect of central corticotropin-releasing factor on carbon tetrachloride-induced acute liver injury in rats. Am J Physiol 1999;276:G622–8. [DOI] [PubMed] [Google Scholar]
- 6.Cucchiaro G, Yamaguchi Y, Milles E, et al. Evaluation of selective liver denervation methods. Am J Physiol 1990;259:G781–5. [DOI] [PubMed] [Google Scholar]
- 7.Wheatley AM, Shaw SG, Stuart ET, et al. Selective sympathectomy of the liver: a comparison of orthotopic liver transplantation and intraportal 6-hydroxydopamine injection. J Auton Pharmacol 1993;13:15–22. [DOI] [PubMed] [Google Scholar]
- 8.Kruszewska B, Felten SY, Moynihan JA. Alteration in cytokines and antibody production following chemical sympathectomy in two strains of mice. J Immunol 1995;155:4613–20. [PubMed] [Google Scholar]
- 9.Lautt WW, Cote MG. The effect of 6-hydroxydopamine-induced hepatic sympathectomy on the early hyperglycemic response to surgical trauma under anesthesia. J Trauma 1977;17:270–4. [DOI] [PubMed] [Google Scholar]
- 10.Tiegs G, Bang R, Neuhuber WL. Requirement of peptidergic sensory innervation for disease activity in murine models immune hepatitis and protection by β-adrenergic stimulation. J Neuroimmunol 1999;96:131–43. [DOI] [PubMed] [Google Scholar]
- 11.Holmin T, Ekelund M, Kullendorff CM, et al. A microsurgical method for denervation of the liver in the rat. Eur Sug Res 1984;16:288–93. [DOI] [PubMed] [Google Scholar]
- 12.Lautt WW. Evaluation of surgical denervation of the liver in cats. Can J Physiol Pharmacol 1989;59:1013–16. [DOI] [PubMed] [Google Scholar]
- 13.Adachi M, Suematsu S, Kondo T, et al. Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nat Genet 1995;11:294–300. [DOI] [PubMed] [Google Scholar]
- 14.Afford SC, Randhawa S, Eliopoulos AG, et al. CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface Fas ligand expression and amplified Fas-mediated hepatocyte death during allograft refection. J Exp Med 1999;189:441–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondo T, Suda T, Fukuyama H, et al. Essential roles of the Fas ligand in the development of hepatitis. Nat Med 1997;3:409–13. [DOI] [PubMed] [Google Scholar]
- 16.Ogasawara J, Watanabe-Fukunaga R, Adachi M, et al. Lethal effect of the anti-Fas antibody in mice. Nature 1993;364:806–9. [DOI] [PubMed] [Google Scholar]
- 17.Tokushige K, Yamaguchi N, Ikeda I, et al. Predominant T helper 1 cells in patients with idiopathic portal hypertension. Am J Gastroenterol 2000;95:2040–6. [PubMed] [Google Scholar]
- 18.Chida Y, Sudo N, Sonoda J, et al. Electric foot-shock stress-induced exacerbation of α-galactosylceramide-triggered apoptosis in the mouse liver. Hepatology 2004;39:1131–40. [DOI] [PubMed] [Google Scholar]
- 19.De Bie JJ, Henricks PAJ, Cruikshank WW, et al. Modulation of airway hyperresponsiveness and eosinophilia by selective histamine and 5-HT receptor antagonists in a mouse model of allergic asthma. Br J Phramacol 1998;124:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovalovich K, Li W, DeAngelis R, et al. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem 2001;276:26605–13. [DOI] [PubMed] [Google Scholar]
- 21.Yu PL, Fijimura M, Okumiya K, et al. Immunohistochemical localization of tryptophan hydroxylase in the human and rat gastrointestinal tracts. J Comp Neurol 1999;411:654–65. [DOI] [PubMed] [Google Scholar]
- 22.Burt AD, Tiniakos D, MacSween RNM, et al. Localization of adrenergic and neuropeptide tyrosin-containing nerves in the mammalian liver. Hepatology 1989;9:839–45. [DOI] [PubMed] [Google Scholar]
- 23.Benarroch EE. Neuropeptides in the sympathetic system: presence, plasticity, modulation, and implication. Ann Neurol 1999;36:6–13. [DOI] [PubMed] [Google Scholar]
- 24.Haga S, Terui K, Zhang HQ, et al. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest 2003;112:989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liedtke C, Trautwein C. A protective role of Stat3 in Fas mediated apoptosis of the liver. J Hepatol 2004;40:874–5. [DOI] [PubMed] [Google Scholar]
- 26.André C, Couton D, Gaston J, et al. β2-adrenaergic receptor-selective agonist clenbuterol prevents Fas-induced liver apoptosis and death in mice. Am J Physiol 1999;276:G647–54. [DOI] [PubMed] [Google Scholar]
- 27.Jacob LS. Sympathomimetic agents. In: Jacob LS, ed. In: Pharmacology, 4th edn. Maryland: Williams and Wilkins, 1996:22–30.
- 28.Ota A, Matsui H, Asakura M, et al. Distribution of β1- and β2-adrenoceptor subtypes in various mouse tissues. Neurosci Lett 1993;160:96–100. [DOI] [PubMed] [Google Scholar]
- 29.Schmelck PH, Hanoune J. The hepatic adrenergic receptors. Mol Cell Biochem 1980;33:35–48. [DOI] [PubMed] [Google Scholar]
- 30.Van Snick J. Interleukin-6: an overview. Annu Rev Immunol 1990;8:253–78. [DOI] [PubMed] [Google Scholar]
- 31.Camargo CA, Madden JF, Gao WF, et al. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocytes proliferation in the rodent. Hepatology 1997;26:1513–20. [DOI] [PubMed] [Google Scholar]
- 32.Yoshizawa K, Naruto M, Ida N. Injection time of interleukin-6 determines fatal outcome in experimental endotoxin shock. J Interferon Cytokine Res 1996;16:995–1000. [DOI] [PubMed] [Google Scholar]
- 33.Fischer M, Goldschmitt J, Peschel C, et al. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol 1997;15:142–5. [DOI] [PubMed] [Google Scholar]
- 34.Kajiyama Y, Ui M. Switching from α1- to β-subtypes in adrenergic response during primary culture of adult-rat hepatocytes as affected by the cell-to-cell interaction through plasma membranes. Biochem J 1994;303:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oben JA, Roskams T, Yang S, et al. Sympathetic nervous system inhibition increases hepatic progenitors and reduces liver injury. Hepatology 2003;38:664–73. [DOI] [PubMed] [Google Scholar]