

Abstract
Spontaneous autoimmune diabetes in nonobese diabetic (NOD) mice is the result of a CD4+ and CD8+ T cell-dependent autoimmune process directed against the pancreatic beta cells. CD8+ T cells play a critical role in the initiation and progression of diabetes, but the specificity and diversity of their antigenic repertoire remain unknown. Here, we define the structure of a peptide mimotope that elicits the proliferation, cytokine secretion, differentiation, and cytotoxicity of a diabetogenic H-2Kd-restricted CD8+ T cell specificity (NY8.3) that uses a T cell receptor α (TCRα) rearrangement frequently expressed by CD8+ T cells propagated from the earliest insulitic lesions of NOD mice (Vα17-Jα42 elements, often joined by the N-region sequence M-R-D/E). Stimulation of splenic CD8+ T cells from single-chain 8.3-TCRβ-transgenic NOD mice with this mimotope leads to preferential expansion of T cells bearing an endogenously derived TCRα chain identical to the one used by their islet-associated CD8+ T cells, which is also identical to the 8.3-TCRα sequence. Cytotoxicity assays using islet-derived CD8+ T cell clones from nontransgenic NOD mice as effectors and peptide-pulsed H-2Kd-transfected RMA-S cells as targets indicate that nearly half of the CD8+ T cells recruited to islets in NOD mice specifically recognize the same peptide/H-2Kd complex. This work demonstrates that beta cell-reactive CD8+ T cells mount a prevalent response against a single peptide/MHC complex and provides one peptide ligand for CD8+ T cells in autoimmune diabetes.
Development of autoimmune insulin-dependent diabetes (IDDM) in nonobese diabetic (NOD) mice is the result of an ill-defined CD4+ and CD8+ T cell-dependent process against the pancreatic beta cells (1, 2). Although the role of T cells as effectors of beta cell destruction in diabetes is well established, the nature of the antigens, cells, and mechanisms that initiate diabetogenesis in susceptible mice remains poorly understood.
Adoptive T cell transfer studies by using spleen cells from prediabetic NOD mice have shown that transfer of diabetes into immunodeficient NOD mice requires both CD4+ and CD8+ T cells (3–6). However, splenic CD4+ T cells from diabetic mice and preactivated beta cell-specific CD4+ T cell clones can home into pancreatic islets and kill beta cells in the absence of CD8+ T cells (6–9). Because beta cells do not express MHC class II molecules (10), it has been proposed that naive autoreactive CD4+ T cells differentiate into effector cells by engaging autoantigens shed from the beta cells by a prior insult, in the context of MHC class II molecules on local antigen-presenting cells (11, 12).
Studies of β2-microglobulin-deficient (β2 m−/−) and anti-CD8 mAb-treated NOD mice (13–16), which do not develop islet inflammation or diabetes, have suggested that the initial insult that triggers the shedding of beta cell autoantigens is mediated by beta cell-cytotoxic CD8+ T cells, which are consistently recruited to islets in NOD mice (17–20). Although there is evidence suggesting that initiation of IDDM is associated with recognition of the autoantigen glutamic acid decarboxylase (GAD65) by CD4+ T cells (21, 22), the need for CD8+ T cells in this process is supported by two additional key observations: that restoring expression of MHC class I molecules on beta cells of β2m−/− NOD mice restores insulitis susceptibility (23) and that splenocytes from prediabetic NOD mice cannot efficiently transfer insulitis into β2m−/− NOD.scid mice (24).
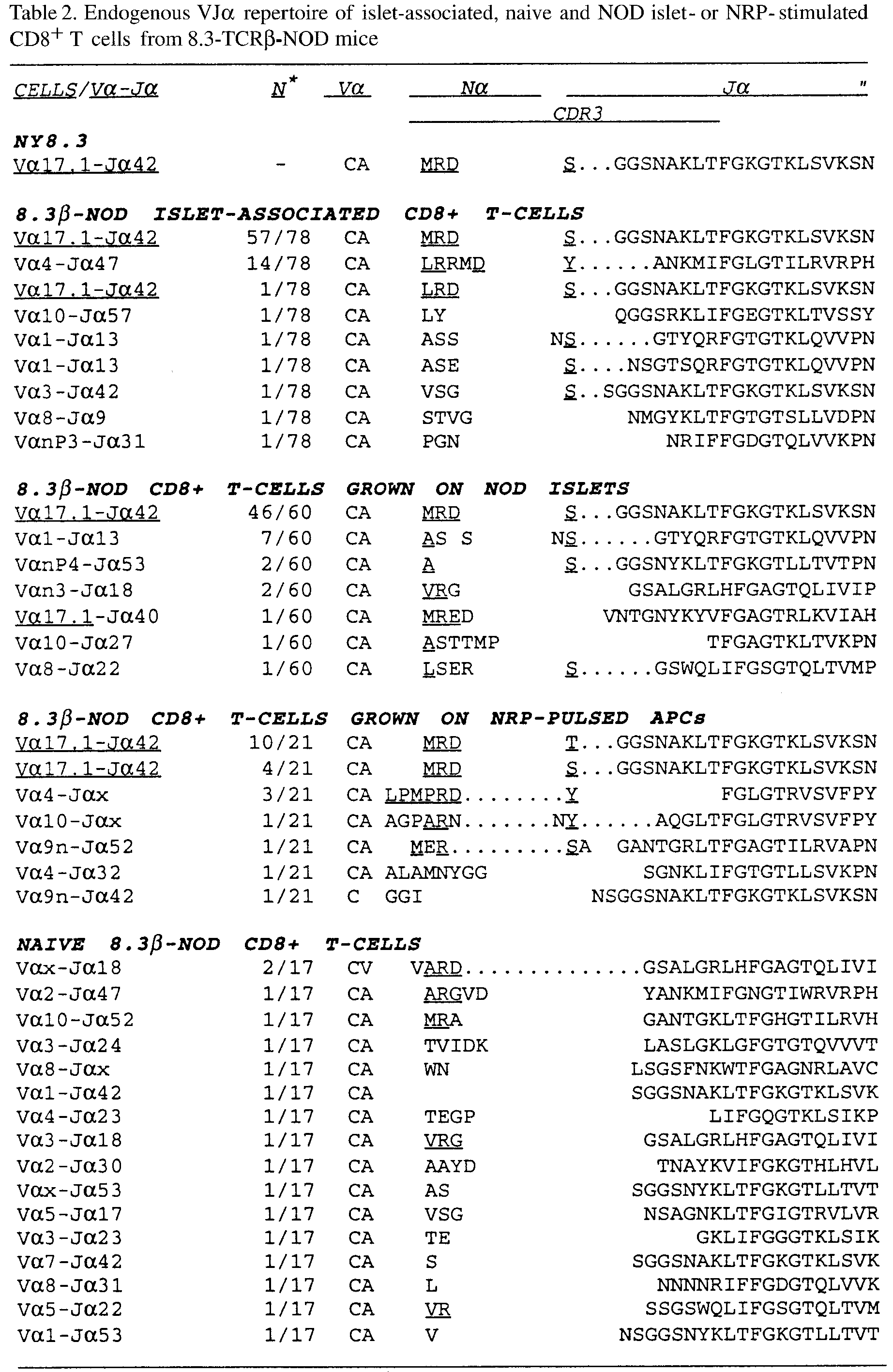
The antigenic specificity(ies) of the CD8+ T cells that are putatively involved in the initiation and progression of IDDM, however, is (are) unknown. Circumstantial lines of evidence have suggested that the antigenic repertoire of these T cells is very restricted. Most of the CD8+ T cells that can be isolated from islets of diabetic NOD mice are cytotoxic to beta cells in the context of H-2Kd and use T cell receptor (TCR)α chains with highly homologous complementarity determining region (CDR)-3 sequences (20). Furthermore, the majority of the islet-associated CD8+ T cells of transgenic NOD mice expressing the TCRβ chain of a CD8+ clonotype that uses a representative TCRα-CDR3 sequence (NY8.3) were found to express a TCRα chain sequence that was identical to the one used by the clonotype donating the TCRβ transgene (25). Strikingly, DiLorenzo et al. (26) have recently shown that a significant fraction of the CD8+ T cells that can be propagated from the earliest insulitic lesions of NOD mice use TCRα chains that are very similar, or even identical, to those used by the CD8+ T cells that we had previously isolated from diabetic NOD mice and 8.3-TCRβ-transgenic NOD mice (Vα17 and Jα42 elements joined by the N-region sequence M-R-D/E) (20, 25). Studies of 8.3-TCRαβ-transgenic NOD mice have demonstrated that this beta cell-reactive TCR heterodimer is in fact highly pathogenic, even in the absence of T cells bearing endogenous TCRs (27).
Taken together, these findings suggest that the CD8+ T cells that are recruited to islets in IDDM predominantly target a restricted set of local peptide/H-2Kd complexes, and that the CD8+ T cell response in diabetes is not subject to determinant spreading, a phenomenon commonly seen in CD4+ T cell and autoantibody responses in this and other autoimmune disorders (1, 2). To test this hypothesis, we have defined the structure of a peptide mimotope capable of eliciting the proliferation, cytokine secretion, differentiation, and cytotoxicity of naive CD8+ T cells from 8.3-TCRαβ-transgenic NOD mice, and have tested the ability of insulitic CD8+ T cells from NOD mice to recognize this peptide. We demonstrate that a large fraction of the islet-associated CD8+ T cell lines (87%) and clones (45%) recruited to pancreatic islets specifically recognize the same peptide/H-2Kd complex. We therefore propose that activation of the CD8+ T cells that contribute to the initiation and progression of IDDM is triggered by recognition of one peptide/MHC class I complex on beta cells or islet-associated antigen-presenting cells.
MATERIALS AND METHODS
Mice, Cell Lines, and Antibodies.
8.3-NOD, recombination activating gene-2 (RAG-2)-deficient 8.3-NOD, and 8.3-TCRβ-NOD mice, expressing the TCRαβ or TCRβ rearrangements of the H-2Kd-restricted beta cell-reactive CD8+ T cell clone NY8.3 have been described (25, 27). NOD/Lt and C57BL/6 mice were bred from stocks purchased from The Jackson Laboratory. RMA-SKd cells were from B. Wipke and M. Bevan (University of Washington, Seattle, WA). The KJ16 (anti-Vβ8.1/8.2) hybridoma was from P. Marrack (National Jewish Center, Denver, CO). The GK1.5 (anti-CD4) and 1411–2C11 (anti-CD3) hybridomas were from the ATCC. Anti-Lyt-2 (CD8α) (53–6.7), anti-L3T4 (IM7), anti-Vβ8.1/8.2 (MR5–2), anti-H-2Kd (SF1–1.1), and anti-H-2Db (KH95) were from PharMingen (San Diego, CA).
Peptides.
The peptide libraries were prepared by using multipin synthesis technology and fluorenylmethoxycarbonyl (Fmoc) chemistry (Chiron). Single peptides (Table 1) were purified by RP-HPLC to >90% purity and their sequence confirmed by ion spray mass spectrometry (Chiron). Peptides were diluted in RPMI medium 1640 containing 0.25% BSA.
Table 1.
Peptide sequences
| Protein | Sequence |
|---|---|
| NRP | KYNKANWFL |
| tum | KYQAVTTTL |
| A1 | AYNKANWFL |
| A3 | KYAKANWFL |
| A4 | KYNAANWFL |
| A6 | KYNKAAWFL |
| A7 | KYNKANAFL |
| A8 | KYNKANWAL |
| ST | KYEKSNWFL |
| LL | KYEKSNWFM |
| PH | KYSKEVWFV |
| ELS | DKGSNKGFE |
ST, Streptococcus thermophylus aminopeptidase C; LL, Lactobacilus lactis aminopeptidase C; PH, Pyrococcus horikoshii hypothetical protein
Generation of Spleen- and Islet-Derived CD8+ T Cells.
Spleen cells from 8.3-NOD mice were depleted of CD4+ T cells (25), adjusted to 104 CD8+ T cells/100 μl of complete medium (RPMI medium 1640 containing 10% fetal bovine serum), stimulated with irradiated NOD islets, peptide-pulsed (1 μg/ml) NOD splenocytes, or plate-bound anti-CD28 and anti-CD3 or anti-Vβ8.1/8.2 mAbs for 3–4 d, and expanded with or without recombinant IL-2 for 7–14 d (27). CD4+ T cell-depleted spleen cells from 8.3-TCRβ-NOD mice were grown by using a similar protocol, except that they were restimulated once (with peptides) or twice (with NOD islets). Islet-derived CD8+ T cells were generated within 2 d of diabetes onset as described (25) and used in cytotoxicity assays or cloned by limiting dilution within 6 d (25). Islet-derived beta cell-cytotoxic clones from 8.3-NOD mice were used to screen the peptide libraries. Clones from NOD/Lt mice were tested for peptide reactivity within 2 wk of plating. The average cloning efficiency was 8.37 ± 2%.
51Cr-Release Assays.
RMA-SKd cells were incubated at 26°C overnight, labeled with sodium [51Cr]chromate for 2 h at 26°C, and resuspended at 105 cells/ml in RPMI-1640, 0.25% BSA. Pancreatic islet cells were prepared from 5- to 8-wk-old NOD/Lt or C57BL/6 mice, labeled with 51Cr for 2 h at 37°C, and seeded at 104 cells/well. 51Cr-labeled RMA-SKd cells (104/well) were pulsed with 60 μg/ml (for libraries) or 1 μg/ml (for peptides) for 1 h at 37°C and used as targets in 51Cr-release assays. Effectors were added to each well in duplicate at different target/effector ratios. Cultures of RMA-SKd cells with peptides but no T cells were used as controls. None of the peptides were toxic to beta cells in the absence of cytotoxic T klymphocyte (CTL). The plates were incubated at 37°C for 8 h and the supernatants collected for determination of 51Cr release [% lysis = 100 × (test cpm − spontaneous cpm)/(total cpm − spontaneous cpm)].
H-2Kd-Stabilization Assay.
RMA-SKd cells that had been cultured overnight at 26°C were seeded, in quadruplicate, at 104 cells/well in 96-well plates, pulsed with peptides in RPMI-1640, 0.25% BSA for 1 h at 26°C, incubated at 37°C for 3 h, washed, stained with anti-H-2Kd-FITC or anti-H-2Db-FITC, and analyzed by flow cytometry. Data were calculated by subtracting the mean fluorescence intensity for H-2Kd or H-2Db on nonpeptide-pulsed cells from that on peptide-pulsed cells.
Proliferation Assays.
Splenic CD8+ T cells (2 × 104/well) were incubated, in triplicate, with irradiated NOD islet cells or peptide-pulsed splenocytes in 96-well plates for 3 d at 37°C, pulsed with 1 μCi (1 Ci = 37 GBq) of [3H]-thymidine during the last 18 h of culture, and harvested (27).
Cytokine Secretion.
Splenic CD8+ T cells (2 × 104/well) were incubated with irradiated NOD islet cells (105/well) or peptide-pulsed (1 μg/ml) splenocytes for 48 h at 37°C and the supernatants assayed for IL-2, IL-4, and IFN-γ by using ELISA kits (Genzyme).
Peptide-Induced Differentiation of CTL Precursors (CTLp) into CTL.
Naive splenic T cells from RAG-2−/− 8.3-NOD mice (105/well) were incubated with peptide-pulsed (1 μg/ml) splenocytes, incubated for 7 d at 37°C, and tested for cytotoxicity against islet cells or peptide-pulsed RMA-SKd cells.
Cloning and Sequencing of TCRα mRNAs.
The TCRα cDNAs of NRP- and islet-stimulated CD8+ T cells from 8.3-TCRβ-NOD mice were amplified by anchor-PCR, cloned, and sequenced as described (20). Islet-stimulated lines were analyzed after two restimulations (at 7-d intervals). Peptide-stimulated lines were studied after one restimulation (with peptide-pulsed NOD splenocytes 10 d after priming).
Statistical Analyses.
Data was compared by Mann–Whitney u test, Student’s t test, or χ2.
RESULTS
NRP, a Mimotope Targeted by a Diabetogenic H-2Kd-Restricted TCR.
To test the hypothesis that the CD8+ T cells putatively involved in the initiation and/or progression of IDDM target a restricted set of autoantigenic determinants on beta cells, it was necessary to determine the nature of peptides targeted by representative CD8+ T cells. To do this, we first attempted to expand H-2Kd-restricted beta cell-cytotoxic CD8+ T cell clones expressing the appropriate TCRα-CDR3 sequences [i.e., with the motif: hydrophobic amino acid-R-D/E or G/N-Y/S (20)] from islets of diabetic NOD mice. Unfortunately, all these clones proliferated slowly, had brief life spans in vitro, and tended to lose cytotoxic activity shortly after isolation (20). This limitation was overcome by using islet-associated CD8+ T cells from 8.3-NOD mice as an indefinite source of clonotypic CTLp. 8.3-NOD mice express a transgenic H-2Kd-restricted beta cell-specific TCR (27) that uses a TCRα rearrangement that, in addition to bearing the canonical CDR3 sequence, is very similar or even identical to TCRα sequences rearranged by a significant fraction of the CD8+ T cells isolated from the earliest insulitic lesions of NOD/Lt mice (Vα17-Jα42 elements often joined by the N-region sequence M-R-D/E) [the current nomenclature for the Vαn1 and Jα34 elements of the 8.3-TCRα chain is Vα17 and Jα42, respectively (28)] (26).
Initial experiments with GAD65 cDNA-transfected and insulin peptide-pulsed targets suggested that the 8.3-TCR does not recognize any of these two primary beta cell autoantigens (unpublished results). Because repeated attempts to define the structure of this TCR’s target antigen by cDNA expression cloning were unsuccessful, we used positional scanning combinatorial peptide libraries (29) to find an agonistic ligand for this TCR. Because H-2Kd-binding peptides are nonamers and are usually, albeit not always, anchored on H-2Kd molecules by Y and L at positions 2 and 9, respectively (30), we designed libraries in which all the peptides were nonamers and had these anchor residues. To generate the libraries, we divided 18 amino acids (all but C and M) into groups of one or two based on the chemical nature of their side groups. We then made 72 libraries in which all the peptides per library had one of two possible amino acids at individual nonanchor positions and a random assortment of 19 amino acids (except C) at the other six positions. Each library (9.4 × 107 peptides) was then tested (at 60 μg/ml) for its ability to sensitize 51Cr-labeled RMA-SKd cells for lysis by islet-derived 8.3-CD8+ CTL clones selected for high proliferative and beta cell-cytotoxic activity. RMA-SKd cells are H-2Kd cDNA-transfected cells that lack the transporter associated with antigen-processing. When incubated at 26°C, these cells transport unstable empty MHC class I molecules to the surface, where they can bind exogenous peptides (31). Because chromium release assays are very sensitive and lysis of targets by CD8+ CTL usually requires recognition of ≤100 peptide/MHC class I complexes (32), 60 μg/ml of a target peptide-containing library was expected to contain sufficient target peptide (3 × 107 molecules) to sensitize RMA-SKd cells for 8.3-CD8+ CTL-induced lysis, assuming that this was not a saturating concentration. In that case, the amino acids occupying the fixed positions in libraries eliciting CD8+ CTL-induced lysis can be regarded as those occupying these positions in the agonistic peptide.
These experiments, which were repeated up to five times, resulted in the identification of candidate residues for six of the seven nonanchor positions of a target peptide. Fig. 1 shows the results of one experiment. Only one library per position was able to reproducibly elicit the cytotoxicity of 8.3-CD8+ CTL; the residues occupying these positions in target peptide-containing libraries are shown at the top in Fig. 1. No residues could be defined for position 5, suggesting that the residue occupying this position in the peptide is either C or M or does not interact with the TCR. Screening of the libraries at lower concentrations gave comparable results, but the differences between positive and negative libraries were smaller. The negative chromium release values obtained with some libraries in the experiment shown in Fig. 1 were not reproducible and were probably caused by consumption of spontaneously released chromium by the effector CTL, which was an actively proliferating clone. Taken together, these results suggested that 8.3-CD8+ CTL target one or more peptides with the following sequence: (K/R)-Y-(N/Q)-(K/R)-X-(N/Q)-(W/I)-(F/Y)-L. Using this information, we designed 12 additional libraries, two for each of the five nonanchor positions defined by using the first library set. All the peptides in each of these libraries had one of the two candidate residues per position and a random assortment of 19 different amino acids at the five remaining nonanchor positions. We then investigated which of these libraries (at 60 μg/ml) were able to sensitize RMA-SKd cells for 8.3-CD8+ CTL-induced cytotoxicity. Because of the relatively high degree of background chromium release obtained with these libraries in initial experiments, we repeated these assays using a negative control (tumor-derived) H-2Kd-binding peptide [tum (33); at 60 μg/ml] as a reference. As shown in Fig. 2, only one library per position yielded 51Cr-release values two SDs above those induced by this negative control. The sequence of the peptide targeted by 8.3-CD8+ CTL was therefore deduced to be K-Y-N-K-X-N-W-F-L and was designated NRP. For subsequent experiments, position 5 was assumed to be occupied by A.
Figure 1.

Screening of the first-generation peptide libraries. 51Cr-labeled RMA-SKd cells were incubated with 60 μg/ml of each of the 72 first-generation libraries at 26°C for 1 h and then challenged with 8.3-CD8+ CTL clones at a 1:10 target/effector ratio for 8 h at 37°C. The amino acids/position that consistently elicited 8.3-CTL-induced lysis of peptide-pulsed targets are shown at the top.
Figure 2.

Screening of the second-generation peptide libraries. RMA-SKd cells were pulsed with each of the 12-sec generation libraries or tum and tested in cytotoxicity assays as in Fig. 1. The amino acids that elicited 51Cr-release values >2 SD above those elicited by tum (horizontal line) are shown at the top. NT, not tested.
Immunological Properties of NRP.
We next asked whether NRP had the same agonistic properties as the natural NOD beta cell autoantigen on naive 8.3-CD8+ T cells. As shown in Fig. 3 (A–C Left), naive splenic CD8+ T cells from 8.3-NOD mice (8.3-CD8+ CTLp) proliferated, secreted IFN-γ (but very low levels of IL-2 and no IL-4; data not shown), and differentiated into T cells capable of killing NRP-pulsed (but not tum-pulsed) RMA-SKd cells, in response to stimulation with irradiated NOD islet cells. Stimulation of 8.3-CD8+ CTLp with NRP-pulsed NOD splenocytes resulted in qualitatively similar responses; as shown in Fig. 3 A and B Right, 8.3-CD8+ CTLp proliferated vigorously and secreted high levels of IFN-γ and some IL-2 in response to NRP-pulsed NOD splenocytes. Because the amount of NRP used in these experiments was probably much greater than the amount of beta cell peptide displayed on islet cells, it is not surprising that NRP was more efficient than islets at inducing the different T cell responses. These responses were NRP-specific and 8.3-TCR-driven because they did not occur when using tum-pulsed NOD splenocytes as antigen-presenting cells or splenic CD8+ T cells from nontransgenic NOD mice as responders. Finally, stimulation of 8.3-CD8+ CTLp from RAG-2−/− 8.3-NOD mice with NRP-pulsed NOD splenocytes for 7 d in the absence of recombinant IL-2 resulted in their differentiation into cells capable of killing NOD islet cells (H-2Kd) and NRP-pulsed RMA-SKd cells, but not C57BL/6 islet cells (H-2Kb) or tum-pulsed RMA-SKd cells (Fig. 3C Center and Right, respectively). A similar pattern of cytotoxicity was obtained when the effectors were 8.3-CD8+ CTL grown on NOD islet cells in the presence of IL-2 (Fig. 3C Left). Thus, like the natural NOD islet cell antigen, NRP is a powerful agonist of the 8.3-TCR.
Figure 3.

Immunological properties of NRP. (A) Proliferation of CTLp from 8.3-NOD and NOD/Lt mice in response to NOD islet-cells (Left) or NOD splenocytes pulsed with NRP or tum (Right) (P < 0.05 for islet cell- or NRP- vs. tum-induced proliferation of 8.3-CTLp, and NRP-induced proliferation of 8.3-CTLp vs. NOD/Lt T cells). (B) Cytokine secretion by 8.3-CTLp in response to NOD islet cells or splenocytes pulsed with 1 μg/ml of NRP or tum (Center and Right) (P < 0.009 for IFN-γ/IL-2 secretion induced by NRP vs. tum). (C) Differentiation of 8.3-CTLp from RAG-2−/− 8.3-NOD mice into CTL. The panel shows 51Cr-release assays by using NRP- or tum-pulsed RMA-SKd cells (Left and Center) or NOD or B6 islet cells as targets, at a 1:10 target/effector ratio (Right) (P < 0.05 for NRP vs. tum reactivity or NOD vs. B6 islet cell reactivity).
TCRα Repertoire of NRP-Stimulated CD8+ Splenocytes from 8.3-TCRβ-NOD Mice.
We next asked whether in vitro stimulation of splenic CD8+ T cells from single-chain transgenic 8.3-TCRβ-NOD mice with NRP would be able to elicit the growth of T cells displaying the highly restricted TCRα repertoires exhibited by the islet-associated (but not peripheral) CD8+ T cells of these mice (most express the 8.3-TCRα sequence) (25). A large fraction of the TCRα sequences used by NRP-stimulated splenic CD8+ T cells from 8.3-TCRβ-NOD mice (one restimulation) were identical to the 8.3-TCRα sequence (14/21 TCRα cDNAs). The same was true when the splenic CD8+ T cells of these mice were grown on irradiated NOD islet cells (two restimulations; 46/60 cDNAs) (see the supplemental data on the PNAS web site, www.pnas.org). This dramatic skewing of the repertoire was induced by NRP or NOD islet cell antigen, respectively, because it was not observed in unstimulated splenic CD8+ T cells from 8.3-TCRβ-NOD mice (0/21 TCRα cDNAs) (see the supplemental data). These results therefore demonstrated that NRP elicits the growth of the same CD8+ clonotypes that are induced to proliferate, both in vitro and in vivo, by the natural beta cell autoantigen.
Immunological Properties of Synthetic and Naturally Occurring NRP Analogs.
Next, we investigated which, if any, of the nonanchor residues of NRP were essential for its immunological properties. To do this, we generated NRP analogs carrying A substitutions at each of the nonanchor positions, except position 5 (already an A) and studied their MHC-binding and 8.3-TCR agonistic properties. We first compared the ability of NRP analogs, NRP, tum, and a non-Kd-binding peptide (ELS) (Table 1) to stabilize Kd or Db molecules on RMA-SKd cells, as determined by flow cytometry. As shown in Fig. 4A Left, the mean fluorescence intensity (mfi) values for Kd on NRP analog-, NRP-, and tum-pulsed RMA-SKd cells were significantly greater than those on ESP-pulsed RMA-SKd cells. These differences were specific for Kd, because the mfi values for Db on peptide-pulsed RMA-SKd cells were not different than baseline values (Fig. 4A Right).
Figure 4.

Alanine-scanning mutagenesis of NRP. (A) H-2Kd-stabilization assay. RMA-SKd cells were pulsed with peptides, stained with anti-Kd or anti-Db mAbs, and analyzed by flow cytometry (P < 0.02 for Kd-binding of all peptides vs. ESP). Data shown correspond to cells pulsed with 1 μg/ml of peptide, but the different peptides displayed comparable differences in their ability to stabilize Kd molecules at lower concentrations. (B) Proliferation of 8.3-CD8+ CTLp in response to peptide-pulsed NOD splenocytes [P < 0.05 for analog peptide- (except A4 and A8) vs. tum-induced]. (C and D) Secretion of IFN-γ (C) and IL-2 (D) by 8.3-CTLp in response to peptide-pulsed NOD splenocytes [P < 0.009 for analog peptide- (all except A4 and A8) vs. tum-induced IFN-γ secretion; P < 0.009 for A1, A6 and A7 vs. tum-induced IL-2 secretion]. (E) Cytotoxicity of NRP-differentiated 8.3-CTL against peptide-pulsed RMA-SKd cells at a 1:10 target/effector ratio (P < 0.05 for A1-, A3-, A6-, and A7- vs. tum-induced).
We then compared the ability of each NRP analog to elicit the proliferation of, and cytokine secretion by, 8.3-CD8+ CTLp. As shown in Fig. 4 B–D, introduction of A substitutions at five of six positions in NRP caused variable reductions in the peptide’s ability to trigger the proliferation of, and IFN-γ and IL-2 secretion by, 8.3-CD8+ CTLp: the A4 and A8 analogs induced no responses in these assays, whereas the A1, A3, and A6 analogs induced partial responses. Cytotoxicity assays using peptide-pulsed RMA-SKd cells as targets and NRP-activated 8.3-CD8+ CTL as effectors revealed that, with the exception of the A3, A4, and A8 analogs, which were not recognized at all (A4 and A8) or only partially (A3), the A1, A6, and A7 analogs were almost as efficient as NRP at eliciting the cytotoxic activity of 8.3-CD8+ CTL (Fig. 4E). It thus appears that the agonistic activity of NRP is highly susceptible to amino acid substitutions at five of its seven nonanchor positions. These data suggest that N at position 3, K at position 4, and F at position 8 are major 8.3-TCR-contact residues, and that K at position 1 and N at position 6 are secondary 8.3-TCR contact residues. Database searches yielded three naturally occurring NRP-homologous peptides (ST, LL, and PH, all from bacteria, Table 1), but none of these peptides was able to elicit 8.3-CD8+ T cell activation (not shown).
NRP Reactivity of Islet-Derived CD8+ T Cells from NOD/Lt Mice.
Once we had shown that NRP was a full agonist of the 8.3-TCR, we proceeded to test the hypothesis that the CD8+ T cell response in spontaneous diabetes in NOD/Lt mice is predominantly directed against one peptide/MHC class I complex. We asked whether CD8+ T cell lines propagated from islets of eight acutely diabetic NOD/Lt mice (in recombinant IL-2 for ≤8 d, to prevent the selective expansion of individual clonotypes) could recognize NRP. As shown in Fig. 5A, most of these lines [seven of eight (87.5%), Fig. 5C] killed NRP-, but not tum-pulsed, RMA-SKd cells in vitro. The NRP reactivity of these lines was not caused by nonspecific recognition of the peptide by irrelevant TCRs, because polyclonal CD8+ and Vβ8.1+CD8+ CTL lines derived from NOD spleens killed neither NRP- nor tum-pulsed targets (Fig. 5A). This observation indicated that the islet-associated CD8+ T cells of most NOD/Lt mice contain clonotypes capable of recognizing the NRP/Kd complex. We then generated 31 CD8+ CTL clones from five CTL lines propagated from islets of nine additional acutely diabetic NOD mice (one or two mice per line) and tested all these clones for their ability to kill NRP- or tum-pulsed RMA-SKd cells (limiting dilution assays are not feasible here because the number of CD8+ CTL that can be isolated from islets of single diabetic mice is often very low). Of the 31 clones tested (at a 1:1 target/effector ratio), 14 were cytotoxic against NRP-pulsed, but not tum-pulsed, RMA-SKd targets (Fig. 5B). The overall percentage of NRP-reactive clones per line was 45% [+clones/line: 2/4, 5/12, 1/7, 2/4, and 4/4] (Fig. 5C). These results thus demonstrate that CD8+ T cells mount a prevalent response against one peptide/MHC class I complex in spontaneous IDDM.
Figure 5.

NRP reactivity of islet-associated CD8+ T cells from NOD/Lt mice. (A) NRP and tum reactivity of islet- and spleen-derived CD8+ T cell lines from 8.3-NOD and/or NOD/Lt mice. CD3-activated and Vβ8.1-act. are control CD8+ CTLs generated by stimulation of NOD splenic CD8+ T cells with plate-bound anti-CD28 and anti-CD3 or anti-Vβ8.1/8.2 mAbs. The figure shows results of cytotoxicity assays obtained with one islet line, two splenic lines, and the average values obtained with seven NRP-reactive and one nonreactive islet lines (at a 1:10 target/effector ratio) (P < 0.03). The tum reactivity of the +lines was caused by one line that displayed some cytotoxicity against tum-pulsed targets. (B) NRP reactivity of islet-derived CD8+ T cell clones from NOD/Lt mice. Assays were done at a ≈1:1 target/effector ratio. A clone was defined as positive if it triggered 51Cr-release values from NRP-pulsed targets at least 2 SD above those triggered by tum-pulsed targets. The figure shows results of assays obtained with one clone and average values corresponding to 14 NRP-reactive clones (P < 0.004 for NRP vs. tum reactivity) and 17 non-NRP-reactive clones. (C) Percentage of NRP-reactive CTL lines and clones from NOD/Lt mice (P < 0.0001 for % of NRP- vs. tum-reactive clones).
DISCUSSION
Although the precise sequence of events leading to IDDM in humans and mice remains ill defined, a growing body of evidence suggests that initiation of the disease requires both CD8+ and CD4+ T cells (13, 16, 23, 24). Temporal studies have shown that the CD4+ T cells involved in the early stages of the disease process recognize a few epitopes on the autoantigen GAD65 and that, as the disease progresses, this response quickly spreads to other epitopes of this and other beta cell autoantigens (21, 22). The antigenic specificity and repertoire diversity of the CD8+ T cells putatively involved in the initiation and progression of IDDM, however, are unknown. Here, we have defined a peptide mimotope that is recognized by a non-GAD65/noninsulin-reactive highly diabetogenic H-2Kd-restricted TCR that is beta cell-specific and uses a TCRα rearrangement frequently used by CD8+ T cells propagated from the earliest insulitic lesions of NOD/Lt mice (27). Using this mimotope as target antigen in cytotoxicity assays, we have shown that a large fraction of the islet-associated CD8+ T cells in diabetic NOD/Lt mice recognize the same peptide/H-2Kd complex. These results therefore demonstrate that CD8+ T cells mount a prevalent response against one peptide/H-2Kd complex in IDDM and provide a valuable new reagent for understanding the biology of pathogenic autoreactive CD8+ T cells in autoimmunity.
Circumstantial lines of evidence have previously suggested that the antigenic repertoire of islet-associated CD8+ T cells at the onset of both insulitis and diabetes in NOD mice is very restricted. We found that most of the CD8+ clonotypes that we could propagate from islets of prediabetic and acutely diabetic NOD/Lt mice were cytotoxic to beta cells in the context of H-2Kd and that these cells used TCRα chains with highly homologous CDR3 sequences (20). Although we found no evidence for restricted Vβ, Vα, Jβ, or Jα gene usage in this study, we noticed that most of the TCRα-CDR3 sequences used by these cells encoded the N-region-encoded motif: hydrophobic residue-R-D/E or N/G-Y/S (20). It is noteworthy that the TCRα chains expressed by CD8+ T cells derived from young prediabetic NOD/Lt mice preferentially used the N-region-encoded M/A-R-D/E motif in the CDR3 region (20). Importantly, DiLorenzo et al. (26) have recently shown that a significant fraction of the CD8+ T cells that can be propagated from the earliest insulitic lesions of NOD/Lt mice (at 4–6 wk of age) also use this N-region encoded motif in the CDR3 of their TCRα chains (7 of 16 TCRα chains used the M-R motif and 5 of these used the M-R-E/D motif). In fact, a significant number of the clonotypes described by DiLorenzo et al. (i.e., 4 of 16 TCRα chains) used a TCRα rearrangement that is very similar, or even identical, to TCRα rearrangements that we had previously cloned from CD8+ T cells isolated from several different NOD/Lt mice (Vα17-M-R-E/D-Jα42) (19, 20). Because NRP was defined by using a clonotype expressing a Vα17-M-R-D-Jα42 TCRα rearrangement, it is tempting to speculate that these early insulitic CD8+ T cells were also NRP reactive, although this remains to be determined.
The hypothesis that NRP-reactive CD8+ T cells using the Vα17-M-R-D-Jα42 TCRα rearrangement might be among those triggering the onset of IDDM (perhaps in concert with GAD65- or insulin-specific CD4+ T cells) is consistent with three other observations. First, transgenic NOD mice expressing an NRP-reactive TCR that uses this TCRα rearrangement (8.3-NOD mice) develop a dramatically accelerated form of diabetes (27). Second, this transgenic TCR can trigger the onset of diabetes in RAG-2−/− 8.3-NOD mice, which express a monoclonal repertoire completely devoid of MHC class II-restricted TCR specificities (27). And third, the vast majority of the islet-associated CD8+ T cells of single-chain 8.3-TCRβ-transgenic NOD mice express endogenously derived TCRα chains with the Vα17-M-R-D-Jα42 sequence (25). Importantly, experiments reported here have demonstrated that this dramatic skewing of the intra-islet CD8+ T cell repertoire in these 8.3-TCRβ-transgenic NOD mice is indeed induced by a single peptide/H-2Kd complex: short-term in vitro stimulation of the peripheral CD8+ T cells of these mice (which express a heterogeneous TCRα repertoire) with either NOD islets or NRP selectively induced the growth of clonotypes bearing the Vα17-M-R-D-Jα42 rearrangement. Because productive collaboration between CD4+ T-helper cells and CD8+ CTLp does not require recognition of epitopes from the same antigen (34), this hypothesis that NRP-reactive clonotypes may play a critical role in disease initiation is perfectly compatible with a primary role for GAD65 and insulin in this process (21, 22, 35): disease-initiating CD4+ and CD8+ T cells may very well collaborate by targeting peptides derived from different autoantigens.
Although we have evidence suggesting that NRP-reactive CD8+ T cells do not recognize peptides from at least two primary beta cell autoantigens, GAD65 and insulin, the nature of the autoantigen donating NRP or a related sequence remains unknown. Peptide sequences deduced by screening combinatorial peptide libraries have usually been remarkably close to naturally occurring peptides (36, 37), but we cannot assume that the NRP sequence is identical or even similar to the naturally occurring autoantigen. Nevertheless, NRP exhibits the very same immunological properties as the natural islet cell autoantigen (proliferation, cytokine secretion, cytotoxicity, and TCRα repertoire selection). In addition, single amino acid substitutions at three of the peptide’s seven nonanchor residues completely abrogate most of its immunological properties, suggesting that the residues occupying these positions are the same ones occupying these positions in the natural antigen.
In conclusion, we have shown that in NOD mice, CD8+ T cells mount a prevalent response against one peptide/MHC class I complex on beta cells. This polarization of the beta cell autoreactive CD8+ T cell response in IDDM is in striking contrast with the TCR heterogeneity of islet-reactive CD4+ T cells isolated from NOD mice, which do not show skewed TCR usage, use heterogeneous CDR3 sequences, and undergo determinant spreading as the disease progresses (1, 2, 38). Availability of NRP, the first peptide ligand for diabetogenic CD8+ T cells, should facilitate studies on the precise role and developmental biology of these T cells in spontaneous IDDM in NOD mice.
Supplementary Material
Acknowledgments
We thank M. Bevan, P. Marrack, B. Wipke, and T. Utsugi for reagents; S. Bou, S. Culp, and K. Rouleau for technical assistance; L. Bryant for flow cytometry; R. Dawson for animal care; and Y. Yang for reading the manuscript. This work was supported by the Natural Sciences and Engineering Research Council of Canada and by the Medical Research Council of Canada. J.V. is supported by a Canadian Diabetes Association Fellowship. P.S. is a Senior Scholar of the Alberta Heritage Foundation for Medical Research (Edmonton, Alberta, Canada).
ABBREVIATIONS
- IDDM
insulin-dependent diabetes
- CDR
complementarity-determining region
- CTL
cytotoxic T lymphocyte
- CTLp
CTL precursors
- NOD
nonobese diabetic
- TCR
T cell receptor
- tum
tumor-derived H-2Kd-binding peptide
References
- 1.Tisch R, McDevitt H O. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 2.Delovitch T L, Singh B. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 3.Bendelac A, Carnaud C, Boitard C, Bach J-F. J Exp Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller B J, Appel M C, O’Neil J J, Wicker L S. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 5.Yagi H, Matsumoto M, Kunimoto K, Kawaguchi J, Makino S, Harada M. Eur J Immunol. 1992;22:2387–2393. doi: 10.1002/eji.1830220931. [DOI] [PubMed] [Google Scholar]
- 6.Christianson S W, Shultz L D, Leiter E H. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 7.Haskins K, McDuffie M. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 8.Katz J D, Benoist C, Mathis D. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 9.Peterson J D, Haskins K. Diabetes. 1996;45:328–336. doi: 10.2337/diab.45.3.328. [DOI] [PubMed] [Google Scholar]
- 10.McInerney M F, Rath S, Janeway C A., Jr Diabetes. 1991;40:648–651. doi: 10.2337/diab.40.5.648. [DOI] [PubMed] [Google Scholar]
- 11.Serreze D V, Leiter E H. Curr Opin Immunol. 1994;6:900–906. doi: 10.1016/0952-7915(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 12.Wong F S, Visintin I, Wen L, Flavell R A, Janeway C A., Jr J Exp Med. 1996;183:67–76. doi: 10.1084/jem.183.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz J, Benoist C, Mathis D. Eur J Immunol. 1993;23:3358–3360. doi: 10.1002/eji.1830231244. [DOI] [PubMed] [Google Scholar]
- 14.Wicker L S, Leiter E H, Todd J A, Renjilian R J, Peterson E, Fisher P A, Podolin P L, Zijlstra M, Jaenisch R, Peterson L B. Diabetes. 1994;43:500–504. doi: 10.2337/diab.43.3.500. [DOI] [PubMed] [Google Scholar]
- 15.Serreze D V, Leiter E H, Christianson G J, Greiner D, Roopenian D C. Diabetes. 1994;43:505–508. doi: 10.2337/diab.43.3.505. [DOI] [PubMed] [Google Scholar]
- 16.Wang B, Gonzalez A, Benoist C, Mathis D. Eur J Immunol. 1996;26:1762–1769. doi: 10.1002/eji.1830260815. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa M, Yokono K, Nagata M, Hatamori N, Ogawa W, Miki A, Mizoguti H, Baba S. Diabetes. 1991;40:1210–1217. doi: 10.2337/diab.40.9.1210. [DOI] [PubMed] [Google Scholar]
- 18.Nagata M, Yoon J-W. Diabetes. 1992;41:998–1008. doi: 10.2337/diab.41.8.998. [DOI] [PubMed] [Google Scholar]
- 19.Nagata M, Santamaria P, Kawamura T, Utsugi T, Yoon J-W. J Immunol. 1994;152:2042–2050. [PubMed] [Google Scholar]
- 20.Santamaria P, Utsugi T, Park B-J, Averill N, Yoon J-W. J Immunol. 1995;154:2494–2503. [PubMed] [Google Scholar]
- 21.Kaufman D, Clare-Salzler M, Tian J, Forsthuber T, Ting G, Robinson P, Atkinson M, Sercarz E, Tobin A, Lehmann P. Nature (London) 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tisch R, Yang X-D, Singer S, Liblau R, Fugger L, McDevitt H O. Nature (London) 1993;366:72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 23.Kay T W H, Parker J L, Stephens L A, Thomas H E, Allison J. J Immunol. 1996;157:3688–3693. [PubMed] [Google Scholar]
- 24.Serreze D V, Chapman H D, Varnum D S, Gerling I, Leiter E H, Shultz L D. J Immunol. 1997;158:3978–3986. [PubMed] [Google Scholar]
- 25.Verdaguer J, Yoon J-W, Anderson B, Averill N, Utsugi T, Park B-J, Santamaria P. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 26.DiLorenzo T P, Graser T R, Ono T, Christianson G J, Chapman H D, Roopenian D C, Nathenson S G, Serreze D V. Proc Natl Acad Sci USA. 1998;95:12538–12542. doi: 10.1073/pnas.95.21.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. J Exp Med. 1997;186:1663–1676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koop B F, Rowen L, Wang K, Kuo C L, Seto D, Lenstra J A, Howard S, Shan W, Deshpande P, Hood L. Genomics. 1994;19:478–493. doi: 10.1006/geno.1994.1097. [DOI] [PubMed] [Google Scholar]
- 29.Blake J, Johnson J V, Hellstrom K E, Marquardt H, Chen L. J Exp Med. 1996;184:121–130. doi: 10.1084/jem.184.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelhard V H. Curr Opin Immunol. 1994;6:13–23. doi: 10.1016/0952-7915(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 31.Ljunggren H-G, Stam N J, Öhlden C, Neefjes J J, Höglund P, Heemels M T, Bastin J, Schumacher T N M, Townsend A, Kärre K, et al. Nature (London) 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 32.Christinck E R, Luscher M A, Barber B H, Williams D B. Nature (London) 1991;352:67–70. doi: 10.1038/352067a0. [DOI] [PubMed] [Google Scholar]
- 33.Wallny H J, Deres K, Faath S, Jung G, Van Pel A, Boon T, Ramensee H-G. Int Immunol. 1992;4:1085–1090. doi: 10.1093/intimm/4.10.1085. [DOI] [PubMed] [Google Scholar]
- 34.Stuhler G, Walden P. Eur J Immunol. 1993;23:2279–2286. doi: 10.1002/eji.1830230934. [DOI] [PubMed] [Google Scholar]
- 35.Wegmann D R, Norbury-Glaser M, Daniel D. Eur J Immunol. 1994;24:1853–1858. doi: 10.1002/eji.1830240820. [DOI] [PubMed] [Google Scholar]
- 36.Hiemstra H S, van Veelen P A, Schloot N C, Geluk A, van Meijgaarden K E, Willemen S J M, Leunissen J A M, Benckhuijsen W E, Amons R, de Vries R R P, et al. J Immunol. 1998;161:4078–4082. [PubMed] [Google Scholar]
- 37.Gundlach B R, Wiesmüller K -H, Junt T, Kienle S, Jung G, Walden P. J Immunol Methods. 1996;192:149–155. doi: 10.1016/0022-1759(96)00040-3. [DOI] [PubMed] [Google Scholar]
- 38.Candeias S, Katz J, Benoist C, Mathis D, Haskins K. Proc Natl Acad Sci USA. 1991;88:6167–6172. doi: 10.1073/pnas.88.14.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}