

Abstract
Selective activation of the peripheral cannabinoid receptor 1 (CB1R) has been shown to suppress neuropathic pain symptoms in rodents. However, relatively little is known about changes in CB1R and its endogenous ligands during development or maintenance of neuropathic pain. Using immunohistochemistry, Western blot, real-time reverse transcription polymerase chain reaction, as well as liquid chromatography/mass spectrometry, we studied the changes in CB1Rs and endocannabinoids N-arachidonoylethanolamine/anandamide (AEA) and 2-arachidonoylglycerol (2-AG) in rat lumbar (L4 and L5) dorsal root ganglia (DRG) after neuropathic pain induction (L5 spinal nerve ligation: SNL). Immunohistochemistry revealed that in control rats, CB1R is expressed in the majority (76–83%) of nociceptive neurons as indicated by co-labeling with isolectin B4 (IB4) or antibodies recognizing transient receptor potential vanilloid (TRPV1), calcitonin gene related peptide (CGRP), and the NR2C/2D subunits of the N-methyl-D-aspartate receptor. After L5 SNL, CB1R mRNA and protein increases in the ipsilateral uninjured L4 DRG whereas the percentages of CB1R immunoreactive (CB1R-ir) neurons remain unchanged in L4 and L5 DRG. However, for these CB1R-ir neurons, we observe significant increases in percentage of TRPV1-ir cells in ipsilateral L4 DRG, and decreases in percentage of IB4- and CGRP-co-labeled cells in ipsilateral L5 DRG. Levels of both AEA and 2-AG increase significantly only in the ipsilateral L5 DRG. These results are consistent with the preserved analgesic effects of cannabinoids in neuropathic pain and provide a rational framework for the development of peripherally acting endocannabinoid-based therapeutic interventions for neuropathic pain.
Keywords: Endocannabinoid system, L5 spinal nerve ligation, Cannabinoid 1 receptors, Anandamide, 2-arachidonoylglycerol
1. Introduction
Synthetic and naturally occurring cannabinoids are a focus of strong social, legal and medical controversy concerning their therapeutic utility, yet studies show that cannabinoids reduce the hyperalgesia and allodynia associated with persistent pain of neuropathic origin in humans (Karst et al., 2003; Berman et al., 2004; Notcutt et al., 2004) and animals (Herzberg et al., 1997; Fox et al., 2001). Furthermore, cannabinoids effectively alleviate neuropathic pain symptoms after repeated treatment (Bridges et al., 2001; Costa et al., 2004), unlike opioids, which have only limited effectiveness (Mao et al., 1995; Ossipov et al., 1995; Rashid et al., 2004).
The targets of the antinociceptive cannabinoids may be defined by the distribution of two cloned subtypes of cannabinoid receptors, CB1R and CB2R (Matsuda et al., 1990; Munro et al., 1993). Both are members of the G protein-coupled receptor (GPCR) superfamily of which CB1R is the most abundant central nervous system (CNS) GPCR expressed at high levels in the hippocampus, cortex, cerebellum and basal ganglia (Matsuda et al., 1990; Tsou et al., 1998), whereas CB2R is primarily expressed in immunocompetent cells (Lynn and Herkenham, 1994; Galiegue et al., 1995). Central CB1Rs are also localized in regions involved in pain transmission and modulation, specifically in the spinal dorsal horn and periaqueductal gray (Lichtman and Martin, 1991; Lichtman et al., 1996).
The endogenous cannabinoids which bind to their receptors are synthesized on demand by neuronal tissues (Di Marzo et al., 1994; Stella et al., 1997). Blockade of peripheral or central CB1Rs leads to hyperalgesia, suggesting tonic activation of CB1Rs by endocannabinoids (Richardson et al., 1997; Calignano et al., 1998; Strangman et al., 1998). In the periaqueductal gray, endocannabinoid concentrations increase in response to peripheral inflammation (Walker et al., 1999). However, changes in endocannabinoid concentrations as a consequence of chronic pain syndromes have not been studied.
Endocannabinoids and CB1Rs have been detected in dorsal root ganglion (DRG) neurons of heterogeneous size, with variable degrees of CB1R mRNA and protein localization to different sensory neuron subtypes (Hohmann and Herkenham, 1999b; Ahluwalia et al., 2000; Ahluwalia et al., 2002; Salio et al., 2002; Bridges et al., 2003; Price et al., 2003). To understand the basis for the persistent effectiveness of CB1R agonists against neuropathic pain symptoms, we studied changes in CB1R expression in lumbar DRG in a rat model of peripheral neuropathy induced by L5 spinal nerve ligation (SNL). We hypothesized that CB1R up-regulation may contribute to the effectiveness of exogenous cannabinoids in alleviating neuropathic pain symptoms. We also measured endocannabinoid levels since they affect neuronal excitability through CB1R or vanilloid receptor activation. The SNL model was chosen to allow comparison of the injured (deafferented) L5 DRG to the uninjured L4 DRG neurons sharing the sciatic nerve and some overlapping receptive fields in the hindpaw (Kim and Chung, 1992). Notably, neurons in both L4 and L5 DRG become hyperexcitable after SNL; such hyperexcitability is widely considered to contribute to the behavioral symptoms of tactile and thermal hyperalgesia in neuropathic pain models (Ali et al., 1999; Ma et al., 2003).
2. Methods
2.1. Subjects
Adult male Sprague–Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) weighing 200–225 g were used. All experimental procedures were carried out in accordance with the National Institute of Health guidelines for the handling and use of laboratory animals and received approval from the Animal Research Committee of the University of California, Los Angeles.
2.2. Behavioral testing
Behavioral responses to thermal and tactile stimuli were determined in naïve and sham control rats as well as in neuropathic rats 1 day before surgery (“pre”), and just prior to surgery (day 0), then daily for the first 5 days after SNL and then on alternate days (7–15 days).
2.2.1. Thermal hypersensitivity
The Hargreaves method was used to assess paw-withdrawal latency to a thermal nociceptive stimulus (Hargreaves et al., 1988). Rats were allowed to acclimate (15 min) within the Plexiglas enclosures (10 × 20 × 20 cm) on a clear glass plate preheated to 30 °C. A radiant heat source consisting of an adjustable infrared lamp and a built-in stopwatch accurate to 0.1 s were used to measure paw-withdrawal latency. Each paw was tested three times at 30% maximal intensity allowing 5 min between each test. This intensity setting resulted in a baseline withdrawal of 8–10 s. The test was performed only when a rat was stationary and standing on all four paws. Care was taken to keep the glass bottom clean and dry during the testing. If the glass required cleaning during the experiment, the rats were allowed 10 min to reacclimatize to the environment. The results of three tests were averaged for each paw for that day. Two-way analysis of variance (ANOVA) with Tukey post hoc comparisons was used to assess significant differences.
2.2.2. Mechanical hypersensitivity
Rats were placed in a plastic-walled cage (10 × 20 × 13 cm) with a metal mesh floor (0.6 × 0.6 cm holes) and allowed to acclimate for 15 min. The paw-withdrawal thresholds were determined in response to pressure from a digital von Frey anesthesiometer (Model 1601C, IITC Instruments, Woodland Hills, CA). The amount of pressure (g) needed to produce a paw-withdrawal response was measured three times on each paw separated by 30-s intervals. The results of three tests were averaged for each paw for that day. Two-way ANOVA with Tukey post hoc comparisons was used to assess group differences.
2.3. L5 spinal nerve ligation (SNL) surgery
Surgery was performed under sterile conditions as described previously (Kim and Chung, 1992) with some modifications. Under sodium pentobarbital anesthesia (50 mg/kg, i.p.), L5 spinal nerve ligation was performed unilaterally on each rat, without contralateral surgical intervention. The paraspinal muscles were separated from the spinous processes at the L5–6 levels and the L6 transverse process was carefully removed. The L5 spinal nerve was isolated and tightly ligated with 4–0 silk threads. The wound was washed with 5 ml of sterile saline and sutured with 3–0 nylon threads. Antibiotic cream containing polymyxin B, bacitracin and neomycin (Kroger) was applied to the suture site. Buprenorphine (0.01 mg/kg, s.c.) was injected once 1 h after surgery to decrease post-surgical pain. Skin sutures were removed 7 days after surgery.
2.4. Immunohistochemistry
Rats were anesthetized with a lethal dose of pentobarbital (100 mg/kg, i.p.), and fixed by intracardiac perfusion of phosphate buffered saline (PBS, 300 ml), pH 7.4, at room temperature followed by an ice-cold fixative (500 ml, 4% paraformaldehyde and 0.14% picric acid in 0.1 M phosphate buffer, pH 7.4). After fixation, a laminectomy was performed to extract the DRG. Tissue was post-fixed and cryoprotected in 30% sucrose in phosphate buffer (PB). Free-floating sections (25 μm thick) of the entire DRG (15–18 sections/DRG) were cut after embedding in Tissue-Tek (Sakura Fine-tek USA, Inc.). Sections were washed three times with PBS and incubated either with an affinity purified rabbit CB1R C-terminal antibody (1:1000) and the co-markers (listed in Table 1) in PBS/Triton X-100 with 5% normal goat serum (NGS) or with 1% normal donkey serum (NDS) in combination with one of the co-markers at room temperature overnight (14–16 h). After washing with PBS, sections were incubated with secondary antibody in PBS/Triton with 5% NGS/1% NDS for 2 h at room temperature. For co-labeling with isolectin B4 (IB4), sections were incubated with 10 μg/ml of biotin-labeled IB4 (Sigma, St. Louis, MO) supplementing the 1% NDS, PBS/Triton solution with CaCl2, MgCl2, and MnCl2 (all 0.1 mM). Post-wash in PBS, NeutrAvidin-Alexa 488 (5 μg/ml; Molecular Probes) was applied for 2 h. For each SNL rat, sections from ipsilateral and contralateral L4/L5 DRG were processed simultaneously. Sections were examined with a fluorescence microscope (Leica, Wetzlar, Germany) and digital images acquired with a charge-coupled device (CCD) camera (Apogee Model: KX32ME, Auburn, CA) using filter sets for FITC (green) and TRITC (red). MaxIm DL™ (Diffraction Limited, Ontario, Canada) software was used to acquire the images. The same range of minimum and maximum intensity was used to acquire all images.
Table 1.
Type/host, sources and dilutions for each primary antibody/lectin and secondary antibody used for immunohistochemistry
| Primary antibody/agent | Type | Source | Dilution | Secondary antibody | Source | Dilution |
|---|---|---|---|---|---|---|
| Cannabinoid 1 receptor (CB1R) | APa rabbit IgG polyclonal | Ken Mackie | 1:1000 | 1. Goat anti-rabbit Alexa488 (co-labeling with TRPV1,CGRP,N52) | Molecular Probes | 1:1000 |
| 2. Donkey anti-rabbit FITC (co-labeling with NR2C/D) | Jackson ImmunoResearch | 1:1000 | ||||
| 3. Goat anti-rabbit Cy3 (co-labeling with IB4) | Jackson ImmunoResearch | 1:1000 | ||||
| Co-markers | ||||||
| Transient receptor potential vanilloid 1 (TRPV1) | Guinea pig IgG polyclonal | Neuromics | 1:1000 | Goat anti-guinea pig Alexa 568 | Molecular Probes | 1:2000 |
| Calcitonin gene related peptide (CGRP) | Mouse monoclonal | CUREb | 1:200 | Goat anti-mouse RRX | Jackson ImmunoResearch | 1:200 |
| Neurofilament 200 kDa (N52) | Mouse monoclonal | Sigma | 1:500 | Goat anti-mouse RRX | Jackson ImmunoResearch | 1:500 |
| N-Methyl-D-aspartate receptor subunit 2C and 2D (NR2C/D) | APa goat IgG polyclonal | Santa Cruz Biotechnology | 1:500 | Donkey anti-goat RRX | Jackson ImmunoResearch | 1:500 |
| Isolectin B4 (IB4) | Plant lectin | Sigma | 5 μg/ml | Streptavidin–Alexa 488 | Molecular Probes | 5 μg/ml |
FITC, fluorescein isothiocyanate; Cy3, cyanine-3 dye; RRX, rhodamine red-X.
AP, affinity-purified.
CURE, Digestive Research Center, Department of Medicine, UCLA.
2.5. Cell counts and size measurements
Cell profiles were classified according to their labels and counted using Adobe PhotoShop CS (Adobe Systems Inc, San Jose, CA). Three sections (125 μm apart) from a given DRG were used. For each section, neurons of interest were outlined and their area measured using Scion Image software (Beta version 4.0.2, Scion Corporation). Only neurons that did not overlap with other cells and with clearly visible nuclei were included in the analysis. Since the diameter of the largest neurons observed in DRG sections did not exceed 60 μm, we are confident that no neuron was counted twice. One-way ANOVA with Tukey post hoc comparison was used to assess differences in labeling after SNL.
2.6. RNA isolation and real-time RT-PCR
After rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) the L4 and L5 DRGs were excised, placed in RNAlater™ (Ambion, Austin, TX) overnight at 4 °C and stored at −20 °C. Using TRIzol (Invitrogen, Carlsbad, CA), total RNA was extracted from each DRG. RNA samples were treated with DNase I (Ambion Inc., Austin, TX) for 30 min, aliquoted and stored at −80 °C. Synthesis of cDNA from total RNA (1 μg) was performed by reverse transcription using RETROscript™Kit (Ambion Inc., Austin, TX). Real-time PCR was carried out using the iCycler system (Bio-Rad). DNA for CB1R genes and for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) reference gene was subjected to 5 min of denaturing at 95 °C followed by 45 cycles of PCR (30 s at 95 °C for denaturing, 20 s at 60 °C for annealing, 30 s at 72 °C for extension of amplification) using iQ™ SYBR Green Supermix (Bio-Rad, Hercules, CA) and gene specific primers. (CB1R: sense primer 5′-CTA CTG GTG CTG TGT GTC ATC-3′, antisense primer: 5′-GCT GTC TTT ACG GTG GAA TAC-3′; GAPDH: sense primer: 5′-ATG GAC TGT GGT CAT GAG CC-3′, antisense primer: 5′-ATT GTC AGC AAT GCA TCC TG-3′). The design of CB1R primers was carried out by Oligo 4.0 software (National Bioscience, Plymouth, MN). Specificity of PCR products was determined by melting curve analysis immediately following amplification. For each real-time PCR experiment, DNA samples were run together with cDNA from hippocampus serving as a calibrator. The amount of CB1R mRNA in each DRG was first normalized to an endogenous reference (GAPDH; ΔCT) and then relative to a calibrator (hippocampus; ΔΔCT), and expressed as 2−ΔΔCT. One-way ANOVA with Tukey post hoc comparison was used to assess differences in CB1R mRNA levels after SNL.
2.7. Protein isolation and immunoblot assay
Two weeks following SNL, L4/L5 DRG ipsilateral and contralateral to SNL and from control rats were excised, frozen in liquid nitrogen and stored at −80 °C. For each blot, 4 DRG were pooled to obtain membrane fractions for L4/L5 ipsilateral and contralateral to SNL, and control L4/L5 DRG. Samples were homogenized with a Dounce homogenizer in ice-cold extraction buffer (Biosource, Camarillo, CA) supplemented with phenylmethylsulfonyl fluoride (PMSF, 1 mM) and a protease inhibitor cocktail (Sigma–Aldrich, Saint Louis, MO). Homogenates were kept on ice for 30 min, sonicated for 3 min and centrifuged at 12,000g for 10 min at 4 °C. The pellet, containing the total membrane fraction, was then re-suspended in the extraction buffer. Protein content was determined using DC Protein Assay (Bio-Rad, Hercules, CA). For immunoblot assay, samples (15–20 μg) were denatured in Laemmli’s sample buffer (Bio-Rad, Hercules, CA) for 10 min at 90 °C. Proteins were fractionated by SDS–PAGE using 4–15% gradient Tris–HCl Ready gels (Bio-Rad) and then electrotransferred at 22 mV to Immun-Blot PVDF membrane (Bio-Rad) overnight at 4 °C. Membranes were blocked in Tris buffer saline with 0.05% Tween® 20 (TBST, Bio-Rad) containing 10% non-fat dried milk (NFDM) overnight at 4 °C before overnight incubation at 4 °C with the primary antibody (rabbit anti-CB1R: L14, 1:200 dilution) diluted in TBST containing 5% normal swine serum (NSS). Blots were washed extensively in TBST and then blocked in TBST containing 5% NFDM for 2 h. After an extensive wash, blots were incubated with biotin-ylated swine anti-rabbit IgG (1:1000; Dako, Glostrup, Denmark) in TBST/5% NSS for 30 min at room temperature, washed extensively and then incubated for 30 min with streptABCComplex/horseradish peroxidase (HRP; Dako), developed in Signal West Dura Extended Duration Chemiluminescence (Pierce, Rockford, IL) and exposed onto Kodak BioMax MR film (Eastman Kodak Company, Rochester, NY). The blots were then incubated in stripping buffer (100 mM 2-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mM Tris–HCl, pH 6.7) at 55 °C for 30 min with intermittent agitation. After extensive washing, the blots were reprobed with ERK2 antibody (1:300; Santa Cruz Biotechnology, Santa Cruz, CA) as a loading control. The intensity of the selected bands was captured and analyzed using Scion Image beta version 4.0.2 (Scion Corporation). The data from ipsilateral and contralateral SNL DRG were expressed as a fold change from control DRG after normalization to loading control. Kruskal–Wallis one-way ANOVA on ranks with Dunn’s post hoc comparison was used to assess changes in CB1R protein expression after SNL.
2.8. Measurements of endocannabinoid concentrations
2.8.1. Tissue preparation for endocannabinoid quantification
The endocannabinoid extraction methods were adapted from previous studies {Patel et al., 2003 937/id}. Frozen (−80 °C) DRG samples were defrosted, weighed and placed into borosilicate glass culture tubes containing 2 ml of acetonitrile with the internal standards [2H8]AEA (25 pmol) and [2H8]2-AG (1 nmol). Tissue was homogenized with a glass rod and sonicated for 30 min. Samples were incubated overnight at −10 °C to precipitate proteins. Samples were centrifuged at 1500g, and supernatants were removed to a new glass tube and evaporated to dryness under N2 gas. The samples were resuspended in 500 μl of methanol and dried again. Finally, lipid extracts were suspended in 40 μl of methanol, 25 μl of which was used for analysis by liquid chromatography/mass spectrometry.
2.8.2. Liquid chromatography/mass spectrometry
The amounts of AEA and 2-AG were determined by combined liquid chromatography–atmospheric pressure chemical ionization (APCI) mass spectrometry using multiple reaction monitoring (MRM) in the tandem mass spectrometric mode (MS/MS). Samples (25 μl) were injected onto a reverse-phase column (Zorbax Eclipse XDB C18, 4.6 × 50 mm, 5 μ particle size) equilibrated in methanol/water (85/15, v/v) containing 1 mM ammonium acetate and 0.005% acetic acid, and eluted (500 μl/min) with an increasing concentration of methanol (min:% methanol, 0:85, 7:100, 10:100). The effluent from the column was passed directly to an APCI source (nebulizer at 450 °C, discharge needle at 4.5 kV, nebulizing gas (3.0 L/min) produced from the vapors of liquid nitrogen) attached to a triple quadrupole mass spectrometer (Perkin-Elmer Sciex, Thornhill, Canada, API III). The mass spectrometer had been previously tuned and calibrated by flow injection (100 μl/min) of a mixture of polypropylene glycol (PPG) 425, 1000 and 2000 (3.3 × 10–5, 1 × 10–4 and 2 × 10–4 M, respectively) in water/methanol (1/1, v/v) containing 2 mM ammonium formate and 0.1% acetonitrile. Calibration was accomplished using the singly charged PPG signals at m/z 59.0, 326.3, 384.3, 906.7, 1254.9, 1545.1, 1603.2, and 1661.2. To lower the limit of detection for AEA and 2-AG, the mass spectrometer was operated under conditions of degraded mass resolution for both Q1 and Q3. Under these conditions the 13C-isotopes of the PPG calibrant ions were not resolved from one another. For the quantitation of AEA and 2-AG the mass spectrometer was operated in the MS/MS mode (orifice voltage 55 V, argon collision gas thickness instrumental setting 250), and recordings were made of MRM signals corresponding to specific MH+ → selected fragment ion transitions: [2H8]AEA, m/z 356 → 61, retention time 7.2 min; AEA, m/z 348 → 60, retention time 7.4 min; [2H8]2-AG, m/z 387 → 294, retention time 7.5 min; 2-AG and 1(3)-AG, m/z 379 → 287, retention time 7.7 and 8.1 min, respectively. In some samples (~10%), 2-AG was observed as a doublet because it isomerizes to 1(3)-AG during extraction (Stella et al., 1997); therefore the areas of both peaks were combined to yield total 2-AG. For quantification of AEA and 2-AG in each sample, the peak area for each transition was measured using Clampfit 8.0 software (Axon Instruments). For each batch of samples a standard response curve was constructed from known concentrations of synthetic AEA and 2-AG and the same quantities of the [2H8]AEA and [2H8]2-AG internal standards as were used for tissue sample preparation. Importantly, AEA or 2-AG peaks were not detected when only [2H8]AEA and [2H8]2-AG were injected into the mass spectrometer. The mole content of AEA, 2-AG including 1(3)-AG in each biological sample was calculated by interpolation from the standard response curves. Statistical differences between the mean endocannabinoid content in control, ipsilateral and contralateral DRG were determined by ANOVA and Tukey post hoc comparison.
3. Results
3.1. CB1-ir distribution and co-localization in control rat DRG
We first studied the distribution of CB1R in DRG by using the C-terminal antibody previously shown to specifically label CB1R in the CNS (Hajos et al., 2000; Coutts et al., 2002). For all co-labeling experiments, DRG from 3 rats (3 sections per rat) were analyzed. CB1R-ir was observed in 89.1 ± 4.5% and 89.8 ± 3.4% of total DRG neurons in L4 and L5, respectively. The distribution of CB1R-positive cell profiles in L4/L5 DRGs is summarized in Fig. 1A–E. CB1R-ir was prevented by pre-incubation of the antibody with the peptide immunogen (1 μg/ml) as shown in Fig. 1F. The majority of the neurons, ranging from 76.4 ± 4.5–82.8 ± 4.7%, which were positive for the nociceptive neuron markers: IB4, TRPV1, CGRP and NR2C/D, were also positive for CB1R. Selectivities of the lectin and antibodies for these markers have been extensively characterized in previous studies (Popper and Micevych, 1989; Silverman and Kruger, 1990; Guo et al., 1999; Marvizon et al., 2002). CB1R-ir was also detected in a substantial number (81%) of neurons immunoreactive for N52; a marker of large-diameter myelinated sensory neurons (Shaw et al., 1986). The percentage of CB1R-ir neurons labeled by the various markers was as follows: IB4, 43.6 ± 1.6%; TRPV1, 25.6 ± 1.7%; CGRP, 19.5 ± 1.7%; NR2C/D, 41.8 ± 2.8%; N52, 21.1 ± 1.5% (Fig. 1A–E).
Fig. 1.
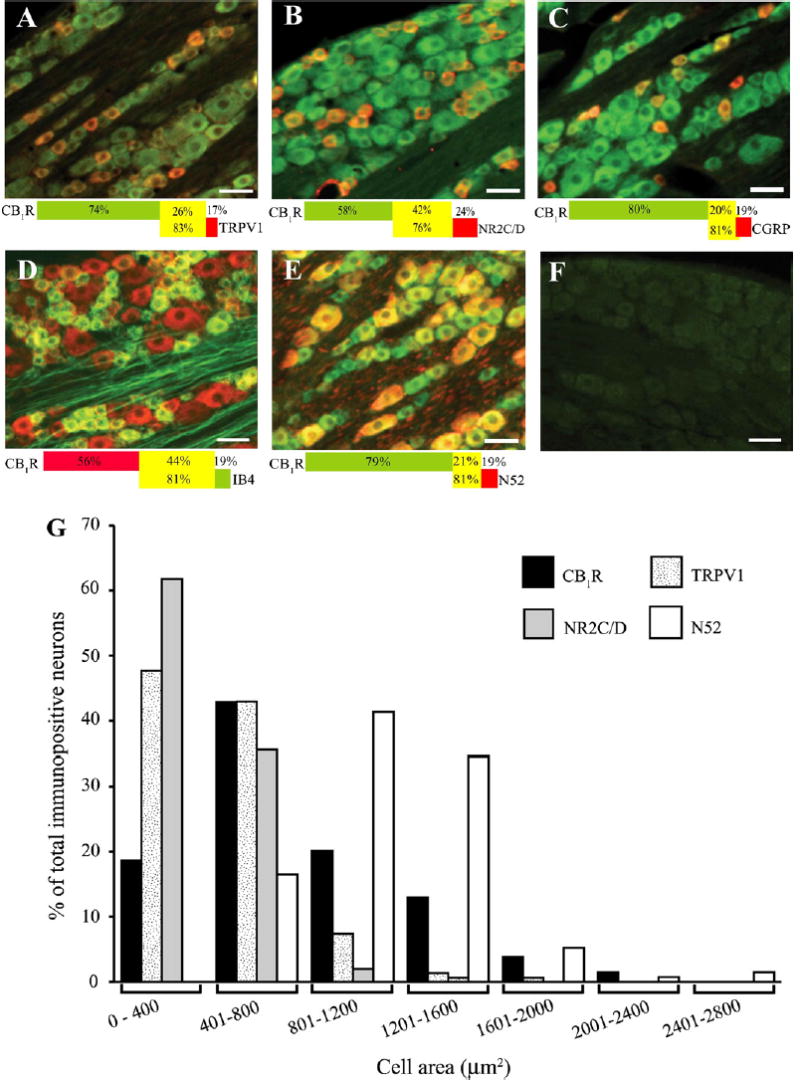
Co-localization of CB1R-ir with TRPV1 (A), NR2C/D (B), CGRP (C), IB4 (D) and N52 (E) in control rats. The CB1R-ir is green for all panels except panel D red. The co-markers are red for all the panels except panel D green. All panels are superimposed images of CB1R-ir and the co-markers, while co-labeling is yellow. The percentage of CB1R co-localization with the corresponding marker is illustrated by the colored bar corresponding to the CB1R and co-marker color. The percentage of CB1R-ir neurons co-labeled with marker is shown in the yellow area of top bar whereas the percentage of marker-positive neurons co-labeled with CB1R-ir is shown in the yellow area of bottom bar. All data were obtained from 3 sections/rat (n = 3 rats). Panel F shows the staining after pre-absorption with the CB1R blocking peptide (1 μg/ml). The scale bar = 50 μm throughout. Panel G shows the size distribution of CB1R-ir, TRPV1-ir, NR2C/D-ir and N52-ir cells in DRG. Note that CB1R-ir can be found throughout the range of cell sizes, whereas co-markers are seen predominantly in small-medium cells (<1200 μm2, TRPV1 and NR2C/D) or large cells (N52).
CB1R-ir was observed in a wide range of cell sizes (Fig. 1G). For L4 cell profiles, the cell area ranged from 280 μm2 to 3931 μm2, with an average cell area (mean ± SEM) of 995 ± 74.8 μm2. For L5 cell profiles, the cell area ranged from 198 μm2 to 3204 μm2, with an average cell area of 857.3 ± 47.8 μm2. For the co-markers, the average cell size of 450 ± 15.7 μm2 was observed for TRPV1, 398 ± 13.4 μm2 for NR2C/D and 1158 ± 31.3 μm2 for N52, illustrating the smaller size of nociceptive neurons compared to non-nociceptors.
3.2. Increased thermal and mechanical sensitivity following L5 SNL
The time course of thermal and mechanical sensitivity changes in naïve and sham control as well as SNL rats is shown in Fig. 2. Pre-operatively, the paw-withdrawal latency to thermal stimuli was 8.3 ± 0.08 s. The baseline threshold for withdrawal from mechanical stimuli was 60 ± 3 g. There were no significant pre-operative differences in withdrawal latency and thresholds among naïve, sham and SNL rats. Post-operatively, measurements revealed that the SNL animals exhibited increases in both thermal (Fig. 2A) and mechanical (Fig. 2B) sensitivity in the ipsilateral hindpaw. There were also signs of spontaneous pain, guarding behavior and changes in the posture of the affected hindpaw including plantar flexion and toe-clenching, typical of this model (Kim and Chung, 1992).
Fig. 2.
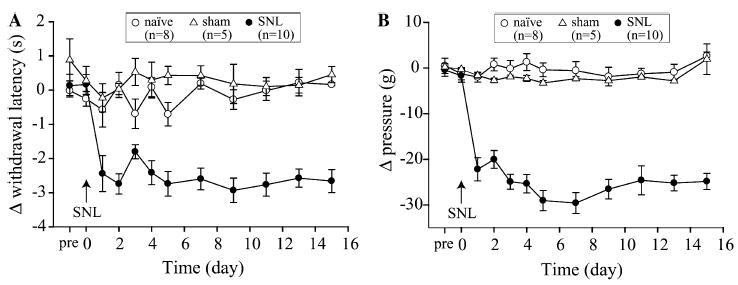
Increased sensitivity to thermal and mechanical stimuli after L5 SNL. Graphs illustrate mean (±SEM) of difference in withdrawal latency (A) or pressure threshold (B) between ipsilateral–contralateral hindpaw of SNL and sham surgery rats or left–right in naïve control rats. Day “pre” and “0” represent the baseline measurements before SNL surgery. There is no significant difference in the baseline value for both thermal (A) and mechanical (B) testings among three groups of rats. All of the post L5 SNL data points for both thermal and mechanical sensitivity are significantly different (P < 0.05, two-way ANOVA with Tukey post hoc comparison) from baseline at time “pre”, and from sham and naïve controls within each post-operative day.
3.3. Changes in CB1R transcripts and protein expression
To elucidate the possible changes in CB1R at the mRNA and protein levels, we examined CB1R mRNA and protein levels in the L4/L5 DRG 2 weeks after ligation of the L5 spinal nerve using real-time RT-PCR and Western blotting. Ipsilateral L4 DRG had a significantly higher level of CB1R mRNA as compared to the contralateral side, naïve and sham controls (Fig. 3). In contrast, there was no significant change in CB1R mRNA levels in either ipsilateral or contralateral L5 SNL DRG compared to both naïve and sham controls. The absence of changes in withdrawal thresholds coupled with the lack of changes in CB1R mRNA levels of sham surgery rats suggested that the up-regulation of CB1R mRNA resulted from L5 SNL. Also, given the similarity between the sham group and naïve controls, in subsequent experiments we compared changes in SNL rats to naïve controls.
Fig. 3.
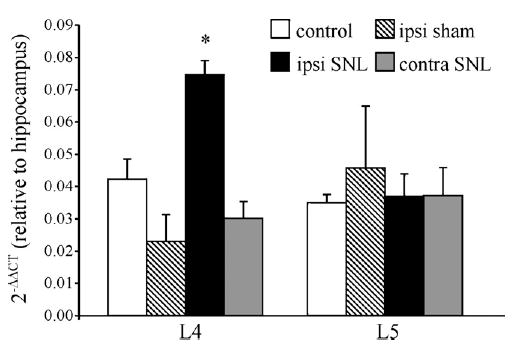
Up-regulation of CB1R transcripts in the L4 DRG following L5 SNL. Ipsilateral L4 DRG showed a significant increase in level of CB1R mRNA expression (normalized to GAPDH) and expressed as a value relative to the independent control (hippocampus) compared to naïve and sham control DRG. *P < 0.05 (n = 3 rats, one-way ANOVA and Tukey post hoc comparison).
For CB1R protein, the ipsilateral L4 DRG displayed a significant up-regulation (1.2-fold) compared to control (Fig. 4). This increase in expression level of CB1R protein was consistent with the increased CB1R mRNA levels found in ipsilateral L4 DRG of SNL rats. In L5 DRG, there was no significant change of CB1R protein in either ipsilateral or contralateral DRG which also agreed with the mRNA findings.
Fig. 4.
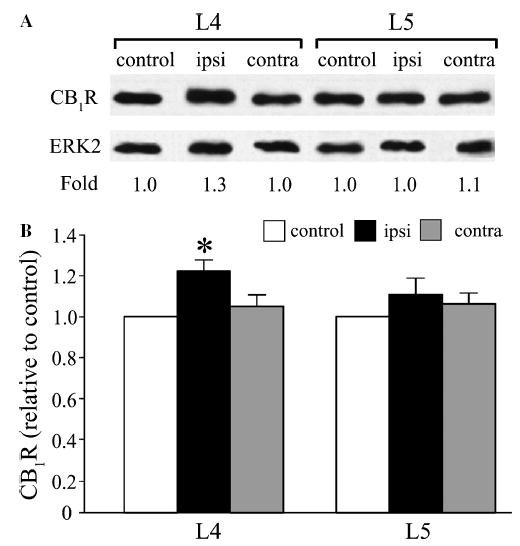
Up-regulation of CB1R protein in the L4 DRG following L5 SNL. (A) Representative results showing Western blotting for CB1R protein in L4 and L5 DRG from control and L5 SNL rats. (B) Summary graph indicates a significant increase in band intensity of CB1R from ipsilateral L4 DRG (normalized to ERK2) compared with control L4 DRG. (n = 6 immunoblots, 4 DRG per lane; *P < 0.05, Kruskal–Wallis one-way ANOVA on ranks with Dunn’s post hoc comparison).
3.4. Changes in CB1R co-localization
We next investigated whether the SNL-induced changes in CB1R expression were due to an increase in the number of CB1R-ir neurons as a consequence of a phenotypic switch (synthesis in neurons that do not normally express CB1R) or due to increased expression levels in the existing CB1R-ir neurons. The total percentage of CB1-ir neurons in contralateral or ipsilateral L4 and L5 DRG was unchanged after SNL (Fig. 5A). However, significant changes were observed in co-labeling with other markers (Fig. 5B and C). The percentage of CB1R-ir neurons co-labeled with TRPV1 increased from 24.8% in control to 58.6% in the ipsilateral L4 DRG without a significant change in contralateral DRG. Co-labeling in ipsilateral or contralateral L4 DRG with IB4, CGRP, N52, and NR2C/D was unchanged. In ipsilateral L5 DRG, the percentage of CB1R-ir neurons co-labeled with IB4 was significantly decreased from 44.2% in control to 20.1%. The CGRP/CB1R co-labeling decreased from 18% in control to 8% on the ipsilateral side. There were also trends to decreased CB1R co-labeling with TRPV1 and NR2C/D in ipsilateral L5 DRG, although these did not reach statistical significance.
Fig. 5.
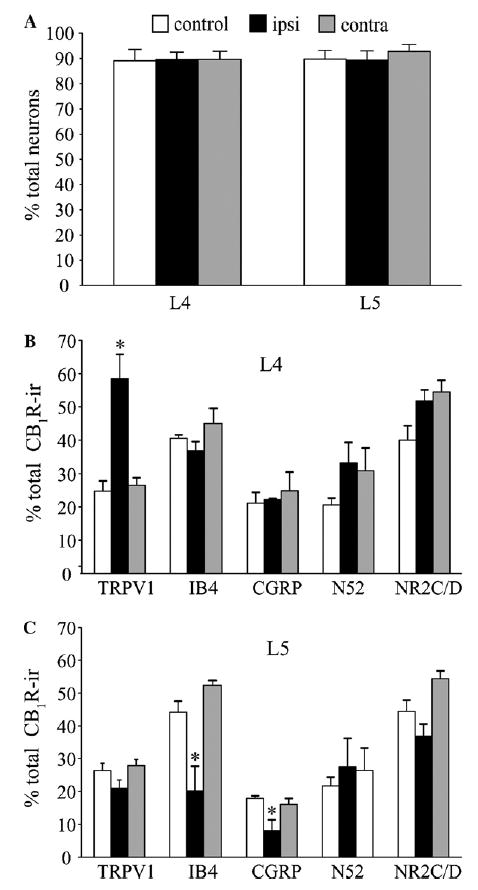
(A) Percentages of CB1R-ir neurons in L4 and L5 remain unchanged after L5 SNL. Bars indicate percentage (±SEM) of CB1R-ir neurons in sections from ipsilateral and contralateral L4 and L5 of SNL and control rats (B). Changes in CB1R co-localization with TRPV1, CGRP, IB4, N52 and NR2C/D in ipsilateral and contralateral L4 (B) and L5 (C) DRG neurons following SNL. Bars represent percentage (±SEM) of co-marker labeling of total CB1R-ir neurons. Note the significant increase of TRPV1-ir in the ipsilateral L4 and the decrease in IB4 positive and CGRP-ir in ipsilateral L5 of SNL rats (n = 3 rats, 3 sections/rat, *P < 0.05, one-way ANOVA with Tukey post hoc comparison).
3.5. Changes in DRG endocannabinoid levels
Both AEA and 2-AG were detected using LC-APCI/MRM-MS/MS. A representative chromatograph depicting the presence of AEA and 2-AG as well as deuterated synthetic standards in a tissue extract is shown in Fig. 6A. In general, the signal-to-noise ratio for detecting native AEA was lower and more variable than that observed for 2-AG. Small day-to-day changes were observed in the sensitivity of analytical hardware to standards; therefore, each experiment included DRG samples from both control and experimental animals, and statistical analyses were confined to comparisons among samples subjected to same-day experimental and analytical procedures. The mean values (±SEM, n = 4) obtained for naïve control L4 DRG were 26.0 ± 2.3 pmol/g tissue weight for AEA and 8.0 ± 0.4 nmol/g tissue for 2-AG. Similarly, mean values obtained for naïve control L5 DRG were 26.5 ± 0.8 pmol/g tissue for AEA and 8.9 ± 0.5 nmol/g tissue for 2-AG, corresponding to more than 300-fold lower DRG content of AEA than 2-AG. The effects of L5 SNL on AEA and 2-AG content were determined 2 weeks after sham or L5 SNL surgery. Significant increases in both AEA and 2-AG were observed in ipsilateral L5 compared to naïve and sham surgery controls and contralateral DRG (Fig. 6B and C). In contrast, AEA and 2-AG content was unchanged in naïve and sham surgery controls or in ipsilateral and contralateral L4 DRG.
Fig. 6.
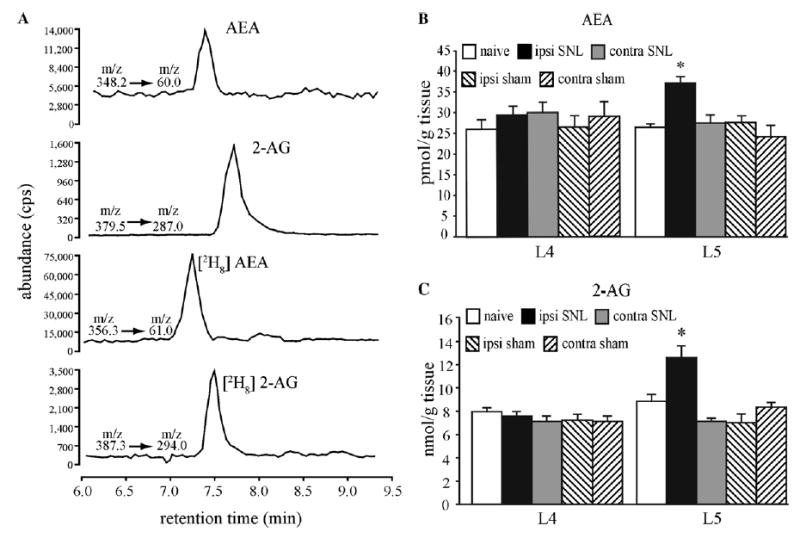
Endocannabinoid content changes in DRG of SNL rats. (A) Representative chromatograph from a tissue extract depicting the abundance (in counts per second) of endocannabinoids (AEA and 2-AG) and deuterated internal standards, [2H8]anandamide (AEA) and [2H8]2-AG together with their respective mass to charge ratio (m/z) transitions. (B) The bar graphs illustrate significant increases in AEA and (C) 2-AG in the ipsilateral L5 DRG of L5 SNL compared to contralateral, sham surgery and naïve control DRG (n = 3–4 ganglia, *P < 0.05; one-way ANOVA with Tukey post hoc comparison). Note the difference in scale between AEA and 2-AG.
4. Discussion
4.1. Expression of CB1R in DRG
Previous studies report considerably smaller percentage (25–57%) of CB1R-positive DRG neurons (Ahluwalia et al., 2000; Bridges et al., 2003) although the first study employing CB1R immunocytochemistry in DRG indicates that virtually all neurons are labeled to some degree (Sanudo-Pena et al., 1999). One explanation for these differences is that CB1R labeling is markedly influenced by the fixation procedures (Coutts et al., 2002). Also, we use a C-terminal antibody raised against the last 72 amino acids of CB1R, the specificity of which has been extensively confirmed (Hajos et al., 2000; Coutts et al., 2002). Others use a C-terminal antibody against the 461–473 amino acid sequence of CB1R (Bridges et al., 2003), or N-terminal antibodies (Ahluwalia et al., 2000; Salio et al., 2002). The N-terminal antibodies do not recognize a putative splice variant of CB1R because it has a truncated N-terminus (Shire et al., 1995; Salio et al., 2002); this could affect the number of CB1-ir neurons detected.
Our findings on the co-labeling of CB1R with various markers are consistent with those of Ahluwalia et al. (2000), although they used a cell culture system. They report 82% of TRPV1-ir neurons exhibiting CB1R-ir, also ~80% of these TRPV1/CB1R-ir neurons express CGRP or IB4, all of which predominantly label nociceptor subpopulations (Silverman and Kruger, 1990; Lawson, 1995; Guo et al., 1999) and consistent with our findings with the NR2C/D antibody, which labels both the IB4 and CGRP nociceptor subpopulations (Marvizon et al., 2002). This contrasts studies which find little CB1R co-localization with CGRP and substance P (Hohmann and Herkenham, 1999b) or those where CB1R is expressed almost exclusively in medium and large diameter neurons (Bridges et al., 2003; Price et al., 2003). Their studies used CB1R mRNA detection with in situ hybridization, which like immunocytochemistry studies are highly technique-sensitive. In agreement with our results, the same studies report a high degree of co-labeling with the large diameter neuron marker, N52 (Shaw et al., 1986).
4.2. Increased CB1R transcripts and protein in uninjured L4 DRG neurons after SNL
The finding of increased CB1R mRNA in ipsilateral uninjured L4 DRG is corroborated by the demonstration of increased CB1R expression. In agreement with our findings, TRPV1 transcripts and protein are selectively increased in uninjured ipsilateral L4 DRG neurons after SNL (Hudson et al., 2001; Fukuoka et al., 2002). In the injured ipsilateral L5 DRG we find significantly decreased IB4 and CGRP labeling and a trend to decreased TRPV1 and NR2C/D. Similar decreases in IB4 and CGRP but not N52 labeling were demonstrated after SNL (Hammond et al., 2004). Profoundly altered gene expression of various receptors, neuropeptides and ion channels occurs in both injured L5 and uninjured L4 DRG (Wang et al., 2002).
Currently, there is no consensus on how uninjured neurons in the adjacent DRG sense the injury and regulate gene expression. However, there might be interactions between the axons and peripheral receptive fields of L4 and L5 DRG neurons. During Wallerian degeneration of the injured L5 DRG neuron axons, electrical and chemical cross-excitation may occur between injured and uninjured axons (Lisney and Devor, 1987; Amir and Devor, 2000). This could send signals affecting immediate early gene (IEG) expression to neuronal somata. IEGs regulate a host of other genes including those affecting neuronal excitability (Wang et al., 2002). IEG activation and expression of many neuronal receptors, neuropeptides and channels appears to be nerve growth factor (NGF)- or glial derived nerve growth factor (GDNF)-dependent (Segal and Greenberg, 1996; Bennett, 2001). Thus, sciatic nerve injury-induced CB1R up-regulation in the spinal cord is partly mediated through tyrosine kinase receptors and the mitogen-activated protein kinase (Lim et al., 2003), although CB1R expression in sensory neurons in culture appears independent of either NGF or GDNF (Ahluwalia et al., 2002), indicating a need to further investigate the mechanisms of CB1R up-regulation.
4.3. Altered endocannabinoid concentrations after SNL
We demonstrate selectively increased AEA and 2-AG levels in the injured L5, but not the uninjured L4, DRG after SNL. Differences in extraction procedures may account for the 20-fold lower 2-AG levels reported for control rat DRG (Huang et al., 1999). The increased endocannabinoid levels are likely related to increased biosynthesis or decreased metabolism and transport because endocannabinoids are produced on demand without any substantial storage (Di Marzo, 1998). AEA is mainly produced by a two-step enzymatic pathway involving calcium-dependent transacylase and phospholipase D (Sugiura et al., 1996; Cadas et al., 1997; Okamoto et al., 2004). Then, AEA either diffuses (Glaser et al., 2003) or is actively transported into cells (Patricelli and Cravatt, 2001) and is rapidly degraded by the membrane-bound fatty acid amide hydrolase (FAAH) to arachidonic acid. 2-AG is synthesized via the diacylglycerol lipase-mediated hydrolysis of diacylglycerol and metabolized by monoacylglycerol lipase (Dinh et al., 2002).
Studies show that peripheral denervation leads to progressive degeneration of ~35% of DRG neurons (Lisney, 1989). In the CNS, neurodegeneration is associated with increased endocannabinoid levels (Panikashvili et al., 2001; Witting et al., 2004). Such increases are proposed to serve as an endogenous neuroprotective mechanism, especially given the neuroprotection afforded by administration of cannabinomimetics (Nagayama et al., 1999; Panikashvili et al., 2001). Thus, selective increases in L5 DRG endocannabinoids may represent an endogenous response to neurodegeneration. Alternatively, endocannabinoids may increase because the L5 DRG neurons become hyperexcitable, although this scenario is less likely as uninjured L4 neurons also become hyperexcitable after L5 SNL (Li et al., 2000).
4.4. Functional and therapeutic implications
Early electrophysiological studies have demonstrated marked hyperexcitability of injured L5 DRG neurons after SNL (Sheen and Chung, 1993; Yoon et al., 1996). Later studies revealed that neurons in the ipsilateral uninjured L4 DRG also become hyperexcitable (Li et al., 2000; Ma et al., 2003). The relative contributions of injured and uninjured afferents to hyperexcitability are a subject of debate (Gold, 2000). Furthermore, abnormal (ectopic) spike discharge within sensory neurons may occur at the site of axonal injury, at the soma, or at the axonal T-junction (Amir et al., 2005). However, there is general agreement that hyperexcitability and ectopic discharge of sensory neurons are the primary contributors to the symptoms of tactile and thermal hyperalgesia and spontaneous pain of the SNL neuropathy (Yoon et al., 1996; Ali et al., 1999; Gold, 2000). Our findings of increased CB1R expression may thus be viewed as an adaptive response to the hyperexcitability of uninjured L4 sensory neurons. Chronic up-regulation of hippocampal CB1Rs is observed in two seizure models (Chen et al., 2003; Wallace et al., 2003). However, it fails to explain the selective increases in L4 and not L5 DRG, given that hyperexcitability is common to both uninjured and injured DRG neurons (Li et al., 2000; Ma et al., 2003). One possibility is that the chronic increases in endocannabinoid levels result in down-regulated CB1R within L5 DRG. Such CB1R down-regulation has been demonstrated in central neurons (Basavarajappa and Hungund, 2002).
Selective AEA increases within the L5 DRG may actually be pronociceptive since high [AEA] directly activates TRPV1 receptors (Zygmunt et al., 1999; Ahluwalia et al., 2003). TRPV1 activation by AEA is significantly enhanced in the presence of the inflammatory mediators, bradykinin and prostaglandin E2 (Singh et al., 2005). Large increases in macrophages and lymphocytes are observed within L4/L5 DRG 10–20 days after tight ligation of the sciatic nerve (Eckert et al., 1999). Inflammatory mediator release from these immunocompetent cells would increase the likelihood of pronociceptive AEA effects in the L5 DRG after SNL.
Our findings provide an explanation for the persistent effectiveness of peripherally administered CB1R agonists in alleviating the painful symptoms of peripheral neuropathy (Bridges et al., 2001; Fox et al., 2001; Costa et al., 2004). Increased CB1R expression in L4 DRG should increase net transport and incorporation of CB1R at peripheral sensory terminals (Hohmann and Herkenham, 1999a). Transport to central terminals is likely, but undetectable by immunocytochemistry (Farquhar-Smith et al., 2000). Activation of CB1Rs at these sites may produce antinociception by decreasing Ca2+-dependent neurotransmitter release from primary afferents (Ross et al., 2001; Ellington et al., 2002). Neurotransmitters are also released from somata of primary afferents (Neubert et al., 2000; Matsuka et al., 2001), however the importance of this release site for nociception is presently unclear. In the spinal cord, CB1Rs are up-regulated ipsilateral to sciatic nerve constriction, consistent with enhanced effectiveness of WIN 55,212-2 in decreasing pain symptoms (Lim et al., 2003). This sharply contrasts with the disappointing control of neuropathy symptoms by opioid analgesics (Mao et al., 1995; Ossipov et al., 1995; Rashid et al., 2004) possibly explained by the drastic down-regulation of μ-opioid receptors in sensory neurons after peripheral nerve injury (Rashid et al., 2004).
Our data also provide a rational framework for the development of novel therapeutics that target the peripheral endocannabinoid system. One strategy might be to develop selective CB1R agonists which do not penetrate the blood–brain barrier, thereby providing pain relief without the side-effects associated with central CB1R activation. Another would be to develop peripherally acting selective inhibitors of endocannabinoid metabolism to elevate endocannabinoid levels, which would result in increased activation of both CB1R and CB2R. Indeed, selective FAAH inhibitors have already been developed (Kathuria et al., 2003), and demonstrated to ameliorate neuropathic pain symptoms (Lichtman et al., 2004). Our hope is that such strategies might assist neuropathy patients who currently resort to the use of cannabinoids of controversial legal status in order to alleviate their pain and suffering.
Acknowledgments
We thank Drs. Juan Carlos Marvizon and Bing Bing Song for help with immunohistochemistry, as well as Drs. Ichiro Nishimura and Sotirios Tetradis for help with PCR and Western analysis. We are also grateful to Dr. Lawrence Kruger for fruitful discussions and critique of the manuscript. This work was supported by the NIH Grants DE14573, DA11322, DA00286 and the Stein Oppenheimer Endowment Award.
References
- Ahluwalia J, Urban L, Bevan S, Capogna M, Nagy I. Cannabinoid 1 receptors are expressed by nerve growth factor- and glial cell-derived neurotrophic factor-responsive primary sensory neurones. Neuroscience. 2002;110:747–53. doi: 10.1016/s0306-4522(01)00601-7. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Bevan S, Nagy I. Anandamide regulates neuropeptide release from capsaicin-sensitive primary sensory neurons by activating both the cannabinoid 1 receptor and the vanilloid receptor 1 in vitro. Eur J Neurosci. 2003;17:2611–8. doi: 10.1046/j.1460-9568.2003.02703.x. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–8. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Ali Z, Ringkamp M, Hartke TV, Chien HF, Flavahan NA, Campbell JN, et al. Uninjured C-fiber nociceptors develop spontaneous activity and alpha-adrenergic sensitivity following L6 spinal nerve ligation in monkey. J Neurophysiol. 1999;81:455–66. doi: 10.1152/jn.1999.81.2.455. [DOI] [PubMed] [Google Scholar]
- Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95:189–95. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- Amir R, Kocsis JD, Devor M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J Neurosci. 2005;25:2576–85. doi: 10.1523/JNEUROSCI.4118-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Hungund BL. Neuromodulatory role of the endocannabinoid signaling system in alcoholism: an overview. Prostaglandins Leukot Essent Fatty Acids. 2002;66:287–99. doi: 10.1054/plef.2001.0352. [DOI] [PubMed] [Google Scholar]
- Bennett DL. Neurotrophic factors: important regulators of nociceptive function. Neuroscientist. 2001;7:13–7. doi: 10.1177/107385840100700105. [DOI] [PubMed] [Google Scholar]
- Berman JS, Symonds C, Birch R. E3cacy of two cannabis based medicinal extracts for relief of central neuropathic pain from brachial plexus avulsion: results of a randomised controlled trial. Pain. 2004;112:299–306. doi: 10.1016/j.pain.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Bridges D, Ahmad K, Rice AS. The synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br J Pharmacol. 2001;133:586–94. doi: 10.1038/sj.bjp.0704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertova M, Elphick MR, Winter J, Michael GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–12. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Cadas H, di Tomaso E, Piomelli D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidyl-ethanolamine, in rat brain. J Neurosci. 1997;17:1226–42. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–81. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyas A, Freund TF, Smith M, et al. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/s0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Costa B, Colleoni M, Conti S, Trovato AE, Bianchi M, Sotgiu ML, et al. Repeated treatment with the synthetic cannabinoid WIN 55,212-2 reduces both hyperalgesia and production of pronociceptive mediators in a rat model of neuropathic pain. Br J Pharmacol. 2004;141:4–8. doi: 10.1038/sj.bjp.0705587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AA, Irving AJ, Mackie K, Pertwee RG, Anavi-Goffer S. Localisation of cannabinoid CB1 receptor immunoreactivity in the guinea pig and rat myenteric plexus. J Comp Neurol. 2002;448:410–22. doi: 10.1002/cne.10270. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. ’Endocannabinoids’ and other fatty acid derivatives with cannabimimetic properties: biochemistry and possible physiopathological relevance. Biochim Biophys Acta. 1998;1392:153–75. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–91. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–24. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Segond vB, Sopper S, Petersen M. Spatio-temporal pattern of induction of bradykinin receptors and inflammation in rat dorsal root ganglia after unilateral nerve ligation. Pain. 1999;83:487–97. doi: 10.1016/S0304-3959(99)00152-9. [DOI] [PubMed] [Google Scholar]
- Ellington HC, Cotter MA, Cameron NE, Ross RA. The effect of cannabinoids on capsaicin-evoked calcitonin gene-related peptide (CGRP) release from the isolated paw skin of diabetic and non-diabetic rats. Neuropharmacol. 2002;42:966–75. doi: 10.1016/s0028-3908(02)00040-0. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Egertova M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR. Cannabinoid CB1 receptor expression in rat spinal cord. Mol Cell Neurosci. 2000;15:510–21. doi: 10.1006/mcne.2000.0844. [DOI] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, et al. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Tokunaga A, Tachibana T, Dai Y, Yamanaka H, Noguchi K. VR1, but not P2X3, increases in the spared L4 DRG in rats with L5 spinal nerve ligation. Pain. 2002;99:111–20. doi: 10.1016/s0304-3959(02)00067-2. [DOI] [PubMed] [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci USA. 2003;100:4269–74. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS. Spinal nerve ligation: what to blame for the pain and why. Pain. 2000;84:117–20. doi: 10.1016/s0304-3959(99)00309-7. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–58. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, et al. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–49. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Hammond DL, Ackerman L, Holdsworth R, Elzey B. E3ects of spinal nerve ligation on immunohistochemically identified neurons in the L4 and L5 dorsal root ganglia of the rat. J Comp Neurol. 2004;475:575–89. doi: 10.1002/cne.20209. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Herzberg U, Eliav E, Bennett GJ, Kopin IJ. The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci Lett. 1997;221:157–60. doi: 10.1016/s0304-3940(96)13308-5. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999a;92:1171–5. doi: 10.1016/s0306-4522(99)00220-1. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999b;90:923–31. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- Huang SM, Strangman NM, Walker JM. Liquid chromatographic–mass spectrometric measurement of the endogenous cannabinoid 2-arachidonylglycerol in the spinal cord and peripheral nervous system. Acta Pharmacol Sin. 1999;20:1098–102. [PubMed] [Google Scholar]
- Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci. 2001;13:2105–14. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- Karst M, Salim K, Burstein S, Conrad I, Hoy L, Schneider U. Analgesic effect of the synthetic cannabinoid CT-3 on chronic neuropathic pain: a randomized controlled trial. JAMA. 2003;290:1757–62. doi: 10.1001/jama.290.13.1757. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Lawson SN. Neuropeptides in morphologically and functionally identified primary afferent neurons in dorsal root ganglia: substance P, CGRP and somatostatin. Prog Brain Res. 1995;104:161–73. doi: 10.1016/s0079-6123(08)61790-2. [DOI] [PubMed] [Google Scholar]
- Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85:493–502. doi: 10.1016/S0304-3959(00)00250-5. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Cook SA, Martin BR. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther. 1996;276:585–93. [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–8. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Martin BR. Spinal and supraspinal components of cannabinoid-induced antinociception. J Pharmacol Exp Ther. 1991;258:517–23. [PubMed] [Google Scholar]
- Lim G, Sung B, Ji RR, Mao J. Upregulation of spinal cannabinoid-1-receptors following nerve injury enhances the effects of Win 55,212-2 on neuropathic pain behaviors in rats. Pain. 2003;105:275–83. doi: 10.1016/s0304-3959(03)00242-2. [DOI] [PubMed] [Google Scholar]
- Lisney SJ. Regeneration of unmyelinated axons after injury of mammalian peripheral nerve. Q J Exp Physiol. 1989;74:757–84. doi: 10.1113/expphysiol.1989.sp003348. [DOI] [PubMed] [Google Scholar]
- Lisney SJ, Devor M. Afterdischarge and interactions among fibers in damaged peripheral nerve in the rat. Brain Res. 1987;415:122–36. doi: 10.1016/0006-8993(87)90275-7. [DOI] [PubMed] [Google Scholar]
- Lynn AB, Herkenham M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J Pharmacol Exp Ther. 1994;268:1612–23. [PubMed] [Google Scholar]
- Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, et al. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–64. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, McRoberts JA, Ennes HS, Song B, Wang X, Jinton L, et al. Two N-methyl-D-aspartate receptors in rat dorsal root ganglia with different subunit composition and localization. J Comp Neurol. 2002;446:325–41. doi: 10.1002/cne.10202. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Matsuka Y, Neubert JK, Maidment NT, Spigelman I. Concurrent release of ATP and SP within guinea pig trigeminal ganglia in vivo. Brain Res. 2001;915:248–55. doi: 10.1016/s0006-8993(01)02888-8. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, et al. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–95. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert JK, Maidment NT, Matsuka Y, Adelson DW, Kruger L, Spigelman I. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal ganglia in vivo. Brain Res. 2000;871:181–91. doi: 10.1016/s0006-8993(00)02440-9. [DOI] [PubMed] [Google Scholar]
- Notcutt W, Price M, Miller R, Newport S, Phillips C, Simmons S, et al. Initial experiences with medicinal extracts of cannabis for chronic pain: results from 34′N of 1′ studies. Anaesthesia. 2004;59:440–52. doi: 10.1111/j.1365-2044.2004.03674.x. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem. 2004;279:5298–305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci Lett. 1995;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben Shabat S, Hanus L, Breuer A, Mechoulam R, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–31. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther. 2003;306:880–8. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Cravatt BF. Proteins regulating the biosynthesis and inactivation of neuromodulatory fatty acid amides. Vitam Horm. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- Popper P, Micevych PE. Localization of calcitonin gene-related peptide and its receptors in a striated muscle. Brain Res. 1989;496:180–6. doi: 10.1016/0006-8993(89)91064-0. [DOI] [PubMed] [Google Scholar]
- Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–62. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid MH, Inoue M, Toda K, Ueda H. Loss of peripheral morphine analgesia contributes to the reduced effectiveness of systemic morphine in neuropathic pain. J Pharmacol Exp Ther. 2004;309:380–7. doi: 10.1124/jpet.103.060582. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Aanonsen L, Hargreaves KM. SR 141716A, a cannabinoid receptor antagonist, produces hyperalgesia in untreated mice. Eur J Pharmacol. 1997;319:R3–4. doi: 10.1016/s0014-2999(96)00952-1. [DOI] [PubMed] [Google Scholar]
- Ross RA, Coutts AA, McFarlane SM, Anavi-Goffer S, Irving AJ, Pertwee RG, et al. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: implications for antinociception. Neuropharmacol. 2001;40:221–32. doi: 10.1016/s0028-3908(00)00135-0. [DOI] [PubMed] [Google Scholar]
- Salio C, Fischer J, Franzoni MF, Conrath M. Pre- and postsynaptic localizations of the CB1 cannabinoid receptor in the dorsal horn of the rat spinal cord. Neuroscience. 2002;110:755–64. doi: 10.1016/s0306-4522(01)00584-x. [DOI] [PubMed] [Google Scholar]
- Sanudo-Pena MC, Strangman NM, Mackie K, Walker JM, Tsou K. CB1 receptor localization in rat spinal cord and roots, dorsal root ganglion, and peripheral nerve. Acta Pharmacol Sin. 1999;20:1115–20. [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–89. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Shaw G, Osborn M, Weber K. Reactivity of a panel of neurofilament antibodies on phosphorylated and dephosphorylated neurofilaments. Eur J Cell Biol. 1986;42:1–9. [PubMed] [Google Scholar]
- Sheen K, Chung JM. Signs of neuropathic pain depend on signals from injured nerve fibers in a rat model. Brain Res. 1993;610:62–8. doi: 10.1016/0006-8993(93)91217-g. [DOI] [PubMed] [Google Scholar]
- Shire D, Carillon C, Kaghad M, Calandra B, Rinaldi-Carmona M, Le Fur G, et al. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J Biol Chem. 1995;270:3726–31. doi: 10.1074/jbc.270.8.3726. [DOI] [PubMed] [Google Scholar]
- Silverman JD, Kruger L. Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. J Neurocytol. 1990;19:789–801. doi: 10.1007/BF01188046. [DOI] [PubMed] [Google Scholar]
- Singh TA, Santha P, Nagy I. Inflammatory mediators convert anandamide into a potent activator of the vanilloid type 1 transient receptor potential receptor in nociceptive primary sensory neurons. Neuroscience. 2005;136:539–48. doi: 10.1016/j.neuroscience.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–8. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Strangman NM, Patrick SL, Hohmann AG, Tsou K, Walker JM. Evidence for a role of endogenous cannabinoids in the modulation of acute and tonic pain sensitivity. Brain Res. 1998;813:323–8. doi: 10.1016/s0006-8993(98)01031-2. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, et al. Transacylase-mediated and phosphodiesterase-mediated synthesis of N-arachidonoylethanolamine, an endogenous cannabinoid-receptor ligand, in rat brain microsomes. Comparison with synthesis from free arachidonic acid and ethanolamine. Eur J Biochem. 1996;240:53–62. doi: 10.1111/j.1432-1033.1996.0053h.x. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci USA. 1999;96:12198–203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther. 2003;307:129–37. doi: 10.1124/jpet.103.051920. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun H, Della PK, Benz RJ, Xu J, Gerhold DL, et al. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–46. doi: 10.1016/s0306-4522(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Witting A, Weydt P, Hong S, Kliot M, Moller T, Stella N. Endocannabinoids accumulate in spinal cord of SOD1 G93A transgenic mice. J Neurochem. 2004;89:1555–7. doi: 10.1111/j.1471-4159.2004.02544.x. [DOI] [PubMed] [Google Scholar]
- Yoon YW, Na HS, Chung JM. Contributions of injured and intact afferents to neuropathic pain in an experimental rat model. Pain. 1996;64:27–36. doi: 10.1016/0304-3959(95)00096-8. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di MV, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–7. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]