

Abstract
Background and aims: Haeme oxygenase could play a role in the pathogenesis of arterial vasodilation in cirrhosis. The aim of this study was to verify the role of haeme oxygenase in the hyporesponsiveness to phenylephrine of small mesenteric arteries in rats with CCl4 induced cirrhosis, with and without ascites.
Methods: Pressurised small resistance mesenteric arteries were challenged with increasing doses of phenylephrine. Dose-response curves were evaluated under basal conditions, after inhibition of haeme oxygenase with chromium-mesoporphyrin, after inhibition of nitric oxide synthase (NOS) with NG-nitro-L-arginine-methyl-ester (L-NAME), and then after inhibition of both NOS and haeme oxygenase. Haeme oxygenase protein expression was also analysed.
Results: Twenty six control rats and 35 rats with cirrhosis (17 with and 18 without ascites) were studied. Response to phenylephrine was lower in non-ascitic and ascitic cirrhosis than in controls. Chromium-mesoporphyrin increased the response to phenylephrine only in ascitic cirrhosis (p<0.001). L-NAME increased the response to phenylephrine in controls (p<0.001) and in ascitic and non-ascitic cirrhosis (p = 0.002, p<0.001, respectively) but the final response in non-ascitic cirrhosis was similar to that of control rats while it remained impaired in ascitic cirrhosis. Addition of chromium-mesoporphyrin to L-NAME improved the response to phenylephrine in ascitic cirrhosis (p<0.01), with final values not different from those of the other two groups. Protein expression of the inducible isoform of haeme oxygenase was increased in the mesenteric vessels of cirrhotic rats.
Conclusion: Haeme oxygenase mediates hyporeactivity to phenylephrine in the mesenteric vessels of experimental cirrhosis with ascites. NOS plays a major role only in the first stage of the disease.
Keywords: portal hypertension, experimental liver cirrhosis, mesenteric resistance arteries, chromium mesoporphyrin, nitric oxide
Haeme oxygenase (HO) is a microsomal enzyme with two main distinct isoforms—namely, inducible (HO-1) and constitutive (HO-2).1–3 It catalyses the rate limiting step in the degradation of haeme into biliverdin, carbon monoxide (CO), and free iron.2,4 CO generated in endothelial and smooth muscle layers of blood vessels by HO modulates vascular tone by inducing relaxation of vascular smooth muscle cells by stimulating soluble guanylyl cyclase, opening large conductance calcium activated K+ channels, and inhibiting the cytochrome P450 dependent monooxygenase system,1,5 with a decrease in 20-hydroxyeicosatetraenoic acid (20-HETE),6 which sustains contractile tone by inhibiting potassium channels.
Increased expression of HO-1 has been reported in the mesenteric artery of rats with prehepatic portal hypertension7 and with common bile duct ligation,8 and increased expression of HO-2 has been reported in rats with CCl4 cirrhosis.9 HO inhibition improved pressure response to vasoconstrictors of the mesenteric system evaluated according to McGregor,10 both in portal hypertensive rats11 and in CCl4 cirrhotic rats,9 and it improved alterations in systemic haemodynamics of rats with secondary biliary cirrhosis.8 Therefore, the HO/CO system may play a role in the mesenteric vasodilation of experimental portal hypertension but a series of questions have yet to been answered. Indeed, the role of HO in the regulation of small resistance mesenteric arteries has not yet been analysed, nor has its involvement in the different stages of experimental cirrhosis. Moreover, the relationship between the HO/CO and nitric oxide synthase (NOS)/nitric oxide (NO) systems in cirrhosis deserves further study.
The aim of the study was to investigate the role of HO in the regulation of small resistance mesenteric arteries in cirrhosis. The effect of the HO inhibitor chromium mesoporphyrin (CrMP) on phenylephrine (PE) induced contraction of small resistance mesenteric arteries was evaluated in rats with experimental cirrhosis with and without ascites. As the vasodilating effect of both NO and CO is mediated, at least in part, by the same mechanisms,12 the effect of CrMP was also evaluated after NOS inhibition with NG-nitro-L-arginine-methyl-ester (L-NAME). Expression of HO and NOS isoforms was also evaluated, both in the main trunk of the mesenteric artery and in the small resistance mesenteric arteries.
MATERIALS AND METHODS
The study was performed on 61 adult male Wistar-Kyoto rats (Charles River, Calco, Italy); body weight was 200–225 g. Cirrhosis was induced using the CCl4 inhalation method in 35 rats drinking phenobarbital (0.30 g/l in drinking water), following a method described elsewhere.13 Treatment was followed for 10–16 weeks, and animals were free of phenobarbital for the last week before the experiment.14 The protocols were approved by the Institutional Animal Care and Use Committee. Under anaesthesia with ketamine hydrochloride (100 mg/kg body weight intramuscularly), a midventral laparotomy was performed and a section of small intestine was removed. The presence of ascites was confirmed by visual examination at laparotomy. After laparotomy, cirrhotic rats were classified as cirrhosis with or without ascites. Rats were then killed with an overdose of ketamine. Age matched animals were used as untreated controls. Two protocols were implemented.
Protocol 1: evaluation of small mesenteric arteries response to phenylephrine in CCl4 cirrhotic rats
Isolated microvessel preparation
The clamped section of the small intestine was placed in a chilled oxygenated modified Krebs bicarbonate buffer (polysaline solution: PSS) containing 118.5 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.8 mM CaCl2, 25 mM NaHCO3, and 11 mM dextrose.
Third/fourth order branches of the superior mesenteric artery (170–350 µm in diameter, 1–2 mm in length) were isolated from surrounding perivascular tissue, removed from the mesenteric vascular bed, and mounted on glass micropipettes in a water jacked perfusion chamber (Living Systems Instrumentation, Burlington, Vermont, USA) in warmed (37°C), oxygenated (95% O2 and 5% CO2) PSS. The vessels were mounted on a proximal micropipette connected to a pressure servo controller.15 Subsequently, the lumen of the vessel was flushed to remove residual blood and the end of the vessel was mounted on a micropipette connected to a three way stopcock. After the stopcock was closed, intraluminal pressure was allowed to increase slowly until it reached 80 mm Hg. The vessel was superfused with PSS (4 ml/min) at 37°C and gassed with 95% O2 and 5% CO2 for a 45 minute period of equilibration.15 Intraluminal pressure was maintained at 80 mm Hg during the experiment. After the equilibration period, the vessels were challenged with a submaximal dose of PE, an α1-adrenoreceptor agonist (10−6 M). An artery was considered unacceptable for experimentation if it demonstrated leaks or failed to constrict to >20% to PE. The presence of a functional endothelium was determined on the basis of relaxation evoked by acetylcholine (10−6 M) in the vessel precontracted with PE (10−6 M). Arteries with less than 60% relaxation of PE induced contractions were discarded.
The effect of increasing doses of PE was evaluated as changes in internal diameter of the vessel.
Evaluation of the haemodynamic effect of inhibition of HO and/or NOS on the response to phenylephrine of small mesenteric arteries
Responses to PE (10−8–10−4 M) were determined in PSS containing vehicles for the inhibitors tested. Inhibitors were added to freshly prepared PSS, and 20–30 minutes of drug-tissue contact time was allowed before retesting the response to the agonist PE in the same vessel. PE was added to the bath (extraluminal application), and cumulative dose-response curves were generated, with 2–3 minutes between doses. After each dose-response test, the tissues were washed with fresh PSS for at least 20 minutes. Vascular diameters were measured 1–3 minutes after addition of PE to the bath with the use of a video system composed of a microscope with a CCD television camera (Eclipse TS100-F; Nikon, Tokyo, Japan), a television monitor (Ultrak Inc., Lewisville, Texas, USA), and a video measuring system (Systems Instrumentation). In both control and cirrhotic rats (ascitic and non-ascitic), dose-response curves to PE were evaluated: (a) before and after 20 minutes of superfusion with the HO inhibitor CrMP (15 µM); (b) before and after 30 minutes of superfusion with the NOS inhibitor L-NAME (1 mM); (c) after a further 20 minutes of CrMP (15 µM) plus L-NAME (1 mM) superfusion in rats already evaluated after L-NAME superfusion alone. In each artery only one experiment was performed.
Chemicals
CrMP was obtained from Porphyrin Products (Logan, Utah, USA). All other chemicals were obtained from Sigma Chemical (St Louis, Missouri, USA). PE and L-NAME were dissolved in deionised water and diluted with PSS. CrMP was dissolved in a solution of 50 mM NaCO3.
Protocol 2: western blot analysis of HO-1, HO-2, endothelial NOS (eNOS), and inducible NOS (iNOS) protein expression in mesenteric arteries of CCl4 cirrhotic rats
Standard techniques were used to evaluate protein expression. After removal of veins and adipose tissue, small mesenteric arteries (30–40 arteries with diameter <500 µm) and the main trunk of the mesenteric artery were separately collected for every rat after residual blood had been removed, snap frozen in liquid N2, and stored at −80°C until analysed. The vessels were homogenised in urea lysis buffer. Protein extracts were assayed for protein content using the BCA protein assay kit (Pierce, Rockford, Illinois, USA). Sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotting were performed on 50 µg of total protein extracts. HO-1, HO-2, iNOS, and eNOS protein expression were detected using polyclonal rabbit anti-HO-2, iNOS, and eNOS antibodies (StressGen Biotechnologies Corp., Victoria, Canada) and a monoclonal murine antibody against HO-1 (StressGen Biotechnologies Corp.). The secondary antibodies antirabbit and antimouse conjugated to horseradish peroxidase, respectively, were diluted 1:1000 in phosphate buffered saline containing 2% non-fat dry milk. Antigenic detection was visualised by standard ECL enhanced chemiluminescence (Amersham, Arlington Heights, Illinois, USA) with exposure to x ray film. Recombinant soluble protein rat Hsp-32 (StressGen Biotechnologies Corp.) was used as the positive control for HO-1, recombinant human HO-2 (StressGen Biotechnologies Corp.) was used as the positive control for HO-2, synthetic peptide corresponding to amino acids 1131–1144 of mouse macrophage NOS (US Biological, Swampscott, Massachusetts, USA) was used as the positive control for iNOS, and synthetic peptide corresponding to amino acids 596–610 of human eNOS (Chemicon International, Temecula, California, USA) was used as the positive control for eNOS. Protein expression was determined by densitometric analysis using the VersaDoc Imaging System (Bio-Rad Laboratories, Hercules, California, USA). After stripping, blots were assayed for β-actin content as standardization of sample loading. Quantitative densitometric values of each protein of interest were normalised to β-actin and displayed in histograms.
Data analysis
Data are expressed as mean (SEM). All responses were measured as percentage of contraction (that is, reduction in vessel diameter relative to baseline diameter before addition of agonist or antagonist). Concentration-response data derived from each vessel were fitted separately to a logistic function by non-linear regression, and EC(50) (molar concentration of PE causing 50% contraction) was calculated and expressed as −log [M]. A two way ANOVA was used to compare dose-response curves between controls and treated groups. Other data were analysed by one way ANOVA or the Student’s t test for paired or unpaired observations when appropriate. The null hypothesis was rejected at p<0.05.
RESULTS
All rats treated with CCl4 included in the study had macronodular or micronodular cirrhosis.
In 17 of 35 cirrhotic rats the presence of ascites was confirmed by visual examination at laparotomy. Control rats had no appreciable alteration in liver appearance.
Duration of treatment (CCl4 inhalation) was 12 (1) weeks in rats without ascites and 15 (1) weeks in ascitic rats. At the time of the study no difference in body weight between cirrhotic (non-ascitic rats 549 (14) g; ascitic rats 532 (14) g) and control rats (539 (17) g) was observed.
Protocol 1: haemodynamic study
Baseline results
Mesenteric vascular response to PE was blunted in cirrhotic rats, both in ascitic and non-ascitic animals (p<0.001, two-way ANOVA) (fig 1 ▶). The response of the ascitic group was not significantly lower than that of non-ascitic animals. EC(50) was 6.14 (0.12) −log[M] in controls (n = 26) versus 5.49 (0.11) −log[M] in cirrhotics (n = 35) (p<0.001). Analysing separately ascitic (n = 17) and non-ascitic (n = 18) rats, EC(50) was lower in controls rats compared with both non-ascitic (5.62 (0.18) −log[M]; p = 0.014) and ascitic (5.35 (0.14) −log[M]; p<0.001) rats. Among cirrhotic rats, EC(50) was not different between ascitic and non-ascitic rats (NS).
Figure 1.
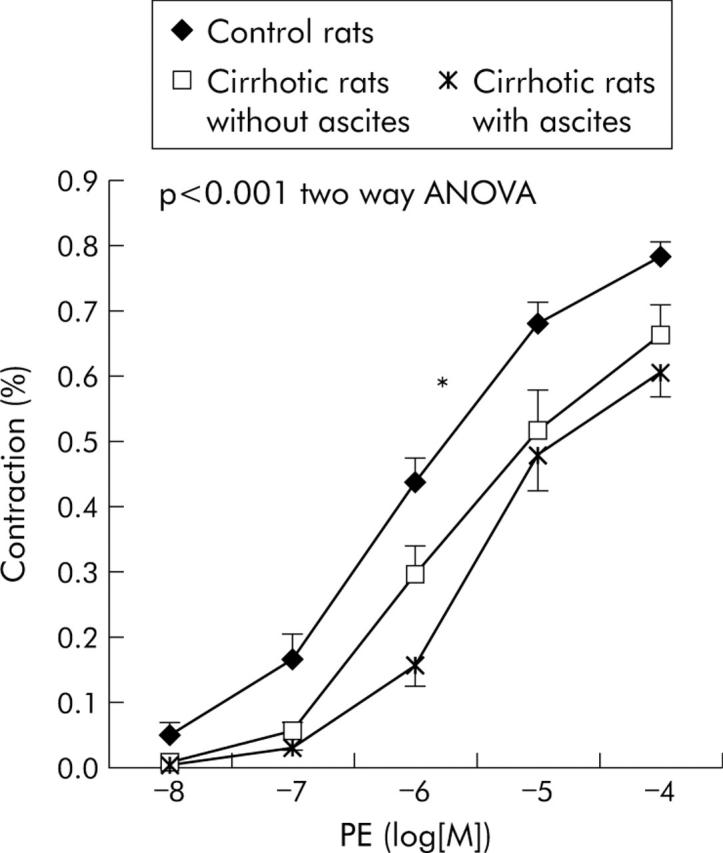
Dose-response curves to phenylephrine (PE) of small resistance mesenteric arteries in the three different groups of rats. *Significantly different (p<0.01) from the other two curves.
Effect of CrMP
CrMP did not modify the dose-response curve to PE in control rats (n = 9) (NS, two way ANOVA); EC(50) to PE was 6.17 (0.17) −log[M] before and 6.17 (0.12) −log[M] after CrMP (NS).
In contrast, a significant decrease in EC(50) was evident in cirrhotic rats after CrMP (n = 14): from 5.71 (0.09) −log[M] to 6.03 (0.12) −log[M] (p = 0.010). However, analysing separately ascitic (n = 6) and non-ascitic (n = 8) rats, we found that CrMP produced a leftward displacement in the concentration-response curve to PE only in ascitic rats (p<0.001, two-way ANOVA) (fig 2 ▶). EC(50) to PE changed form 5.75 (0.22) −log[M] to 5.96 (0.18) −log[M] (NS) in cirrhotic rats without ascites while it decreased significantly from 5.64 (0.13) −log[M] to 6.12 (0.18) −log[M] (p = 0.010) in rats with cirrhosis and ascites.
Figure 2.
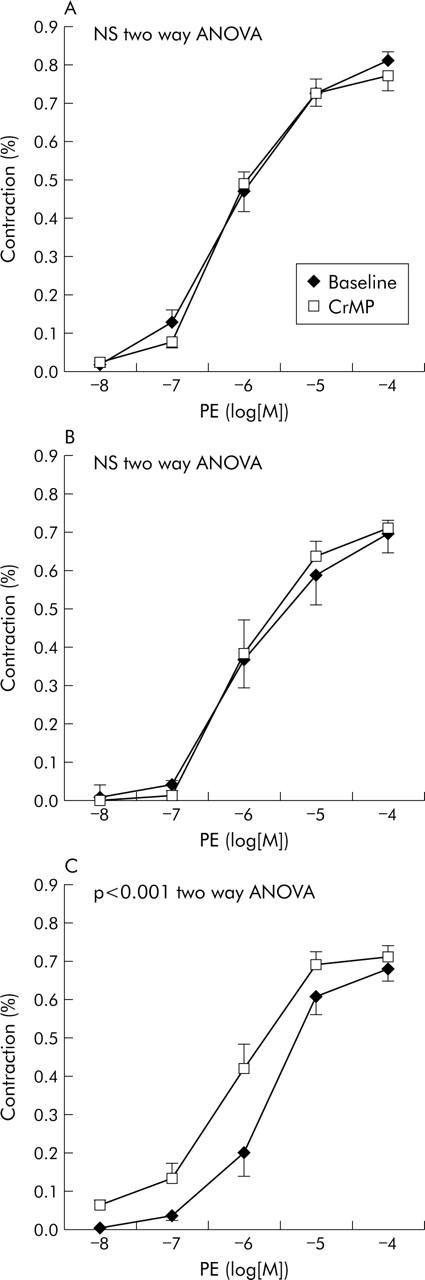
Effect of haeme oxygenase inhibition with chromium mesoporphyrin (CrMP) on mesenteric vascular response to phenylephrine (PE) in (A) control rats (n = 9), (B) cirrhotic rats without ascites (n = 8), and (C) cirrhotic rats with ascites (n = 6).
Effect of L-NAME
L-NAME caused an increase in the vascular response to PE in both control rats (n = 14) and cirrhotic rats, with (n = 7) or without (n = 6) ascites, as demonstrated by the leftward shift of the dose-response curves (p<0.001, p = 0.002, p<0.001, respectively, two-way ANOVA) (fig 3 ▶) and by the significant decrease in EC(50): from 6.10 (0.18) −log[M] to 6.47 (0.17) −log[M] (p = 0.010) in control rats; from 5.68 (0.22) −log[M] to 6.45 (0.18) −log[M] (p = 0.009) in non-ascitic rats; and from 5.45 (0.15) −log[M] to 5.83 (0.19) −log[M] (p = 0.008) in ascitic rats.
Figure 3.
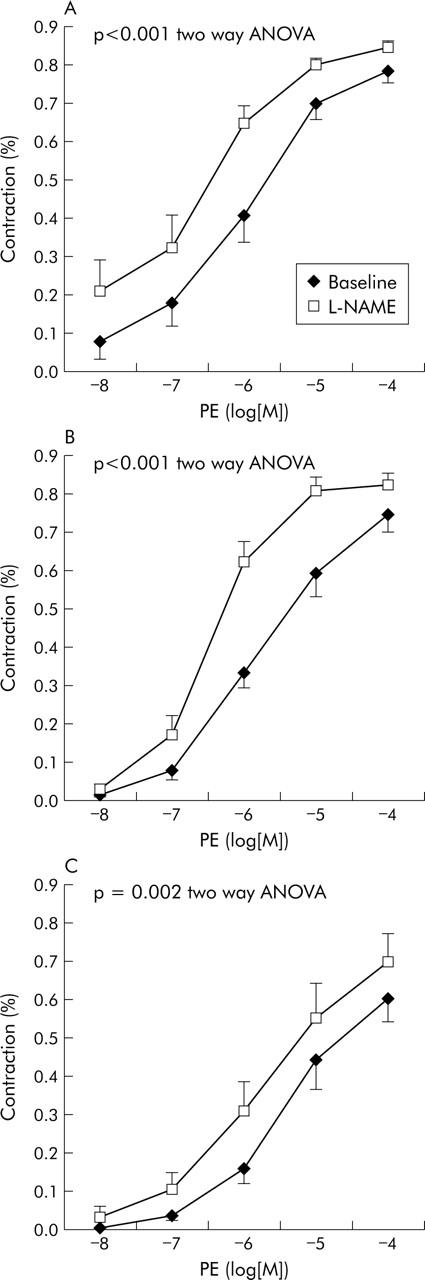
Effect of nitric oxide synthase inhibition with NG-nitro-L-arginine-methyl-ester (L-NAME) on mesenteric vascular response to phenylephrine (PE) in (A) control rats (n = 14), (B) cirrhotic rats without ascites (n = 6), and (C) cirrhotic rats with ascites (n = 7).
However, after L-NAME, EC(50) to PE was similar in controls and in non-ascitic cirrhotic rats (NS) while in ascitic cirrhotic rats it remained significantly higher compared with both controls (p = 0.030) and non-ascitic cirrhotic rats (p = 0.027).
Effect of addition of CrMP to L-NAME
In control rats (n = 5) and in rats with cirrhosis without ascites (n = 6), addition of CrMP to L-NAME did not modify the dose-response curve to PE obtained after L-NAME administration (NS, two way ANOVA). EC(50) changed from 6.32 (0.15) −log[M] to 6.33 (0.16) −log[M] (NS) in control rats and from 6.27 (0.16) −log[M] to 6.32 (0.13) −log[M] (NS) in cirrhotic rats without ascites. In contrast, in rats with cirrhosis and ascites (n = 6), addition of CrMP to L-NAME caused a significant leftward shift of the dose-response curve to PE compared with the curve obtained after L-NAME alone (p<0.01 compared with the L-NAME curve; p<0.001 compared with the baseline curve; two way ANOVA) (fig 4 ▶). In ascitic rats, the EC(50) decreased from 5.65 (0.21) −log[M] after L-NAME to 6.32 (0.21) −log[M] after L-NAME+CrMP (p = 0.05). After inhibiting both NOS and HO, the vascular response to PE was similar in control and cirrhotic rats, both with and without ascites (NS).
Figure 4.
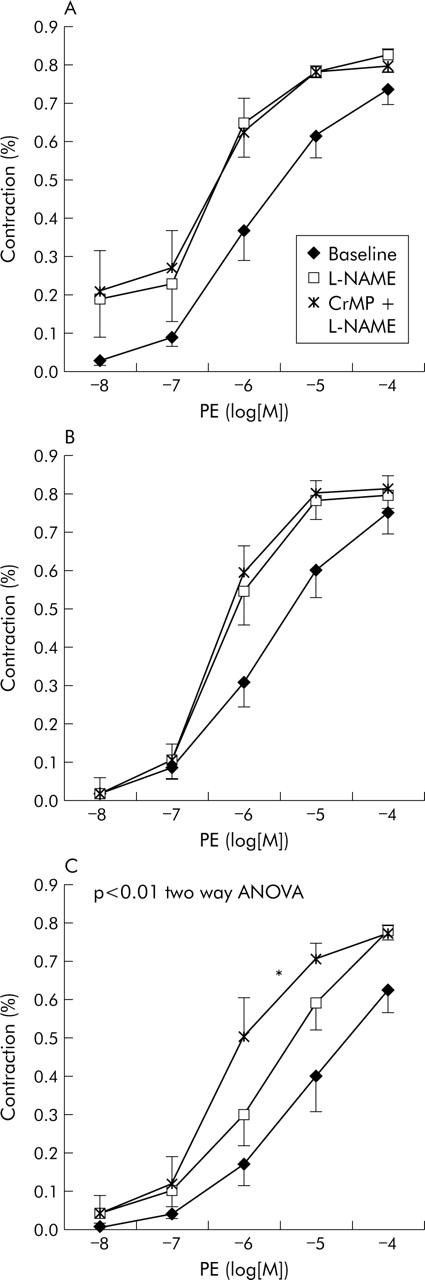
Effect of haeme oxygenase inhibition with chromium mesoporphyrin (CrMP) on mesenteric vascular response to phenylephrine (PE), in arteries treated with nitric oxide synthase inhibition with NG-nitro-L-arginine-methyl-ester (L-NAME). (A) Control rats (n = 5); (B) cirrhotic rats without ascites (n = 6); and (C) cirrhotic rats with ascites (n = 6). *Significantly higher (p<0.01) than baseline and L-NAME alone.
Protocol 2: western Blot analysis
In mesenteric arteries of control rats, eNOS and HO-2 protein expression were evident, while iNOS protein expression was absent and that of HO-1 was low. eNOS protein expression was greater in the small mesenteric arteries compared with the main trunk of the artery (p = 0.006) while HO-2 protein was equally expressed in the main trunk of the artery and in the small vessels (figs 5 ▶, 6 ▶).
Figure 5.

Western blot analysis of inducible haeme oxygenase (HO-1), constitutive haeme oxygenase (HO-2), inducible nitric oxide synthase (iNOS), and endothelial nitric oxide synthase (eNOS) in the main trunk of the mesenteric artery and in the small resistance mesenteric vessels of controls rats and rats with and without ascites. The reported blots are representative of 4–7 experiments. Examples of β-actin expression, analysed as an index of the adequacy of sample loading, are also displayed.
Figure 6.

Quantitative densitometric evaluation, normalised to β-actin, of inducible nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), inducible haeme oxygenase (HO-1), and constitutive haeme oxygenase (HO-2) in the main trunk of the mesenteric artery and in the small resistance mesenteric vessels of control rats and of rats with and without ascites. Protein expression in the main trunk of the mesenteric artery and in the small resistance mesenteric arteries is shown. Results are from 4–7 experiments. *p = 0.039, **p = 0.027, ***p = 0.007 compared with control rats; †p = 0.006 compared with the main trunk of the artery.
In cirrhosis without ascites, there was in increase in protein expression of eNOS (particularly in the main trunk of the mesenteric artery) and of HO-1 (particularly in the small vessels) (p = 0.039).
In rats with cirrhosis and ascites, HO-1 protein expression further increased compared with rats without ascites in the main trunk of the artery while, surprisingly, eNOS protein expression decreased compared with cirrhotic rats without ascites (figs 5 ▶, 6 ▶).
In cirrhosis, very weak iNOS expression was detected in only one sample of six non-ascitic rats and in only one sample of five rats with ascites (fig 6 ▶). HO-2 protein expression was not modified in cirrhosis (fig 6 ▶).
DISCUSSION
This study provides information on the influence of HO on the response to PE of small mesenteric arteries in experimental cirrhosis. The salient conclusion derived from the study is that overexpression of HO participates in the decreased mesenteric response to PE only in the advanced stage of the disease while NOS overexpression mainly participates in the first stage of the disease.
Vascular resistance is more dependent on small rather than large vessels16 and there is evidence that small and large arteries have different physiological regulatory systems.16 Low splanchnic vascular resistance observed in portal hypertension depends mostly on mesenteric resistance arteries, which are precapillary resistance arteries with diameters of less than 500 µm.16 Therefore, we explored the vascular response and protein expression directly in small resistance mesenteric arteries of less than 500 µm. The no-flow model was chosen to avoid interference by the shear stress phenomenon.
Small resistance mesenteric arteries of cirrhotic rats were hyporesponsive to PE. This result is in keeping with previous studies which have demonstrated a blunted pressure response of the perfused superior mesenteric arterial bed to KCl in cirrhotic rats with ascites14 and to methoxamine in portal hypertensive rats,17 and an impaired response to PE and KCl in mesenteric resistance arterial rings from portal hypertensive rats.18 Hyporesponsiveness was present in both ascitic and non-ascitic rats, highlighting that there is a decreased response of the mesenteric artery to vasoconstrictors from the first stage of the disease.
To evaluate the role of HO on the mesenteric response to PE, we analysed the effect of CrMP, a potent non-selective HO inhibitor with no significant effect on NOS activity.19 HO inhibition did not modify the vascular response to PE in control rats, in keeping with the lack of effect of HO inhibitors on perfusion pressure of the mesenteric arterial bed of controls rats.9 In contrast, HO inhibition improved the vascular response in cirrhotic rats, as already suggested by Fernandez and colleagues,11 who studied portal hypertensive rats, and by Sacerdoti and colleagues,9 who investigated cirrhotic rats, both using the McGregor preparation.10 But in our study the improvement was evident only in ascitic rats. Higher HO-1 protein expression was evident in cirrhotic rats, particularly in those with ascites.
NOS inhibition caused an increase in the mesenteric response to PE both in control rats and in cirrhotic rats, according to the study of Sieber and colleagues,14 who verified the pressure response to KCl in the perfused mesenteric arterial bed. However, it is of particular interest that in our study L-NAME completely reversed mesenteric hyporesponsiveness to PE in compensated cirrhosis but was not as effective in ascitic cirrhosis. Similar results were recently obtained in our laboratory analysing the splanchnic haemodynamics of cirrhotic rats in vivo by a perivascular ultrasonic flow probe applied to the main trunk of the mesenteric artery.20 In this study, L-NAME decreased blood flow and increased resistance in the superior mesenteric artery in cirrhotic rats but the effect was much less intense in rats with ascites. These data suggest that NOS activation is the main factor responsible for mesenteric vasodilation in the first stage but is not the only factor in the advanced stage of the disease. This hypothesis is in agreement with the findings of Forrest and colleagues21 who reported that L-NAME improved heart rate and systemic arterial pressure in compensated but not in decompensated cirrhotic patients. These authors hypothesised that in the advanced stage of the disease, NO plays a minor role in the pathogenesis of hyperdynamic circulation, overcome by other vasoactive systems. Our analysis of NOS protein expression supports this interpretation. Indeed, eNOS protein expression in small mesenteric arteries was increased in cirrhosis without ascites, in accordance with other studies that analysed eNOS protein expression in the superior mesenteric artery vascular bed of portal hypertensive rats22,23 and in the proximal 1 cm of the main trunk of the same artery of cirrhotic rats, both with and without ascites.24
But surprisingly, in the advanced stage of the disease (ascitic phase), mesenteric eNOS protein expression was not increased, in accordance with the studies of Morales-Ruiz and colleagues25 who analysed the mesenteric arterial bed of cirrhotic rats with ascites.
Mesenteric iNOS expression was absent in control rats and almost negligible in cirrhotic rats. Therefore, very low expression of iNOS could not be excluded in cirrhotic animals, even though such low levels are probably not significant.
Considering that (a) CO and NO cause smooth muscle cell relaxation interplaying on the same mechanisms and (b) NO seems to be primarily responsible for mesenteric hyporesponsiveness to vasoconstrictors in cirrhosis, we also decided to evaluate the effect of CrMP in rats previously treated with L-NAME. In control rats and cirrhotic rats without ascites, addition of CrMP to L-NAME did not further increase the vascular response compared with the effect of L-NAME alone, while in cirrhotic rats with ascites, HO inhibition was effective in improving mesenteric response to PE. By inhibiting both NOS and HO, the mesenteric response to PE was similar in control rats and cirrhotic rats, with and without ascites. Therefore, in the advanced stage of experimental cirrhosis, increased expression of HO and production of CO could participate in maintaining and worsening mesenteric vasodilation. This hypothesis is indirectly supported by the finding of an increased CO concentration in exhaled air and blood carboxyhaemoglobin reported in human cirrhosis, particularly in patients with ascites.26
Analysis of HO and NOS protein expression provided some interesting data. Firstly, contemporary analysis of the constitutive and inducible forms of the two enzymes in the two different stages of evolution of cirrhosis (non-ascitic and ascitic) allowed us to show that expression varies with progression of disease. In particular, in the ascitic phase of the disease, higher expression of HO-1 and lower expression of eNOS were evident. Secondly, when we analysed protein expression separately in the small resistance branches and in the main trunk of the mesenteric artery, we were able to discover that HO and NOS expression was different in the two regions. This emphasises the importance of analysing selectively the small resistance arteries when the aim is to evaluate regulation of mesenteric resistance. Increased expression of HO-1 has also been reported in mesenteric arteries of rats with prehepatic portal hypertension7 and with common bile duct ligation.8 In contrast, only increased expression of HO-2 has been reported in the mesentery of Sprague-Dawley rats with CCl4 cirrhosis by Sacerdoti et al.9 The difference may be explained by the different sites of protein expression analysis and by the different stages of cirrhosis. The different experimental model (Sprague-Dawley instead of Wistar-Kyoto rats) may also have played a role.
Differences in expression of eNOS and HO-1 in mesenteric vessels of rats with and without ascites may be explained by the relationship between the two systems. Indeed, NO is known to induce expression of HO-1,12,27–29 leading to formation of endogenous CO.27,29 Increased levels of HO in turn have been shown to decrease NO concentration.3 The mechanisms by which induction of HO-1 impairs local NO generation have been identified as follows: competitive consumption of NADPH between the two enzyme systems, degradation of the prosthetic haeme required for assembly of NOS, and CO binding to the NOS haeme.30,31
Hence a role for the HO/CO system in the mesenteric hyporesponsiveness to PE in experimental cirrhosis can be hypothesised. In the first stage of the disease, an increase in eNOS expression has been demonstrated,24 responsible for the early mesenteric hyporesponsiveness to vasoconstrictors. The chronic increase in NO levels might induce HO-1 expression, together with other mechanisms, such as high levels of oxidative stress, glucagon, and angiotensin II.28,32 An increasing role of the HO/CO system may therefore become evident in the advanced stage of the disease, and the interfering action of HO on NOS might contribute in shifting the balance towards the HO system. Indeed, activation of HO-1 may lead to a deficiency in intracellular haeme required as a coenzyme for NOS.30
Further studies are necessary to confirm our results. Indeed, the pathophysiological significance of our findings in isolated vessels will be enhanced if confirmed by measuring CO levels in the mesenteric circulation and by in vivo experiments assessing the effect of CrMP in the mesenteric circulation of cirrhotic rats with ascites.
In conclusion, HO plays a role in the mesenteric hyporesponsiveness of cirrhotic rats. In the early stages of cirrhosis, the NO/NOS system plays a major role in splanchnic vasodilation whereas in the late stages HO-1 derived CO seems to mediate further aggravation.
Abbreviations
CO, carbon monoxide
CrMP, chromium mesoporphyrin
EC(50), molar concentration of phenylephrine causing 50% contraction
eNOS, endothelial nitric oxide synthase
HO, haeme oxygenase
HO-1, inducible haeme oxygenase
HO-2, constitutive haeme oxygenase
iNOS, inducible nitric oxide synthase
L-NAME, NG-nitro-L-arginine methyl ester
NO, nitric oxide
NOS, nitric oxide synthase
PE, phenylephrine
PSS, polysalin solution
Published online first 10 June 2005
Conflict of interest: None declared.
REFERENCES
- 1.Zhang F, Kaide JI, Rodriguez-Mulero F, et al. Vasoregulatory function of heme-heme oxygenase-carbon monoxide system. Am J Hypertens 2001;14:62S–7. [DOI] [PubMed] [Google Scholar]
- 2.Ndisang JF, Zhao W, Wang R. Selective regulation of blood pressure by heme oxygenase-1 in hypertension. Hypertension 2002;40:315–21. [DOI] [PubMed] [Google Scholar]
- 3.Johnson FK, Durante W, Peyton KJ, et al. Heme oxygenase inhibitor restores arteriolar nitric oxide function in Dahl rats. Hypertension 2003;41:149–55. [DOI] [PubMed] [Google Scholar]
- 4.Motterlini R, Gonzales A, Foresti R, et al. Heme oxygenase-1-derived carbon monoxide contributes to the suppression of acute hypertensive responses in vivo. Circ Res 1998;83:568–77. [DOI] [PubMed] [Google Scholar]
- 5.Roman RJ. Gene therapy and heme oxygenase coming of age. Hypertension 2004;43:173–4. [DOI] [PubMed] [Google Scholar]
- 6.Kaide JI, Zhang F, Wei Y, et al. Vascular CO counterbalances the sensitizing influence of 20-HETE on agonist-induced vasoconstriction. Hypertension 2004;44:1–7. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez M, Bonkovsky H. Increased heme oxygenase-1 gene expression in liver cells and splanchnic organs from portal hypertensive rats. Hepatology 1999;29:1672–9. [DOI] [PubMed] [Google Scholar]
- 8.Chen YC, Gines P, Yang J, et al. Increased vascular heme oxygenase-1 expression contributes to arterial vasodilation in experimental cirrhosis in rats. Hepatology 2004;39:1075–87. [DOI] [PubMed] [Google Scholar]
- 9.Sacerdoti D, Abraham NG, Oyekan AO, et al. Role of the heme oxygenases in abnormalities of the mesenteric circulation in cirrhotic rats. J Pharmacol Exp Ther 2004;308:636–43. [DOI] [PubMed] [Google Scholar]
- 10.McGregor D. The effect of sympathetic nerve stimulation on vasoconstrictor responses in perfused mesenteric vessels of the rat. J Physiol (Lond) 1965;177:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez M, Lambrecht RW, Bonkovsky HL. Increased heme oxygenase activity in splanchnic organs from portal hypertensive rats: role in modulating mesenteric vascular reactivity. J Hepatol 2001;34:812–17. [DOI] [PubMed] [Google Scholar]
- 12.Hartsfield CL, Alam J, Cook JL, et al. Regulation of heme oxygenase-1 gene expression in vascular smooth muscle cells by nitric oxide. Am J Physiol Lung Cell Mol Physiol 1997;273:L980–8. [DOI] [PubMed] [Google Scholar]
- 13.Angeli P, Jimenez W, Veggian R, et al. Increased activity of guanosine 3–5-cyclic monophosphate phosphodiesterase in the renal tissue of cirrhotic rats with ascites. Hepatology 2000;31:304–10. [DOI] [PubMed]
- 14.Sieber C, Lopez-Talavera JC, Groszmann RJ. Role of nitric oxide in the in vitro splanchnic hyporeactivity in ascitic cirrhotic rats. Gastroenterology 1993;104:1750–4. [DOI] [PubMed] [Google Scholar]
- 15.Wang MH, Zhang F, Marji J, et al. CYP4A1 antisense oligonucleotide reduces mesenteric vascular reactivity and blood pressure in SHR. Am J Physiol Regul Integr Comp Physiol 2001;280:R255–61. [DOI] [PubMed] [Google Scholar]
- 16.Mulvany MJ, Aalkiaer C. Structure and function of small arteries. Physiol Rev 1990;70:921–61. [DOI] [PubMed] [Google Scholar]
- 17.Sieber CC, Groszmann RJ. In vitro hyporeactivity to methoxamine in portal hypertensive rats: reversal by nitric oxide blockade. Am J Physiol Gastrointest Liver Physiol 1992;262:996–1001. [DOI] [PubMed] [Google Scholar]
- 18.Sogni P, Sabry S, Moreau R, et al. Hyporeactivity of mesenteric resistance arteries in portal hypertensive rats. J Hepatol 1996;24:487–90. [DOI] [PubMed] [Google Scholar]
- 19.Appleton SD, Chretien ML, McLaughlin BE, et al. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metab Dispos 1999;27:1214–19. [PubMed] [Google Scholar]
- 20.Angeli P, Fernanez-Varo G, Dalla Libera V, et al. The role of nitric oxide in the pathogenesis of systemic and splanchnic vasodilation in cirrhotic rats before and after the onset of ascites. Liver Int 2005;25:429–37. [DOI] [PubMed] [Google Scholar]
- 21.Forrest EH, Jones AL, Dillon JF, et al. The effect of nitric synthase inhibition on portal pressure and azygos blood flow in patients with cirrhosis. J Hepatol 1995;23:254–8. [DOI] [PubMed] [Google Scholar]
- 22.Cahill PA, Redmond EM, Hodges R, et al. Increased endothelial nitric oxide synthase activity in the hyperemic vessels of portal hypertensive rats. J Hepatol 1996;25:370–8. [DOI] [PubMed] [Google Scholar]
- 23.Wiest R, Shah V, Sessa WC, et al. NO overproduction by eNOS precedes hyperdynamic splanchnic circulation in portal hypertensive rats. Am J Physiol Gastrointest Liver Physiol 1999;276:G1043–51. [DOI] [PubMed] [Google Scholar]
- 24.Martin PY, Xu DL, Niederberger M, et al. Upregulation of endothelial constitutive NOS: a major role in the increased NO production in cirrhotic rats. Am J Physiol Renal Fluid Electrolyte Physiol 1996;270:F494–9. [DOI] [PubMed] [Google Scholar]
- 25.Morales-Ruiz M, Jimenez W, Perez-Sala D, et al. Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology 1996;24:1481–6. [DOI] [PubMed] [Google Scholar]
- 26.De Las Heras D, Fernandez J, Gines P, et al. Increased carbon monoxide production in patients with cirrhosis with and without spontaneous bacterial peritonitis. Hepatology 2003;38:452–9. [DOI] [PubMed] [Google Scholar]
- 27.Durante W, Kroll MH, Christodoulides N, et al. Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth cells. Circ Res 1997;80:557–64. [DOI] [PubMed] [Google Scholar]
- 28.Moreau R. Heme oxygenase: protective enzyme or portal hypertensive molecule? J Hepatol 2001;34:936–9. [DOI] [PubMed] [Google Scholar]
- 29.Naughton P, Foresti R, Bains SK, et al. Induction of heme oxygenase 1 by nitrosative stress. A role for nitroxyl anion. J Biol Chem 2002;277:40666–74. [DOI] [PubMed] [Google Scholar]
- 30.Siow RCM, Sato H, Mann GE. Heme oxygenase-carbon monoxide signalling pathway in atherosclerosis: anti-atherogenic actions of bilirubin and carbon monoxide? Cardiovasc Res 1999;41:385–94. [DOI] [PubMed] [Google Scholar]
- 31.Suematsu M, Ishimra Y. The heme oxygenase-carbon monoxide system: a regulator of hepatobiliary functions. Hepatology 2000;31:3–6. [DOI] [PubMed] [Google Scholar]
- 32.Ishizaka N, Aizawa T, Mori I, et al. Heme oxygenase-1 is upregulated in the rat heart in response to chronic administration of angiotensin II. Am J Physiol Heart Circ Physiol 2000;279:H672–8. [DOI] [PubMed] [Google Scholar]